

Abstract
The Escherichia coli AlkB protein and human homologs hABH2 and hABH3 are 2-oxoglutarate (2OG)/Fe(II)-dependent DNA/RNA demethylases that repair 1-methyladenine and 3-methylcytosine residues. Surprisingly, hABH1, which displays the strongest homology to AlkB, failed to show repair activity in two independent studies. Here, we show that hABH1 is a mitochondrial protein, as demonstrated using fluorescent fusion protein expression, immunocytochemistry, and Western blot analysis. A fraction is apparently nuclear and this fraction increases strongly if the fluorescent tag is placed at the N-terminal end of the protein, thus interfering with mitochondrial targeting. Molecular modeling of hABH1 based upon the sequence and known structures of AlkB and hABH3 suggested an active site almost identical to these enzymes. hABH1 decarboxylates 2OG in the absence of a prime substrate, and the activity is stimulated by methylated nucleotides. Employing three different methods we demonstrate that hABH1 demethylates 3-methylcytosine in single-stranded DNA and RNA in vitro. Site-specific mutagenesis confirmed that the putative Fe(II) and 2OG binding residues are essential for activity. In conclusion, hABH1 is a functional mitochondrial AlkB homolog that repairs 3-methylcytosine in single-stranded DNA and RNA.
Alkylating compounds are ubiquitous and modify cellular macromolecules such as DNA and RNA. In the human environment, tobacco-specific nitrosamines are probably the major source (1). Various cellular defense mechanisms counteract the deleterious effects of alkylating agents (2). For instance, Escherichia coli induces the expression of a set of genes involved in DNA repair after exposure to low doses of alkylating agents (3). As part of this adaptive response, the inducible E. coli AlkB protein protects the cells against harmful effects of methylating agents (4).
AlkB and functional human homologs thereof were demonstrated to be oxidative demethylases that repair the cytotoxic adducts 1-methyladenine (1-meA)3 and 3-methylcytosine (3-meC) in DNA and RNA. The repair reaction requires Fe(II) and molecular oxygen and is coupled to simultaneous decarboxylation of 2OG. The methyl group is hydroxylated and spontaneously released as formaldehyde (5-8) as displayed in Fig. 1. Importantly, tRNA and rRNA carry numerous natural N-methylations that may be important for structure, stability, and function (9). Many of these, e.g. 3-methylcytosine, are chemically identical to aberrant methylations. However, unlike aberrant methylations, natural methylations are located in distinct sequences. It is unknown whether demethylation has a role in regulating functions of RNA. In addition, numerous N-methylations in Lys and Arg residues of proteins, e.g. histone 3 and histone 4, are also known and these are important in regulation of transcriptional activities. Recently, oxidative demethylation of arginine in histones was demonstrated to be carried out by an enzyme related to those in the AlkB family (10). Different demethylases in this dioxygenase family may therefore have functions both in nucleic acid repair and epigenetic regulation of gene functions.
FIGURE 1.

Oxidative demethylation of 3-meC in DNA/RNA catalyzed by AlkB. The co-substrate 2OG is decarboxylated, whereas the methyl group is oxidized to hydroxymethyl and subsequently released as formaldehyde.
Among the eight putative human AlkB homologs identified (hABH1-8) (8, 11, 12) only hABH2 and hABH3 have been conclusively shown to be functional homologs. hABH2, but not hABH3, was shown to co-localize with proliferating cell nuclear antigen (PCNA) in replication foci during S-phase (8), suggesting a role in genome maintenance near replication forks. Interestingly, hABH3, like AlkB, repairs both DNA and RNA, as demonstrated by in vivo repair of damaged genomic MS2 bacteriophage RNA (8) and tRNA in E. coli (13), as well as in vitro repair of methylated RNA (8, 13, 14). However, the biological significance of RNA repair in vivo remains uncertain.
hABH1 was identified by homology search and reported to partially protect AlkB-deficient E. coli against the toxicity of methyl methanesulfonate (MMS) (15). In more recent studies, His-tagged hABH1 failed to show an enzymatic activity in vitro (8, 11). Neither was in vivo reactivation of chemically methylated DNA or RNA bacteriophages detected when expressed in alkB mutants of E. coli (8). The lack of AlkB-like activity of hABH1 would seem paradoxical, given the high similarity of these proteins (7). However, recent results from mAbh1-/- mice indicate that the murine Abh1 protein may have a role in development through participation in transcriptional regulation although no enzymatic activity was demonstrated (16). In the present study, we show that the widely expressed hABH1 is a mitochondrial demethylase that demethylates 3-meC, but not 1-meA, in DNA and RNA. The repair activity is lower than that of AlkB, hABH2, and hABH3, and it cannot be ruled out that hABH1 may have additional substrates. Earlier failure to detect demethylase activity may be due to difficulties in purifying an active form of the enzyme. Recently, results from a mouse model deficient in mAbh1 demonstrated a role of Abh1 in development, probably at the epigenetic level (16). Our results demonstrate that hABH1 also is a mitochondrial DNA/RNA demethylase, indicating that mammalian ABH1 may have dual roles in nuclei and mitochondria.
EXPERIMENTAL PROCEDURES
Molecular Cloning—hABH1 cDNA (IMAGE ID 3854233) was amplified by PCR using primers containing appropriate restriction sites. The open reading frame of the IMAGE 3854233 clone lacked the codons for the three N-terminal amino acids (MGK), see GenBank™ accession number AC008044 and cDNA data base entries (e.g. NM_006020). Therefore, the protein product is referred to as hABH1(Δ3) (hABH1-(4-389)). A second primer was used to amplify the full-length hABH1. hABH1 and hABH1(Δ3) were subcloned into NdeI/NotI sites of pET28a (Novagen) and pTYB12 (Clontech), and EcoRI/XmaI-blunted pEYFP-N1 (Clontech). hABH1-(11-389) was amplified using appropriate primers and subcloned in pEYFP-N1 at AgeI/EcoRI site. To generate hABH1-(1-119)-EYFP, hABH1-pEYFP-N1 was cleaved with AflII, blunted, and recleaved with HindIII, and subcloned into Xma-blunted/HindIII site of pEYFP-N1. For site-directed mutagenesis we used the QuikChange kit (Stratagene).
Northern Hybridization—Multiple tissue Northern blots (Clontech, number 7759-1/7760-1) were hybridized with 32P-end-labeled full-length cDNA probes of hABH1 or β-actin, according to the manufacturer's instructions and bands were visualized by phosphorimaging.
Confocal Laser Scanning Microscopy—HeLa cells were grown in Dulbecco's modified Eagle's medium, 10% fetal calf serum, 1% gentamicin, and 0.5% Fungizone. For transfections we used a calcium phosphate-based transfection kit (Promega) or FuGENE 6 (Roche), and analyzed cells in a laser scanning microscope (Zeiss LSM 510 or Meta) as described (8). Alternatively, HeLa cells were fixed in 2% paraformaldehyde on ice for 10 min prior to addition of ice-cold methanol and incubation at -20 °C for 20 min, washed with phosphate-buffered saline containing 2% fetal calf serum, monoclonal anti-hABH1 (Sigma) or polyclonal rabbit anti-human mitochondrial antiviral signaling protein antibodies (a marker of the outer mitochondrial membrane, Abcam) (17) were added and incubated at 37 °C for 1 h. After 3 washes, antibodies for double staining were added with further incubation for 1 h. Finally, the secondary antibodies Alexa Fluor goat anti-mouse (GAM) 546 and Alexa Fluor goat anti-rabbit (GAR) 647 (Invitrogen), were added alone (single-staining) or together (double-staining). The cells were analyzed in a laser scanning microscope (Zeiss LSM Meta) with excitation at 546 nm and detection between 560 and 615 nm (GAM), or excitation at 633 nm and detection at >650 nm (GAR).
Subcellular Fractionation—Whole cell protein extract from HeLa cells was prepared as described (18). To isolate nuclei, harvested cells were re-suspended in hypotonic buffer (20 mm HEPES-NaOH, pH 7.4, 5 mm MgCl2, 5 mm KCl, 1 mm dithothreitol (DTT) and EDTA-free protease inhibitor Complete) and disrupted by a Dounce homogenizer. The nuclei were pelleted by centrifugation at 680 × g for 5 min, resuspended in 2 volumes of extraction buffer (10 mm Tris-HCl, pH 7.8, 200 mm KCl, 20% glycerol, 0.25% Nonidet P-40, 1 mm DTT, Complete®, Phosphatase inhibitor mixtures 1 and 2 (Sigma)) and proteins extracted at 4 °C for 2 h. The extract was centrifuged at 16,000 × g for 20 min. The supernatant was collected, snap-frozen in liquid N2, and stored at -80 °C.
Mitochondrial fractions were isolated essentially as described (19), except that cell debris was removed by centrifugation 3 times at 2000 × g for 10 min before the crude mitochondrial fraction was obtained by centrifugation at 10,000 × g for 20 min and further purified by MSH (10 mm HEPES-KOH, pH 7.8, 1 mm EDTA, 1 mm EGTA, 0.21 m mannitol, 0.07 m sucrose, 0.15 mm spermine, and 0.75 mm spenmidine)/Percoll gradient centrifugation. The mitochondrial fraction was washed and resuspended in 1 ml of MSH buffer (19) and treated with proteinase K or digitonin, as described below. To obtain crude mitochondrial protein extract, the pellet was resuspended in the extraction buffer and proteins extracted as described above.
Freshly prepared mitochondria were treated with Proteinase K(0-3 μg/μg protein) at 37 °C for 30 min, then phenylmethylsulfonyl fluoride to 5 mm and the protease inhibitor Complete® were added to inactivate proteinase K, denatured in NuPAGE LDS gel loading buffer (Invitrogen) at 90 °C for 15 min, and subjected to Western blotting. Alternatively, mitochondria were treated with digitonin (0-1 μg/μg of protein) on ice for 30 min, centrifuged at 16,000 × g, and the supernatant collected. The pellet was washed once and resuspended in MSH. The samples were subjected to Western blot analysis as described below.
Western Blot Analysis—Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Immobilon™, Millipore) (19). Membranes were incubated with monoclonal anti-hABH1 (Sigma) or anti-hABH2 (Sigma), anti-PCNA (Santa Cruz), polyclonal anti-VDAC (Abcam), or anti-COX antibodies (Abcam) at room temperature for 1 h. Washed membranes were incubated with secondary antibodies, either peroxidase-labeled polyclonal rabbit anti-mouse or swine anti-rabbit IgG/horseradish peroxidase. Chemiluminescence reagent (SuperSignal® West Femto Maximum, Pierce) was added and blots were developed in Image Station 2000R (Eastman Kodak Co.).
Protein Purification—His-tagged AlkB, hABH1, and hABH3 were purified as described (8). Intein-tagged hABH1(Δ3) and mutant proteins were purified from E. coli BL21(DE3) RIPL or DE3 lysogens of HK82 (AlkB-deficient) (11) (generously provided by Dr. B. Sedgwick, Clare Hall Laboratories, United Kingdom) using the IMPACT-CN system (New England BioLabs). Cells were cultured at 37 °C, induced by 0.6 mm isopropyl β-d-thiogalactopyranoside at A600 0.5-0.8, and further incubated at 18 °C for 16 h. Harvested cells were resuspended in lysis buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 1 mm EDTA, 0.1% Triton X-100, 20 μm phenylmethylsulfonyl fluoride, Complete), and disrupted by sonication. The centrifuged lysate was loaded onto a chitin column. The Intein tag was cleaved off on-column with 50 mm DTT at 4 °C for 40 h. The effluent containing hABH1 was filtered and dialyzed against 50 mm MES, pH 6.0, containing 30 mm NaCl and 1 mm DTT prior to FPLC separation on a 1-ml Hitrap SP cation exchange column (Amersham Biosciences), using a linear salt gradient (0.05-1 m). rhABH1(Δ3) eluted at ∼0.15-0.3 m NaCl. One-liter cultures yielded ∼0.2 mg of >95% pure recombinant protein as confirmed by SDS-PAGE. The identity of the protein was confirmed both by Western blot analysis and mass spectrometry.
Assay for Decarboxylation of 2OG—Decarboxylation of 2OG was assessed by monitoring the release of 14CO2 from 1-[14C]2OG (Amersham Biosciences) essentially as described (20). Apart from the enzyme, the reaction mixture contained 100 μm 2OG containing 10% 1-[14C]2OG (∼24,000 dpm, specific activity 54.5 mCi/mmol), 80 μm FeSO4(NH4)2SO4, 4 mm ascorbic acid, and 0.5 mg/ml catalase in 50 mm Tris-HCl, pH 7.0. In each vial, a small tube containing 2 m hyamine hydroxide (150 μl) was placed without contact with the sample. The samples were incubated at 37 °C for 1 h, following addition of 15 μl of trifluoroacetic acid (50%) and incubation on ice for 1 h. The released 14CO2 was collected in hyamine hydroxide and analyzed by scintillation counting.
Chemical Methylation of Polynucleotides—[3H]Methyl-labeled poly(dA), poly(dC), poly(rA), or poly(rC) were prepared as described (5). Methylnitrosourea-treated poly(rC) was prone to degradation, whereas the other substrates were stable. Alternatively, 0.3-1 mg of polynucleotides were dissolved in 50 mm Tris-HCl, pH 8.0, containing 1 mm EDTA and treated with 500 mm MMS at 37 °C for 1 h. Samples were ethanol-precipitated, washed three times in 70% ethanol, and dissolved in 50 mm Tris-HCl, pH 8.0.
Demethylase Assays—The proteins were incubated with methylated substrates in 50 mm Tris-HCl, pH 7.0, containing 100 μm FeSO4(NH4)2SO4, 100 μm 2OG, and 4 mm ascorbic acid or DTT at 37 °C for 20-60 min, unless otherwise stated.
Demethylation of Radiolabeled Substrates—[3H]Methyl-homopolymers poly(dA), poly(rA), and poly(dC) were used as substrates. After the reaction, the substrate was ethanol-precipitated and the ethanol-soluble supernatant analyzed by scintillation counting. The pellet was hydrolyzed in either 0.1 m HCl (poly(dA)) or 1 m HCl (poly(rA)) at 95 °C for 1 and 4 h, respectively. Poly(dC) was decomposed in formic acid at 170 °C for 30 min. The hydrolyzed substrates were analyzed using a cation exchange column (SCX-Partisil 10, Pharmacia Biosciences) connected to an HPLC system (Agilent) and run isocratically in 0.1 m ammonium formate, pH 3.6, containing 8% methanol (6) and fractions analyzed by scintillation counting. The elution pattern was compared with that of non-labeled 1-meA or 3-meC detected by absorption at 260 nm.
LC/MS/MS Analysis for Measuring Demethylation of DNA and RNA—MMS-treated DNA and RNA were incubated with hABH1(Δ3), hABH3, or AlkB as described above except using 10 μm Fe(II). The substrates were degraded enzymatically to deoxynucleosides or nucleosides and analyzed by LC/MS/MS using an Agilent HP1100 HPLC system interfaced an Applied Biosystems API4000 hybrid triple quadrupole/linear ion trap mass spectrometer, essentially as described (21).
Fluorescent oligonucleotide substrates: 49-mer oligode-oxynucleotides containing 3-meC or 1-meA in the recognition site for the methylation sensitive restriction enzyme DpnII with the sequence as described in Ref. 21, were labeled with 6-carboxyfluorescein at the 5′ end, and synthesized with phosphorothioate linkages (5 bases) at the 3′ and 5′ ends. hABH1(Δ3) was incubated with 1 pmol of single-stranded or double-stranded substrate in 10 mm Tris-HCl, pH 6.6, 10 mm KCl, 100 μg/ml bovine serum albumin, 4 mm DTT, 100 μm 2OG, and 10 μm FeSO4(NH4)2SO4 at 37 °C for 15 min. The oligodeoxynucleotide was ethanol precipitated and resuspended in DpnII reaction buffer (New England Biolabs), annealed to the complementary oligodeoxynucleotide and treated with 10 units of DpnII at 37 °C for 1 h. Substrate and product bands (49- and 22-mer) were separated by PAGE and fluorescence visualized using a Typhoon TRIO GE scanner.
RESULTS
hABH1 Is a Widely Expressed Mitochondrial Protein—Northern blot analysis revealed that hABH1 is widely expressed in different human tissues at the mRNA level (Fig. 2A). The highest levels of mRNA were detected in skeletal muscle and heart, which are both rich in mitochondria. A major band of 2.3 kb was detected in all tissues, although at very low levels in peripheral blood leukocytes. In skeletal muscle three bands of sizes ∼0.9, 2.3, and 3 kb were detected. The weaker band of 3 kb was detected in several other organs. We are currently investigating whether the two extra transcripts observed in skeletal muscle and heart may represent cell-specific isoforms having particular functions in these organs.
FIGURE 2.

Tissue distribution and subcellular localization of hABH1. A, multiple tissue Northern blots (Clontech, numbers 7759-1/7760-1) were hybridized with 32P-end-labeled full-length cDNA probes for hABH1 and β-actin. In heart and skeletal muscle there are two transcripts of β-actin. B, hABH1-EYFP co-expressed with mitochondrial UNG1-ECFP in HeLa cells. C, subcellular distribution patterns of hABH1-(1-119)-EYFP, hABH1-(27-389)-EYFP, and EYFP-hABH1. D, immunostaining of HeLa cells with monoclonal anti-hABH1 and polyclonal anti-mitochondrial antiviral signaling protein (MAVS) antibodies.
HeLa cells transiently expressing fluorescent hABH1-EYFP displayed accumulation of fluorescence in “rod-like” structures almost exclusively in the cytoplasm. Moreover, co-transfection experiments revealed that hABH1-EYFP co-localized with UNG1-ECFP (Fig. 2B). This form of uracil-DNA glycosylase (22), as well as the endogenous form (19), have previously been demonstrated to be located in mitochondria. C-terminal truncated hABH1-(1-119)-EYFP also sorted to mitochondria (Fig. 2C) as did hABH1-(4-389)-EYFP (data not shown), whereas a more fuzzy cytoplasmic localization was seen when the first 26 N-terminal amino acids were removed (Fig. 2C, hABH1-(27-389)-EYFP). These results indicate that the mitochondrial targeting signal is located in the N-terminal region of hABH1. Programs used to identify mitochondrial targeting signals failed to predict mitochondrial localization. However, when disregarding the 21 N-terminal residues, MitoProt II and Predotar predicted 87.5 and 66% probability for mitochondrial targeting, respectively. Furthermore, the N-terminal region 22AFRKLFR- FYRQSR34 is predicted to form an amphipathic helix with one positively charged and one hydrophobic face and may be part of the mitochondrial targeting signal.
Furthermore, when the fluorescent tag was placed at the N-terminal end of hABH1, cytoplasmic fluorescence was diffuse and the largest fraction was found in the nucleus (EYFP-hABH1, Fig. 2C), probably due to negative interference with mitochondrial targeting. The C-terminal end contains a cluster of positive residues that may constitute a nuclear targeting signal (381KRAR384, see Fig. 4A).
FIGURE 4.

Structure sequence-based alignment of AlkB, hABH1, and hABH3 and three-dimensional model prediction for hABH1 active site residues. A, sequence-structure alignment of AlkB, hABH1, and hABH3 generated using ClustalX (43) based on crystal structures of AlkB (2FD8) and hABH3 (2IUW) and secondary structure of hABH1, predicted by the program JNet (44). The alignment was edited with JalView (45) and the sequence colored according to the ClustalX color scheme showing the degree of conservation. The active site residues subjected to analyses in the present study comprise the iron-binding motif (asterisks), 2OG-binding motif (circles), as well as Ile218 in hABH1 (arrowhead). α-Helices (cylinders) and β-strands (arrows) are colored green and blue, respectively, for conserved regions, and red for the variable subdomain forming part of the DNA/RNA-binding groove. A putative α-helix only present in hABH1 is colored gray. B, close-up view of the predicted active site of hABH1 (orange) superimposed onto hABH3 (2IUW) (green) and AlkB (2FD8) (blue). A three-dimensional model of the central catalytic core domain of hABH1 was predicted by the automated comparative homology modeling server SWISS_MODEL (46) based on the structure-sequence alignment presented in Fig. 4A using the crystal structure of AlkB as template. The side chains of the iron- and 2OG-binding motifs of the respective protein, as well as 2OG (hABH3), are represented as balls and sticks. The partly oxidized Leu177 in hABH3 is denoted with an asterisk and the polar contacts between active site atoms as dotted lines. The iron ion (magenta) and the iron-bound water (red) are shown as spheres.
Immunocytochemistry using a monoclonal mouse anti-hABH1 antibody demonstrated that endogenous hABH1 colocalizes with mitochondrial antiviral signaling protein (Fig. 2D). Furthermore, Western blot analysis of mitochondrial and nuclear protein extracts confirmed that the endogenous hABH1 is apparently mitochondrial, in contrast to the nuclear protein hABH2 analyzed as a control (Fig. 3A). However, a faint band in the nuclear fraction indicated that a small fraction of hABH1 may be nuclear. After treatment of the cells for 4 h with 0.6 mm MMS, hABH1 remained predominantly mitochondrial (data not shown).
FIGURE 3.

Western blot analyses of subcellular fractions. The same amount of protein extracts (typically 20 μg) from different fractions as indicated were subjected to Western blot analysis using monoclonal antibodies against hABH1, hABH2, and PCNA, and polyclonal antibodies against VDAC (outer mitochondrial membrane marker) and COX (mitochondrial matrix marker) as indicated. A, subcellular fractionation and identification of hABH1 and hABH2 in mitochondria and nuclei, respectively. B, freshly prepared mitochondrial fractions were treated with various amounts of Proteinase K, centrifuged to remove degraded proteins, and the pellet was subjected to Western blot analysis. As a control recombinant hABH1(Δ3) was subjected to the same treatment and analyzed by SDS-PAGE. C, the mitochondria were treated with digitonin, and centrifuged to separate extracted components. The same amounts of supernatant (S) and pellet (P) were analyzed by Western blotting.
To ensure that hABH1 is a genuine mitochondrial protein and not merely unspecifically attached to the outer surface, isolated mitochondria were treated with either Proteinase K (0-3.0 μg/μg of protein) to degrade proteins attached to the outer membrane, or digitonin (0-1.0 μg/μg of protein) to remove the mitochondrial outer membrane. Similar to the well established mitochondrial proteins VDAC and COX, endogenous hABH1 is quite resistant to treatment of the mitochondrial fraction with Proteinase K. However, whereas VDAC and COX are completely resistant, hABH1 in the mitochondrial fraction is slightly reduced by the treatment. In contrast, contaminating PCNA, as well as recombinant hABH1, were rapidly digested by the lowest concentrations of Proteinase K (Fig. 3B). Similarly, treatment with digitonin did not extract hABH1, VDAC, or COX, but readily solubilized PCNA (Fig. 3C). The amount of contaminating PCNA reflects its abundance in the cell and preponderance for unspecific binding to proteins.
Together, these three approaches (transfection, immunocytochemistry, and subcellular fractionation) demonstrate that hABH1 is a genuine mitochondrial protein in HeLa cells and not just a contaminant attached unspecifically to the surface of mitochondria. It may be misleading to determine localization of a protein by transfection of fusion protein constructs alone.
Comparison of AlkB and Human Homologs and Modeling of hABH1 Active Site—hABH1 contains the five perfectly conserved amino acids in the AlkB family that constitute the iron- and 2OG-binding motifs. In hABH1 the predicted iron-binding motif His231-X-Asp233-XnHis287 corresponds to His131-X-Asp133-Xn-His187 in AlkB (asterisks). Furthermore, in hABH1 and AlkB the predicted 2OG binding sequences are 338RXXXXXR344 and 204RXXXXXR210 (filled circles), respectively (Fig. 4A). The second Arg residue in the 2OG motif is only conserved in the AlkB family of dioxygenases (7, 8, 11, 12, 23), and was previously suggested to be involved in recognition of the prime substrate (23). The co-crystallization of AlkB and the trinucleotide substrate dT-(1-me-dA)-dT in the presence of the co-substrate suggested that Arg210 interacts with the C1 carboxylate of 2OG and makes three van der Waals contacts with the prime substrate (24). Furthermore, crystal structure and mutational analyses of hABH3 revealed that the Leu177 residue plays an important role in substrate binding. Whereas this residue is perfectly conserved in AlkB, hABH2, hABH3, and hABH6 (25) it is replaced by Ile (arrowhead) in hABH1 (Fig. 4A).
The structure-sequence alignment of AlkB, hABH1, and hABH3 indicated that hABH1 (1, 12) likely contains the central jellyroll-fold shared by most members of the Fe(II)/2OG-dependent dioxygenase superfamily (7, 26). Based on this alignment and using the crystal structure of AlkB (2FD8) as template, a putative three-dimensional structure was generated for the central catalytic AlkB core domain of hABH1. Like in the crystal structures of AlkB (24) and hABH3 (Protein Data Bank 2IUW) (25), this region of hABH1 may adopt a double-stranded β-helix jellyroll-fold (data not shown) with the conserved amino acid residues implicated in iron and 2OG binding located at nearly identical positions in the active site (Fig. 4B). Significant differences are observed in the region between the two conserved α-helices (in green) in the N-terminal part of the enzymes (Fig. 4A).
Purification of the hABH1 Protein and Characterization by Mass Spectrometry—Purification of soluble His-tagged hABH1 proved to be difficult, because a major fraction of the protein was recovered as insoluble aggregates of partially degraded protein, and was further degraded during incubation with thrombin to remove the tag. Intein-tagged hABH1(Δ3), in which 3 amino acids at the N-terminal region were deleted, was obtained with better yield and was also more stable after removal of the intein tag, when compared with full-length hABH1. The yield of active hABH1(Δ3) was greatly improved by expression at low temperature (18 °C), but remained low compared with hABH2 and hABH3. In contrast, the yield of full-length hABH1 was not significantly improved by lowering the temperature (data not shown). Due to better recovery, stability, and activity, purified hABH1(Δ3) was used in subsequent in vitro studies. The purification procedure resulted in apparently near homogenous proteins of the expected molecular masses (Fig. 5A).
FIGURE 5.
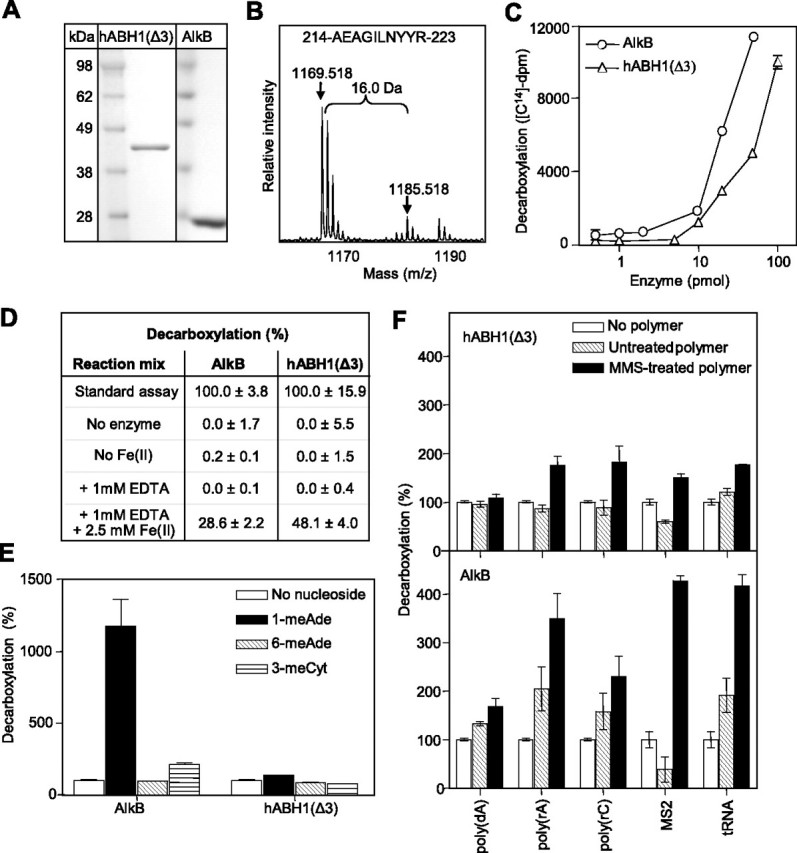
2OG decarboxylase activity of hABH1(Δ3). A, SDS-PAGE of recombinant hABH1(Δ3) and His-tagged AlkB purified to apparent homogeneity. B, mass shift of 16 Da for the peptide 214AEAGILNYYR223 of hABH1. C, AlkB or hABH1(Δ3) were incubated with 1-[14C]2OG (∼24,000 dpm). The released 14CO2 was collected and analyzed by scintillation counting. D, AlkB (10 pmol) and hABH1(Δ3) (30 pmol) were assayed for release of 14CO2 from 1-[14C]2OG when Fe(II) was either absent or chelated by adding 1 mm EDTA. E, effect of the methylated nucleosides 1-methyladenosine (1-meAde), 6-methyladenosine (6-meAde), and 3-methylcytidine (3-meCyt) on 2OG decarboxylation by hABH1(Δ3) and AlkB (15 pmol). F, effect of MMS-treated and untreated DNA and RNA on decarboxylation of 2OG by hABH1(Δ3) and AlkB (15 pmol).
Tryptic digestion of hABH1 and analysis of the resulting peptides by mass spectrometry (matrix-assisted laser desorption ionization time-of-flight) revealed a 16-Da mass shift of some peptides, suggesting that the enzyme is subject to self-hydroxylation, as previously described for AlkB and hABH3 similar to bacterial AlkB than hABH2 and hABH3 (25, 27). As an example, the peptide 214AEAGILNYYR223 resulting from tryptic digestion of hABH1 is presented in Fig. 5B. This peptide contains the Ile218 residue, which corresponds to Leu177 in hABH3. Leu177 was previously demonstrated to be important for catalytic activity, but is subject to self-hydroxylation (25). A mass shift increase of 16 Da of this hABH1 decapeptide was seen in 3 of 4 independent experiments. In comparison, a mass shift in this fragment was seen in only 1 of 6 independent experiments in site-directed mutants that inactivated the enzyme by affecting either iron binding or 2OG binding. Another peptide, 284LLN-HAVPR291, containing the His287 residue of the active site, displayed a mass shift of 16 in 4 of 4 independent experiments of wild type hABH1 (data not shown). Taken together, these results suggest that hABH1 is subject to modifications that are compatible with self-hydroxylation. When in the vicinity of the active site, such modifications are likely to affect the catalytic activity of the enzyme. However, it should be underlined that we have not quantified the degree of self-hydroxylation in individual hABH1 enzyme batches, neither have we directly demonstrated that it inactivates the enzyme.
hABH1(Δ3) Decarboxylates 2-Oxoglutarate—Like several other 2OG/Fe(II)-dependent dioxygenases, AlkB converts 2OG to succinate and releases CO2 also in the absence of the prime substrate, although less efficiently (26, 28). To examine the functionality of the predicted 2OG/Fe(II) binding motifs in hABH1, we studied the capacity of hABH1(Δ3) to carry out “uncoupled” decarboxylation of 2OG. Purified protein was incubated with 1-[14C]2OG and release of 14CO2 was measured. We found that purified hABH1(Δ3) decarboxylates 2OG at a significant rate, although somewhat lower than that of AlkB (Fig. 5C). hABH1(Δ3) displayed highest activity at pH 6.5-7.5. As expected, the decarboxylation reaction required Fe(II) (Fig. 5D). Chelating Fe(II) by adding EDTA abrogated the activity, whereas addition of Fe(II) partially restored the enzymatic activity of the respective enzyme (Fig. 5D). Decarboxylation of 2OG by AlkB was previously shown to be stimulated by methylated nucleosides (28). In agreement with this, we found that 1-methyladenosine (1-meAde) and 3-methylcytidine (3-meCyt) enhanced the AlkB-catalyzed release of CO2 from 2OG markedly. By contrast, neither 1-meAde nor 3-meCyt affected the activity of hABH1(Δ3) (Fig. 5E). However, decarboxylation was stimulated 1.3-2-fold by MMS-treated poly(rC), poly(rA), MS2 bacteriophage RNA, and tRNA, whereas the untreated polymers did not stimulate. AlkB-catalyzed decarboxylation of 2OG was more strongly stimulated by the polymers, including some that were unmethylated (Fig. 5F).
hABH1(Δ3) Demethylates 3-meC in DNA and RNA—The decarboxylation studies indicate that hABH1(Δ3) interacts with methylated nucleic acids. We therefore searched for a possible demethylase activity using [3H]methyl-labeled poly(dA), poly(rA), and poly(dC) as substrates. [3H]Methyl-labeled poly(rC) appeared unstable and prone to degradation, resulting in high background in our assays. Among the substrates tested we identified poly(dC) as the best substrate for hABH1(Δ3). However, the catalytic efficiency is significantly lower than that of hABH3. Furthermore, the substrate preference is different from that of AlkB (Fig. 6A). These results demonstrate that hABH1(Δ3) released ethanol-soluble radioactivity from [3H]methyl-poly(dC), albeit at lower rate than AlkB (Fig. 6B). There was no additional radioactivity released by addition of AlkB and hABH1(Δ3) together, suggesting that hABH1(Δ3) and AlkB recognize the same damage (3-meC) in methylated poly(dC). Furthermore, hABH1(Δ3) did not repair methylated poly(dA) or poly(rA) detectably under the conditions used here (data not shown). Polyclonal antibodies to hABH1 specifically inhibited the demethylase activity of hABH1 (Fig. 6C).
FIGURE 6.
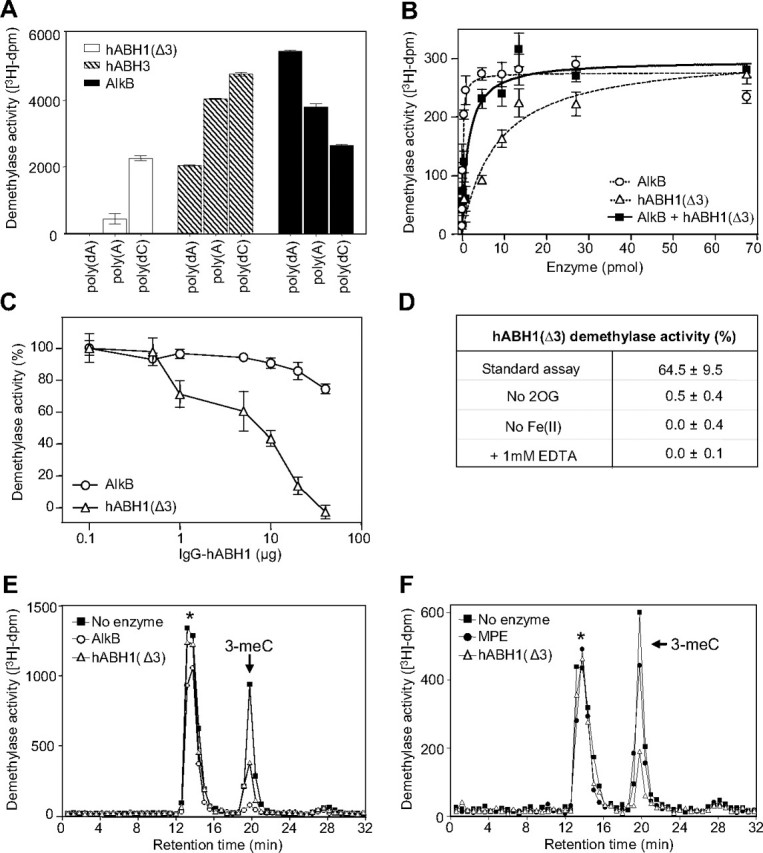
Demethylase activity of hABH1(Δ3). A, demethylase activity of hABH1(Δ3) (10 pmol), hABH3 (10 pmol), and AlkB (5 pmol) on different [3H]methylnitrosourea-methylated polymers of DNA and RNA. B, [3H]methyl-poly(dC) was incubated with AlkB or hABH1(Δ3) or with both proteins for 1 h, followed by ethanol precipitation. The ethanol-soluble radioactivity was quantified by scintillation counting. C, inhibition by polyclonal anti-hABH1 antibody of hABH1(Δ3) (15 pmol) demethylase activity using [3H]methyl-poly(dC) as substrate. AlkB (15 pmol) was used as control. D, demethylase activity of hABH1(Δ3) (20 pmol), on single-stranded oligonucleotide substrate (1 pmol) containing 3-meC, using the fluorescent oligonucleotide substrate assay. The standard reaction mixture was modified as depicted. E, HPLC elution profile of the products generated by acid hydrolysis of the ethanol-precipitated [3H]methyl-poly(dC) after incubation with AlkB or hABH1(Δ3) (10 pmol), or without enzyme. The arrow indicates retention time for unlabeled 3-meC standard as detected by UV absorbance at 260 nm. F, as in E but including whole mitochondria cell extract (MPE) (50 μg), hABH1(Δ3) (10 pmol), or without enzyme.
Omission of both 2OG and Fe(II), or inclusion of EDTA, abolished the release of radioactivity from the 3H-methylated substrate. Excluding either 2OG or Fe(II) and addition of EDTA resulted in nearly complete loss of activity (Fig. 6D), essentially similar to results for AlkB (29). To identify the substrate of hABH1(Δ3) better, the residual [3H]methylpoly(dC) substrate (after demethylase assay) was subjected to acid hydrolysis to yield free bases and then analyzed by cation exchange chromatography. Analysis of the intact substrate (without incubation with enzyme) revealed that ∼20% of the radioactivity eluted at the same position as the unlabeled 3-meC used as standard. The content of 3-meC in the substrate decreased markedly upon treatment with hABH1(Δ3), and was essentially undetectable when treated with AlkB (Fig. 6E). A substantial part of the radioactivity (presumably the [3H]methylated backbone) eluted earlier and contained essentially the same amount of total radioactivity in the hABH1(Δ3)-treated, AlkB-treated, and untreated substrate (Fig. 6E). Using mitochondrial protein extract in the demethylase assay, we observed a very slight decrease in the content of 3-meC using [3H]methyl-poly(dC) as substrate. Although this trend was reproducible, the significance of the observation is not clear (Fig. 6F).
To further verify the nucleic acid demethylase activity of hABH1 and to include methylated poly(rC) as a substrate, we used MMS-treated DNA and RNA homopolymers as substrates. The methylated polymers were enzymatically digested to deoxyribonucleosides and ribonucleosides, respectively. We quantitated reduction in specific methylations by LC/MS/MS, using the effects of hABH3 or AlkB as positive controls. This analysis verified that 3-meC was repaired in DNA and also in RNA by hABH1(Δ3), although less efficiently than by hABH3 or AlkB (Fig. 7, A and B). This analysis also revealed a weak activity toward 1-meA in RNA and an insignificant activity toward 1-meA in DNA (Fig. 7B).
FIGURE 7.

Repair of 1-meA and 3-meC by hABH1(Δ3). A, DNA oligonucleotides containing 1-me(dA) or 3-me(dC) were incubated with hABH3 or hABH1(Δ3) (20 pmol) for 15 min, followed by decomposition to nucleosides and LC/MS/MS quantization. The values are means of triplicates. B, methyl-poly(rA) or methylpoly(rC) were incubated with AlkB or hABH1(Δ3) and treated as in A. The values are means of duplicates. C, repair of carboxyfluorescein end-labeled single-stranded or double-stranded 49-mer DNA oligonucleotides containing 3-me(dC) or a 1-me(dA). The substrates were incubated with hABH1(Δ3) (0-20 pmol) or AlkB (10 pmol) and subsequently cleaved with DpnII restriction enzyme, run on PAGE, and analyzed for fluorescence.
Finally, we extended these studies using a fluorescent assay in which a restriction enzyme site was generated by removal of the methyl group from 3-meC or 1-meA in a fluorescent oligodeoxynucleotide. This cleavage assay demonstrated that hABH1(Δ3) demethylates 3-meC, and that enzyme is strictly single strand specific, whereas AlkB also efficiently removes the methyl group from double-stranded substrate for both 3-meC and 1-meA (Fig. 7C). The weak band observed in the absence of enzyme was due to a small amount of unmethylated C in the relevant position.
Analysis of Predicted Active Site Amino Acid Residues—We generated point mutations in the predicted Fe(II) binding sequence His231-X-Asp233-Xn-His287 in hABH1 and the corresponding sequence His131-X-Asp133-Xn-His187 in AlkB, as well as in the predicted 2OG binding sequences 338RXXXXXR344 in hABH1 and 204RXXXXXR210 in AlkB. Single point mutation (substitution with Ala) in each of the amino acids in the Fe(II) or 2OG binding motifs completely abrogated the activity of the respective protein in terms of CO2-release in the uncoupled reaction (Fig. 8A). These mutations also eliminated or reduced the demethylase activity toward 3-meC in poly(dC) in hABH1(Δ3) and AlkB, although the second Arg residue (Arg210 in AlkB and Arg344 in hABH1) is less critical (Fig. 8B). The second Arg in the 2OG binding motif is perfectly conserved in the AlkB family, but not in the other 2OG/Fe(II) dioxygenases.
FIGURE 8.
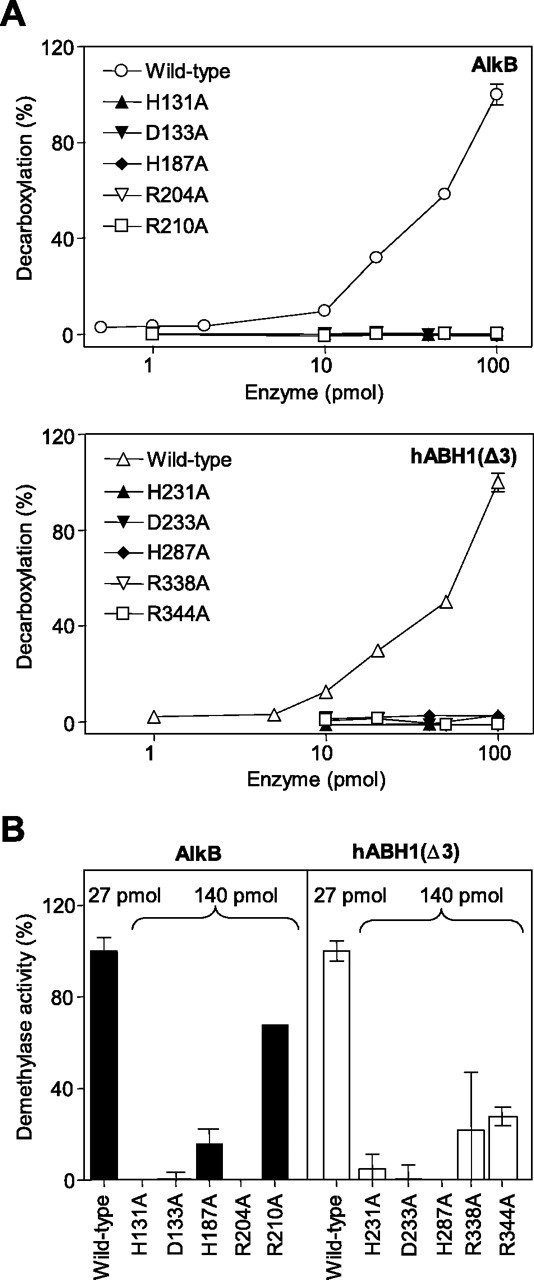
Analysis of active site residues in AlkB and hABH1(Δ3) involved in iron and 2OG binding. A, 2OG decarboxylase activity of AlkB, hABH1(Δ3), and mutants in the iron-binding motif (H131A/H233A, D131A/D233A, H187A/H287A) and 2OG-binding motif (R204A/2338A, R210A/R344A). B, demethylase activity of wild-type and various mutants of AlkB and hABH1(Δ3) on [3H]methyl-poly(dC) substrate. Note that the mutant proteins were added in excess (∼5-fold) as compared with the wild-type proteins.
Finally, Leu118 in AlkB is conserved in hABH2 (Leu157) and hABH3 (Leu177), whereas hABH1 contains Ile in the corresponding position (Ile218). The crystal structures of AlkB and hABH3 (24, 25), as well as the predicted active site structure for hABH1 (Fig. 4B), suggest that an oxidized form of residue may bind the C1 carboxylate of 2OG, possibly perturbing its function. At least in hABH3 Leu177 is essential for demethylase activity (25). However, the I218L variant of hABH1 did not demethylate 1-meA and displayed more than 2-fold decreased ability to repair 3-meC (data not shown), demonstrating the significance of Ile218 in the catalysis and that Leu substitution does not confer novel demethylase activity. In conclusion, all the residues predicted to be involved in binding of Fe(II) or 2OG were shown to be important for both the decarboxylase and demethylase activity of hABH1.
DISCUSSION
We have found that hABH1 is a functional AlkB homolog that is apparently predominantly located in mitochondria. The mitochondrial localization signal is located in the very N-terminal part of the protein. When a fluorescent tag is placed at the N-terminal end this blocks mitochondrial targeting and allows nuclear targeting, possibly by a putative nuclear localization signal in the C-terminal region. hABH1 catalyzes decarboxylation of 2OG to succinate and demethylation of 3-meC in single-stranded DNA and RNA. To our knowledge this is the first distinct mitochondrial alkylation-repair protein identified, although repair of O6-methylguanine by a less well characterized activity has been described (30). Although we so far have not detected demethylase activity toward substrates other than DNA and RNA, it is possible that hABH1, similar to certain other 2OG-dependent dioxygenases, may demethylate other substrates, such as specific methylations in lysine or arginine in protein (31). An epigenetic function of hABH1 in nuclei was indicated by the recently published data on gene-targeted mice lacking mAbh1. However, no enzymatic activity of mAbh1 was reported in this study, although a chromatin-modulating effect involving histone deacetylase was indicated (16). In addition, there may be cell-specific or species-specific variations. In fact, we have previously observed that human UNG1 is strictly mitochondrial, whereas the murine Ung1 homolog sorts to mitochondria and to a smaller extent to nuclei (32). We also considered the possibility that hABH1 might sort to nuclei upon DNA damage, but MMS did not alter the localization. The localization of hABH1 in mitochondria is intriguing, considering the apparent nuclear epigenetic role reported for the mouse ortholog of hABH1 (16). However, this apparent paradox is not without precedence: the RNA-binding protein SLIRP (SRA stem-loop interacting RNA binding protein) is predominantly mitochondrial, but it is a co-repressor of a nuclear steroid receptor (33). Although possibly fortuitous, it is interesting to notice that the gene encoding SLIRP is located next to hABH1 in chromosomal position 14q24. They are organized head-to-head and separated only by a few hundred nucleotides, suggesting that they might be co-regulated. Recently, hABH1 was reported to be essentially unsorted; that is, the authors observed homogenous fluorescence all over the cell (34). However, in that study the resolution was low and the fluorescent protein was placed at the N terminus of hABH1, which could interfere with sorting to mitochondria.
Mitochondrial dysfunction has been associated with aging. Mitochondrial DNA damage, particularly oxidative base lesions, as well as accumulation of mutations, have been important areas of exploration in this respect. DNA repair in mitochondria is apparently limited to direct base repair (this paper), BER, and non-homologous recombination (35). However, the relationship between rate of mutations in mitochondrial DNA and aging remains unsettled (36). Previously, mitochondria have been shown to have a capacity for removal of N-methylpurines probably as part of a BER process (37). This suggests that a form of methylpurine glycosylase may be present in mitochondria, although not experimentally demonstrated. The significance of mitochondrial repair of drug-induced alkylation lesions in reduction of cytotoxicity was recently demonstrated in glia cells transfected with a vector directing methylpurine glycosylase to mitochondria (38). This is significant, because cell-specific differences in repair of mitochondrial N-methylpurines have been observed (39). Whether a significant level of alkylation lesions in mitochondria may be caused by endogenous processes is not known, although this has been demonstrated for total cellular DNA (21). To our knowledge, hABH1 represents the first example of a defined mitochondrial enzyme repairing methylated bases in DNA. Furthermore, search in a mitochondrial protein data base (bioinfo.nist.gov/hmpd/index.html) reveals trimethyllysine hydroxylase (involved in the biosynthesis of carnitine) as the only mitochondrial dioxygenase identified previously.
Earlier studies (8, 11) failed to demonstrate an enzymatic activity for hABH1, the first human AlkB homolog discovered (15). This has been rather puzzling because hABH1 is more similar to bacterial AlkB than hABH2 and hABH3. However, AlkB homologs are subject to self-hydroxylation, as reported here for hABH1 and previously for hABH3 (25). Expression of hABH1 could not reactivate MMS-treated single-stranded DNA or single-stranded RNA phage in an alkB mutant E. coli (8, 11). Unlike a previous study, where expression of hABH1 slightly increased survival of AlkB-deficient E. coli after exposure to MMS (15), we failed to observe increased survival in a similar experiment (data not shown). Unlike AlkB, hABH1 at best repairs 1-meA only very inefficiently, which may in part explain some of the previous results as these studies examined the impact of host cell expression of hABH1 on in vivo repair of MMS-treated bacteriophages, and the in vitro repair of 1-meA. This is a major cytotoxic adduct generated by Sn2 methylating agents (e.g. MMS), and it is recognized by AlkB (and not by hABH1) as a preferred substrate. 3-MeC is less abundant in MMS-treated polynucleotides (approximately half the amount of 1-meA), and is up to 30% mutagenic (40, 41). Hence, the lack of reactivation of MMS-treated phages by hABH1 may be due to the higher level of the cytotoxic 1-meA adduct that is apparently not recognized by the protein.
As evident from the present and previous studies, the three functionally active human AlkB homologs, hABH1, hABH2, and hABH3, have different subcellular localizations and properties. hABH1 is at least in part mitochondrial and repairs 3-meC in DNA and RNA. hABH2 is strictly nuclear and located in replication foci during S-phase, suggesting a role in repair of 1-meA and 3-meC near the replication fork. Furthermore, among the functional human AlkB homologs hABH2 is the only one that is up-regulated in the S-phase.4 hABH3 is found in nuclei and cytoplasm and repairs 1-meA and 3-meC in both DNA and RNA. It does not relocate to replication foci, but displays some accumulation in nucleoli (8). Interestingly, the obesity-associated FTO gene was reported to encode a nuclear 2OG-dependent nucleic acid oxidative demethylase that repairs 3-methylthymine in single-stranded DNA. Expression of this distantly related homolog of AlkB is highest in the brain, particularly hypothalamic nuclei governing energy balance, and FTO expression is regulated by feeding and fasting (42).
In conclusion, we have demonstrated that hABH1 is a mitochondrial protein that demethylates 3-meC in DNA and RNA by oxidative demethylation, presumably carrying out base repair. However, this does not exclude the possibility that hABH1 may also have a role in regulation of the natural level of 3-meC in RNA species such as tRNA and rRNA. Furthermore, these studies do not exclude a role of hABH1 in demethylation of other substrates.
This work was supported by the National Programme for Research in Functional Genomics in Norway (FUGE) in the Research Council of Norway, European Community Integrated Project DNA Repair Grant LSHG-CT-2005-512113, The Norwegian Cancer Association, The Cancer Fund at St. Olav's Hospital, Trondheim, and the Svanhild and Arne Must Fund for Medical Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: 1-meA, 1-methyladenine; 3-meC, 3-methylcytosine; 2OG, 2-oxoglutarate; hABH, human AlkB homolog; PCNA, proliferating cell nuclear antigen; MMS, methyl methanesulfonate; DTT, dithiothreitol; MES, 4-morpholineethanesulfonic acid; HPLC, high pressure liquid chromatography; MS, mass spectrometry; EYFP, enhanced yellow fluorescent protein; me, methyl.
J. Peña-Diaz, F. Drablos, and H. E. Krokan, unpublished results.
References
- 1.Drabløs, F., Feyzi, E., Aas, P. A., Vaagbø, C. B., Kavli, B., Bratlie, M. S., Peña-Diaz, J., Otterlei, M., Slupphaug, G., and Krokan, H. E. (2004) DNA Repair 31389 -1407 [DOI] [PubMed] [Google Scholar]
- 2.Sedgwick, B. (2004) Nat. Rev. Mol. Cell Biol. 5148 -157 [DOI] [PubMed] [Google Scholar]
- 3.Sedgwick, B., and Lindahl, T. (2002) Oncogene 218886 -8894 [DOI] [PubMed] [Google Scholar]
- 4.Kataoka, H., Yamamoto, Y., and Sekiguchi, M. (1983) J. Bacteriol. 1531301 -1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falnes, P. O., Johansen, R. F., and Seeberg, E. (2002) Nature 419178 -182 [DOI] [PubMed] [Google Scholar]
- 6.Trewick, S. C., Henshaw, T. F., Hausinger, R. P., Lindahl, T., and Sedgwick, B. (2002) Nature 419174 -178 [DOI] [PubMed] [Google Scholar]
- 7.Aravind, L., and Koonin, E. V. (2001) Genome Biol. 20007.1 -0007.8 [Google Scholar]
- 8.Aas, P. A., Otterlei, M., Falnes, P. O., Vaagbø, C. B., Skorpen, F., Akbari, M., Sundheim, O., Bjørås, M., Slupphaug, G., Seeberg, E., and Krokan, H. E. (2003) Nature 421859 -863 [DOI] [PubMed] [Google Scholar]
- 9.Rozenski, J., Crain, P. F., and McCloskey, J. A. (1999) Nucleic Acids Res. 27 196-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, B., Chen, Y., Zhao, Y., and Bruick, R. K. (2007) Science 318444 -447 [DOI] [PubMed] [Google Scholar]
- 11.Duncan, T., Trewick, S. C., Koivisto, P., Bates, P. A., Lindahl, T., and Sedgwick, B. (2002) Proc. Natl. Acad. Sci. U. S. A. 9916660 -16665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurowski, M. A., Bhagwat, A. S., Papaj, G., and Bujnicki, J. M. (2003) BMC Genomics 4 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ougland, R., Zhang, C. M., Liiv, A., Johansen, R. F., Seeberg, E., Hou, Y. M., Remme, J., and Falnes, P. O. (2004) Mol. Cell 16107 -116 [DOI] [PubMed] [Google Scholar]
- 14.Lee, D. H., Jin, S. G., Cai, S., Chen, Y., Pfeifer, G. P., and O'Connor, T. R. (2005) J. Biol. Chem. 28039448 -39459 [DOI] [PubMed] [Google Scholar]
- 15.Wei, Y. F., Carter, K. C., Wang, R. P., and Shell, B. K. (1996) Nucleic Acids Res. 24 931-937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan, Z., Sikandar, S., Witherspoon, M., Dizon, D., Nguyen, T., Benirschke, K., Wiley, C., Vrana, P., and Lipkin, S. M. (2008) Dev. Dyn. 237316 -327 [DOI] [PubMed] [Google Scholar]
- 17.Yang, Y., Liang, Y., Qu, L., Chen, Z., Yi, M., Li, K., and Lemon, S. M. (2007) Proc. Natl. Acad. Sci. U. S. A. 1047253 -7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akbari, M., Otterlei, M., Peña-Diaz, J., Aas, P. A., Kavli, B., Liabakk, N. B., Hagen, L., Imai, K., Durandy, A., Slupphaug, G., and Krokan, H. E. (2004) Nucleic Acids Res. 325486 -5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akbari, M., Otterlei, M., Peña-Diaz, J., and Krokan, H. E. (2007) Neuroscience 1451201 -1212 [DOI] [PubMed] [Google Scholar]
- 20.Sabourin, P. J., and Bieber, L. L. (1982) J. Biol. Chem. 2577468 -7471 [PubMed] [Google Scholar]
- 21.Ringvoll, J., Nordstrand, L. M., Vaagbø, C. B., Talstad, V., Reite, K., Aas, P. A., Lauritzen, K. H., Liabakk, N. B., Bjørk, A., Doughty, R. W., Falnes, P. O., Krokan, H. E., and Klungland, A. (2006) EMBO J. 252189 -2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsen, H., Otterlei, M., Haug, T., Solum, K., Nagelhus, T. A., Skorpen, F., and Krokan, H. E. (1997) Nucleic Acids Res. 25750 -755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bratlie, M. S., and Drabløs, F. (2005) BMC Genomics 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu, B., Edström, W. C., Benach, J., Hamuro, Y., Weber, P. C., Gibney, B. R., and Hunt, J. F. (2006) Nature 439879 -884 [DOI] [PubMed] [Google Scholar]
- 25.Sundheim, O., Vaagbø, C. B., Bjørås, M., Sousa, M. M., Talstad, V., Aas, P. A., Drabløs, F., Krokan, H. E., Tainer, J. A., and Slupphaug, G. (2006) EMBO J. 253389 -3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hausinger, R. P. (2004) Crit. Rev. Biochem. Mol. Biol. 3921 -68 [DOI] [PubMed] [Google Scholar]
- 27.Henshaw, T. F., Feig, M., and Hausinger, R. P. (2004) J. Inorg. Biochem. 98856 -861 [DOI] [PubMed] [Google Scholar]
- 28.Welford, R. W., Schlemminger, I., McNeill, L. A., Hewitson, K. S., and Schofield, C. J. (2003) J. Biol. Chem. 27810157 -10161 [DOI] [PubMed] [Google Scholar]
- 29.Mishina, Y., Chen, L. X., and He, C. (2004) J. Am. Chem. Soc. 12616930 -16936 [DOI] [PubMed] [Google Scholar]
- 30.Sawyer, D. E., and Van Houten, B. (1999) Mutat. Res. 434161 -176 [DOI] [PubMed] [Google Scholar]
- 31.Bannister, A. J., and Kouzarides, T. (2005) Nature 4361103 -1106 [DOI] [PubMed] [Google Scholar]
- 32.Nilsen, H., Steinsbekk, K. S., Otterlei, M., Slupphaug, G., Aas, P. A., and Krokan, H. E. (2000) Nucleic Acids Res. 282277 -2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatchell, E. C., Colley, S. M., Beveridge, D. J., Epis, M. R., Stuart, L. M., Giles, K. M., Redfern, A. D., Miles, L. E., Barker, A., MacDonald, L. M., Arthur, P. G., Lui, J. C., Golding, J. L., McCulloch, R. K., Metcalf, C. B., Wilce, J. A., Wilce, M. C., Lanz, R. B., O'Malley, B. W., and Leedman, P. J. (2006) Mol. Cell 22 657-668 [DOI] [PubMed] [Google Scholar]
- 34.Tsujikawa, K., Koike, K., Kitae, K., Shinkawa, A., Arima, H., Suzuki, T., Tsuchiya, M., Makino, Y., Furukawa, T., Konishi, N., and Yamamoto, H. (2007) J. Cell Mol. Med. 111105 -1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson, D. M., 3rd, and Bohr, V. A. (2007) DNA Repair 6544 -559 [DOI] [PubMed] [Google Scholar]
- 36.Lambert, A. J., and Brand, M. D. (2007) Aging Cell 6417 -420 [DOI] [PubMed] [Google Scholar]
- 37.LeDoux, S. P., Wilson, G. L., Beecham, E. J., Stevnsner, T., Wassermann, K., and Bohr, V. A. (1992) Carcinogenesis 131967 -1973 [DOI] [PubMed] [Google Scholar]
- 38.Harrison, J. F., Rinne, M. L., Kelley, M. R., Druzhyna, N. M., Wilson, G. L., and Ledoux, S. P. (2007) Glia 551416 -1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LeDoux, S. P., Druzhyna, N. M., Hollensworth, S. B., Harrison, J. F., and Wilson, G. L. (2007) Neuroscience 1451249 -1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singer, B., and Grunberger, D. (1983) Molecular Biology of Mutagens and Carcinogens: Reactions of Directly Acting Agents with Nucleic Acids, Plenum Press, New York
- 41.Delaney, J. C., and Essigmann, J. M. (2004) Proc. Natl. Acad. Sci. U. S. A. 10114051 -14056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerken, T., Girard, C. A., Tung, Y. C., Webby, C. J., Saudek, V., Hewitson, K. S., Yeo, G. S., McDonough, M. A., Cunliffe, S., McNeill, L. A., Galvanovskis, J., Rorsman, P., Robins, P., Prieur, X., Coll, A. P., Ma, M., Jovanovic, Z., Farooqi, I. S., Sedgwick, B., Barroso, I., Lindahl, T., Ponting, C. P., Ashcroft, F. M., O'Rahilly, S., and Schofield, C. J. (2007) Science 3181469 -1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997) Nucleic Acids Res. 254876 -4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuff, J. A., and Barton, G. J. (2000) Proteins 40502 -511 [DOI] [PubMed] [Google Scholar]
- 45.Clamp, M., Cuff, J., Searle, S. M., and Barton, G. J. (2004) Bioinformatics 20 426-427 [DOI] [PubMed] [Google Scholar]
- 46.Schwede, T., Kopp, J., Guex, N., and Peitsch, M. C. (2003) Nucleic Acids Res. 313381 -3385 [DOI] [PMC free article] [PubMed] [Google Scholar]