

Abstract
Mcm4/6/7 forms a complex possessing DNA helicase activity, suggesting that Mcm may be a central component for the replicative helicase. Although Cdt1 is known to be essential for loading of Mcm onto the chromatin, its precise role in pre-RC formation and replication initiation is unknown. Using purified proteins, we show that Cdt1 forms a complex with Mcm4/6/7, Mcm2/3/4/5/6/7, and Mcm2/4/6/7 in glycerol gradient fractionation through interaction with Mcm2 and Mcm4/6. In the glycerol gradient fractionation, Mcm4/6/7-Cdt1 forms a complex (speculated to be a (Mcm4/6/7)2-Cdt13 assembly) in the presence of ATP, which is significantly larger than the Mcm4/6/7-Cdt1 complex generated in its absence. Furthermore, DNA binding and helicase activities of Mcm4/6/7 are significantly stimulated by Cdt1 protein in vitro. We generd a Cdt1 mutant, which fails to stimulate DNA binding and helicase activities of4/6/7. This mutant Cdt1 showed reduced interaction with Mcm and is deficient in the formation of a high molecular weight complex with Mcm. Thus, a productive interaction between Cdt1 and MCM appears to be essential for efficient loading of MCM onto template DNA, as well as for the efficient unwinding reaction.
To license the origins for DNA replication, multiprotein complexes, called pre-replicative complexes (pre-RCs),2 need to be formed. Cdc6 and Cdt1 are both required for pre-RC formation (1–4). They associate with the chromatin in a manner dependent on the origin recognition complex (ORC) bound to the origins throughout the cell cycle and facilitate the loading of the hexameric Mcm2–7 complex onto chromatin at late mitosis (telophase) through early G1 (5, 6). Mcm2–7 are members of the AAA family proteins and share the conserved ATP binding domain (7, 8). The Cdt1-interacting protein, Geminin, together with CDK activity, prevents the formation of pre-RC during S-G2 phases, thereby assuring that the genome is replicated once and only once per cell cycle (9–15).
In Escherichia coli, the initiator, DnaA, binds to oriC and unwinds the origin. The DnaB helicase complexed with ATP-bound DnaC is then recruited to the origin to form a prepriming complex. DnaC, the DnaB-loading factor, forms a tight complex with DnaB protein at a 1:1 molar ratio in the presence of ATP (16–19). Cryo-electron microscopy studies of the DnaC-DnaB complex have revealed that DnaC polypeptides in the forms of three dumbbell-shaped dimers interlock into one of the faces of the hexameric DnaB helicase (20). Upon loading onto the origin DNA, ATP bound to DnaC is hydrolyzed, resulting in release of DnaC and activation of the DnaB helicase (21).
In eukaryotes, Mcm4/6/7 complexes were found to possess DNA helicase activity in vitro, and it was proposed that MCM may play a role in unwinding of the duplex DNA and progression of the replication forks (22, 23). All the six Mcm2–7 polypeptides contain highly conserved DNA-dependent ATPase motifs, and the presence of the intact ATPase motifs in all the six subunits is essential for viability (8, 24). All six Mcm subunits are required for elongation of DNA chains in Saccharomyces cerevisiae, supporting the above proposal (5, 25). Whereas all Mcm subunits are required for the elongation stage of DNA replication in vivo, DNA helicase activity has been identified only in the Mcm4/6/7 complex (23, 26–29).
Cdt1 has been shown to be an essential component of the pre-RC, required for the recruitment of the Mcm proteins to origins. It interacts with Mcm (30) and binds both single-stranded and double-stranded DNA (31). In this report, we have purified and characterized mouse Mcm4/6/7 and Cdt1 proteins. We report here that Mcm4/6/7 forms a complex with multiple molecules of Cdt1 in the presence of ATP, and Cdt1 protein significantly stimulates the DNA binding and helicase activities of Mcm4/6/7. We generated a Cdt1 mutant deficient in the activation of Mcm4/6/7 helicase and DNA binding. Results described in this report suggest a role for Cdt1 protein not only in the loading of MCM onto chromatin but also in execution of its helicase activity.
EXPERIMENTAL PROCEDURES
Cloning of Wild-type and Mutant Forms of Cdt1 Gene into a Baculovirus Transfer Vector and an E. coli Expression Vector—A DNA fragment containing the full-length mouse Cdt1 gene (Gene ID 67177, GI: 31982545) was cloned into the plasmid pFastBac1 (Invitrogen) and pET24a (Novagen). To facilitate the purification of Cdt1 protein in High5 insect cells and E. coli, sequences encoding a six histidine + FLAG tag were added to the C terminus of the Cdt1 gene by PCR. Site-directed mutagenesis of the Cdt1 gene was conducted with the QuikChange site-directed mutagenesis kit (Stratagene). The oligonucleotide 5′-TAGAGCGGATAAGGGCCGCAGAGGTCCAGGCACAGCTGGCAAGGATGACA-3′ was used as the primer to prepare the Cdt1 KKAA mutant.
Expression and Purification of Recombinant Protein in Insect Cells and E. coli—Sf9 and High five insect cells were cultured at 27 °C in Sf-900 II SFM (Invitrogen) and EX-CELL 405 medium (JRH Biosciences), respectively. For expression of the Mcm4/6/7 protein, cells were coinfected with recombinant baculoviruses carrying the Mcm4-His6+Mcm6 genes and those carrying the Mcm7-FLAG at a multiplicity of infection of ∼10 for each virus and then were collected at 48-h postinfection. For expression of the Mcm2∼7 complex, Sf9 cells were coinfected with the combination of Mcm2+Mcm7-His6, Mcm4-His6+Mcm6, and Mcm3+Mcm5-His6-FLAG baculoviruses, and were collected at 48-h postinfection. The recombinant Mcm4/6/7 and Mcm2∼7 complexes in infected High 5 cell lysates were purified with consecutive steps involving nickel-agarose affinity chromatography, anti-FLAG M2 antibody-agarose affinity chromatography, and glycerol gradient sedimentation as previously described (27).
The highly purified recombinant Cdt1 protein was prepared from insect cells and E. coli as follows. To generate recombinant baculovirus expressing the Cdt1 protein, the recombinant bacmid was constructed and used for transfection of the insect cell, according to the manufacturer's protocol (BAC-TO-BAC baculovirus expression system, Invitrogen). For expression of the Cdt1 protein, High five cells were infected with recombinant baculoviruses and were collected at 60-h post-infection. The recombinant Cdt1 protein in the infected cell lysate was purified as follows. The infected insect cells were washed once in cold phosphate-buffered saline and then suspended in lysis buffer (1 ml per 1.5 × 107 High 5 cells) consisting of 10 mm Tris-HCl, pH 7.5, 130 mm NaCl, 1% Triton X-100, 10 mm NaF, 10 mm sodium phosphate buffer, 10 mm Na4P2O7, and Complete protease inhibitor mixture (EDTA-free, Roche Applied Sciences). After incubation for 40 min on ice, insoluble materials were removed by centrifugation at 40,000 rpm (50.2 Ti rotor; Beckman) for 40 min at 4 °C. To 1 volume of the clarified lysate, 1:10 volume of Ni-NTA (nitrilotriacetic acid)-agarose (Qiagen) pre-equilibrated with buffer A (50 mm sodium phosphate buffer, pH 6.0, 300 mm NaCl, 10% glycerol, 0.01% Triton X-100, and 0.1 mm phenylmethylsulfonyl fluoride) was added. After incubation at 4 °C for 2 h on a rocking platform, the beads were collected and washed six times with 10 bed volumes of buffer A containing 10 mm imidazole. Bound proteins were eluted stepwise in four bed volumes of buffer A containing 200 mm imidazole. The eluted fractions containing Cdt1 was mixed with anti-FLAG M2 Ab-agarose beads (Sigma) equilibrated with buffer A (same as above except that the pH was adjusted at 7.5), and incubated at 4 °C for 1 h on a rocking platform. After washing four times with 10 bed volumes of buffer A (pH 7.5), the Cdt1 protein was eluted stepwise with three bed volumes of buffer A (pH 7.5) containing 0.2 mg/ml of the FLAG peptide. Further purification of the Cdt1 protein was carried out by 15–35% or 15–30% linear glycerol gradient (2 ml) centrifugation at 38,000 rpm for 17 h (TLS55 rotor; Beckman). The buffers used in the gradient was 0.15 m NaCl, 20 mm Tris-HCl, pH 7.5, 0.5 mm EDTA, 1 mm DTT, 0.1 mm phenylmethylsulfonyl fluoride, and 0.01% Triton X-100. Fractions (130 μl each) were collected and were analyzed by SDS-PAGE, followed by staining with silver.
His-FLAG or T7-tagged Cdt1 was overproduced in the E. coli strain BL21 (DE3) codon plus grown in LB medium with 30 μg/ml kanamycin and 50 μg/ml chloroamphenicol. Isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.5 mm, and the cells were incubated for an additional 2 h at 28 °C. Cells were suspended in lysis buffer and incubated at 4 °C for 30 min on a rocking platform after addition of 0.2 mg/ml lysozyme. The suspension was sonicated to reduce viscosity. The procedures for purification using the NTA column, FLAG antibody resin, and glycerol gradient centrifugation were the same as described above. After NTA and FLAG antibody purification, the elute was applied to a Mono Q PC 1.6/5 SMART column (GE Healthcare LifeScience) equilibrated with Buffer Q (25 mm Tris-HCl, pH 7.5, 10% glycerol, 0.5 mm EDTA, 1 mm DTT, and 0.01% Triton X-100) with 60 mm NaCl. The elution was carried out with Buffer Q containing a linear gradient from 0 to 1 m NaCl. Further purification was performed using T7-tag antibody-agarose beads according to the manufacturer's instructions (Novagen). Finally, glycerol gradient centrifugation was carried out.
Reagents—Labeled and unlabeled dNTPs/rNTPs were purchased from GE Healthcare LifeScience. M13mp18 single-stranded circular DNA (ssDNA) and T4 polynucleotide kinase were from TAKARA, and anti-FLAG M2 antibody-agarose and FLAG peptide were from Sigma. Oligonucleotides were purchased from Hokkaido System Science Co., Ltd. (Hokkaido, Japan), and were purified on polyacrylamide gel, followed by purification by Sep-Pak Plus C18 cartridge column (Waters).
Construction of Partial Heteroduplex Substrates for Helicase Assays—The oligonucleotide [5′-GTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGT-(dT)40-3′] (carrying the 40-mer oligo(dT) tail at the 3′-end of the hybridizing 37-mer sequence) was first labeled with [γ-32P]ATP and T4 polynucleotide kinase. The labeled dT40-37-mer (37-mer region hybridizing at positions 6289–6326 of M13mp18 DNA) was annealed to the single-stranded circular M13mp18 DNA in a reaction mixture containing 20 mm Tris-HCl, pH 7.5, and 10 mm MgCl2, which were heated to 95 °C, kept at 67 °C for 1 h, and then allowed to slowly cool down to 37 °C. The labeled substrates were purified by Sepharose CL4B column chromatography (GE Healthcare LifeScience).
Gel Shift and DNA Helicase Assays—In gel shift assays, the 77-mer oligonucleotide [5′-GTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGT-(dT)40-3′], the 5′-terminus of which was labeled with [γ-32P]ATP and T4 polynucleotide kinase, was used as a substrate. Mcm4/6/7 protein was incubated at 30 °C for 30 min in reaction mixtures (15 μl) containing 25 mm Hepes-NaOH, pH 7.5 or 25 mm Tris-HCl, pH 7.5, 50 mm sodium acetate, 10 mm Mg(CH3COO)2, 20 mm 2-mercaptoethanol, 0.25 mg/ml bovine serum albumin, 10 mm ATP, and labeled substrates of the amount indicated. After addition of 2 μl of 50% glycerol, the reaction mixtures were directly applied on a nondenaturing polyacrylamide gel in 1× Tris borate-EDTA (TBE), and electrophoresis was conducted at room temperature. For DNA helicase assays, the reaction mixtures were incubated at 37 °C for 1 h. The reactions were terminated by addition of EDTA (20 mm), SDS (0.1%), and 2 μg of proteinase K, and were incubated for an additional 15 min. The samples were separated by electrophoresis on a nondenaturing polyacrylamide gel in 1× TBE.
Pull-down Assays using Biotinylated Oligonucleotide—A 5′-biotinylated oligonucleotide (bio-oligo) (5′-GTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGT-dT40-3′) was purchased from Hokkaido System Science Co., Ltd. (Hokkaido, Japan). Mcm4/6/7 and/or Cdt1 and bio-oligo (50 pmol) were incubated at 30 °C for 30 min in buffer (20 μl) containing 20 mm Tris-HCl, pH 7.9, 50 mm sodium acetate, 10 mm Mg(CH3COO)2, 1 mm DTT, 0.25 mg/ml casein, 2 mm ATP, and 0.01% Triton X-100. Streptavidin-coated M-280 magnetic beads (Dynal Biotech ASA) was added to the mixture and incubated for 30 min at 4 °C. After the removal of the supernatant, the beads were washed in buffer (200 μl) containing of 20 mm Tris-HCl, pH 7.9, 50 mm sodium acetate, 10 mm Mg(CH3COO)2, 1 mm DTT, 0.01% Triton X-100, and 10% glycerol, resuspended in 15 μl of SDS sample buffer, and were analyzed on 4–20% SDS-PAGE.
Immunoprecipitation—Immunoprecipitation experiments were performed using T7-antibody beads (Novengen). Pre-washed T7-antibody beads were mixed with indicated proteins in buffer containing 10 mm Hepes-NaOH, pH 7.5, 100 mm sodium acetate, 2 mm Mg(CH3COO)2, 2 mm ATP, 10% glycerol, and 0.01% Triton X. After 1 h of incubation, beads were washed with the above buffer, and bound proteins were eluted with 0.1 m citric acid, pH 2.2. The samples were run on SDS-PAGE, followed by silver staining.
RESULTS
Mcm2/3/4/5/6/7, Mcm4/6/7, and Mcm2/4/6/7 Form Complexes with Cdt1—During the loading and translocation on the template DNA, the Mcm helicase is expected to interact with many factors including Cdt1, a loading factor for Mcm. We examined the interaction between Cdt1 and various forms of Mcm complexes by glycerol gradient centrifugation. As shown in Fig. 1A, the purified Cdt1 protein sedimented predominantly in the fractions 12–13, at a molecular weight of about 70,000. The Mcm4/6/7 complex sedimented at around fractions 5– 6, expected for a dimer of two trimers as previously reported (Fig. 1B) (23). When Cdt1 was mixed with Mcm4/6/7, the peak moved to around fraction 4 corresponding to a molecular mass of 670 kDa (Fig. 1C). The Mcm2/3/4/5/6/7 or Mcm2/4/6/7 complex also cosedimented with a portion of Cdt1 in glycerol gradient centrifugation (Fig. 1, D and E).
FIGURE 1.
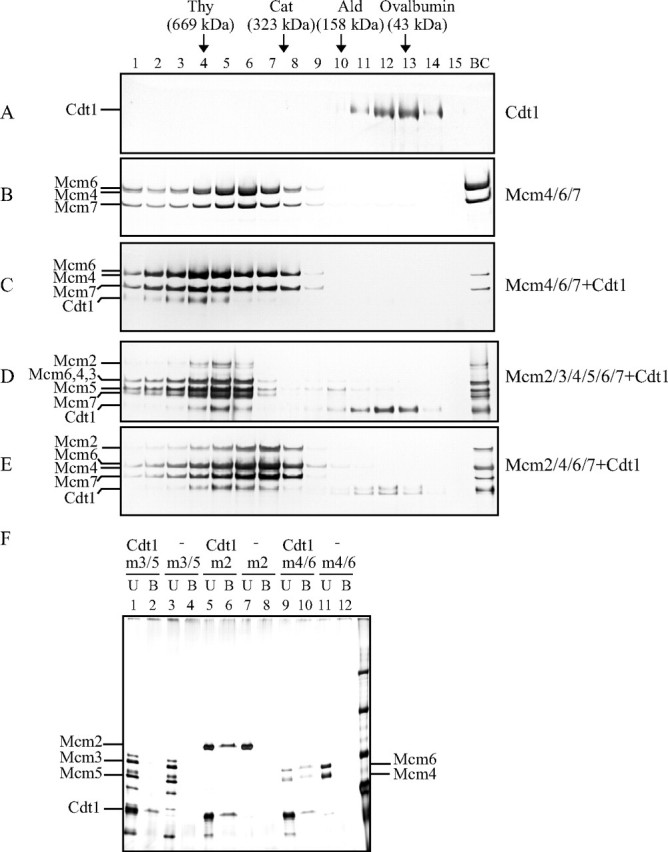
Interaction between Mcm and Cdt1. A–E, purified proteins, singly or in combination, were fractionated by centrifugation at 36,000 rpm for 17 h on 15–35% glycerol gradient. Each fraction was run on 4–20% SDS-PAGE, and proteins were stained with silver. The buffer used in the gradient was 50 mm sodium acetate, 50 mm Hepes-Na, pH 7.5, 0.5 mm EDTA, 1 mm DTT, 0.1 mm phenylmethylsulfonyl fluoride, 0.01% Triton X-100, 2 mm Mg-acetate, and 1 mm ATP. A, Cdt1; B, Mcm4/6/7; C, Mcm4/6/7+Cdt1; D, Mcm2/3/4/5/6/7+Cdt1; E, Mcm2/4/6/7+Cdt1. The positions of molecular weight markers are indicated along with their molecular sizes. BC, samples applied. F, purified Mcm3/5 (m3/5), Mcm4/6 (m4/6), and Mcm2 (m2) proteins were mixed with T7-tagged Cdt1 purifiedfromE. coli, and immunoprecipitation was performed using T7-antibody beads. Unbound (U) and bound (B) fractions were analyzed on 7.5% SDS-PAGE, and proteins were visualized by silver staining.
To find out which subunit of Mcm directly interacts with Cdt1, purified Mcm4/6 complex, Mcm3/5 complex, or Mcm2 was mixed with T7-tagged Cdt1 protein, and immunoprecipitation using T7-antibody beads was performed. Mcm2 and Mcm4/6 complex, but not Mcm3/5 complex, was pulled down with the beads, indicating that Cdt1 can interact with the Mcm2 and Mcm4/6 complex (Fig. 1F).
Cdt1 Stimulates DNA Binding of Mcm—Cdt1 protein can bind both single-stranded and double-stranded DNA, and Mcm4/6/7 binds single-stranded DNA (23, 31). When Cdt1 protein alone was incubated with ssDNA, it generated smeared shifted bands in electrophoretic mobility shift assays on polyacrylamide gel (Fig. 2A, lane 16). On the other hand, Mcm4/6/7 alone incubated with ssDNA formed a single sharp mobility-shifted band along with fast-migrating smeared bands, which may have arisen from dissociation of the complex during the electrophoresis (Fig. 2A, lane 2). When Cdt1 was added to the reaction mixtures containing a fixed amount of Mcm4/6/7, the mobility of the complex was retarded, indicative of formation of a trimolecular complex containing Mcm4/6/7, Cdt1, and DNA. The amount of this complex increased, as more Cdt1 protein was added, suggesting a possibility that Cdt1 promotes binding of the Mcm complex to DNA. Quantification indicated that the stimulation was as much as 7-fold (Fig. 2A, right). Cdt1 did not significantly affect DNA binding of T-antigen helicase under the identical condition (Fig. 2A, right). Similar results were obtained in DNA binding assays using Cdt1 purified from E. coli (Fig. 2C).
FIGURE 2.
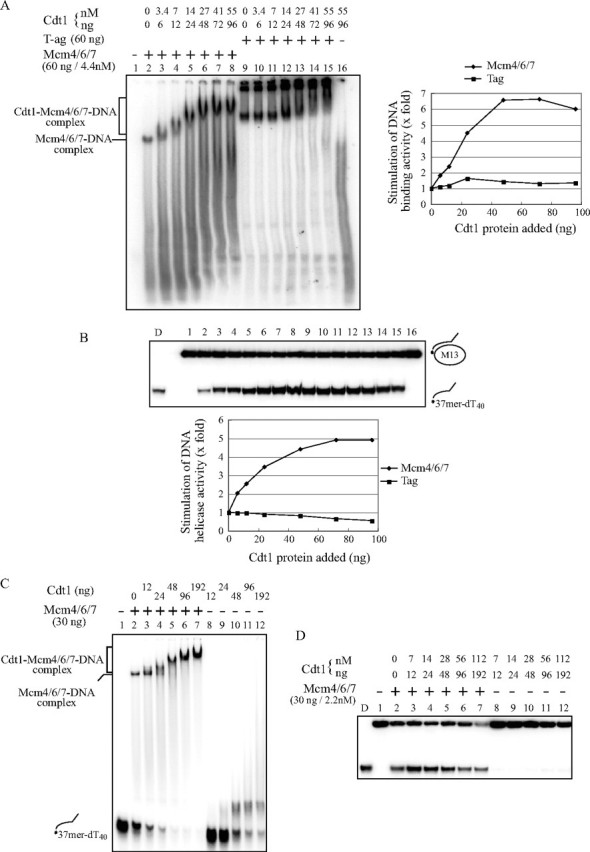
Cdt1 stimulates DNA binding and helicase activities of Mcm4/6/7. DNA binding (A and C) and helicase (B and D) activities were examined with a constant amount of Mcm4/6/7 (60 ng/4.4 nm (A and B) or 30 ng/2.2 nm (C and D)) and various amounts of Cdt1 protein purified from insect cells (A and B) or from E. coli (C and D). A and C, DNA binding activity was measured in gel shift assays using ssDNA (20 fmol) as substrates. Complexes were separated on a 5% native polyacrylamide gel. The bands indicated by brackets in lanes 3– 8 (A) or lanes 3–7 (C) represent the Cdt1-Mcm4/6/7-DNA complex. B and D, DNA helicase assays were conducted as in A and C, respectively, except that the partial heteroduplex substrate (10 fmol) was used. Because the purified Cdt1 protein from the insect cell contained 2 mm ATP, complement buffer was added to maintain the final reaction mix as follows: 50 mm Tris-HCl, pH 7.6, 60 mm sodium acetate, 6% glycerol, 0.3 mm EDTA, 0.6 mm DTT, 20 mm 2-mercaptoethanol, 0.006% Triton X-100, 0.25 mg/ml bovine serum albumin, 11.2 mm MgOAc, and 11.2 mm ATP. Half of the reaction was incubated at 30 °C for the gel-shift assay and the remainder was incubated at 37 °C for the DNA helicase assay (A and B). In A, the extent of DNA binding with Mcm or Tag alone was taken as 1, and the relative efficiency of DNA binding in the presence of various amounts of Cdt1 protein is presented for each helicase. In B, the level of the displaced oligonucleotide was quantified, and that with Mcm or Tag alone was taken as 1. The relative level of DNA unwinding in the presence of various amounts of Cdt1 protein is presented for each helicase. D, heat-denatured substrates.
We also conducted pull-down assays using a biotin-labeled ssDNA to examine the effect of Cdt1 on DNA binding of Mcm. Almost all the Cdt1 protein bound to the biotin-labeled DNA (supplemental Fig. S1, lane 4), suggesting that the Cdt1 protein possesses strong ssDNA binding activity under this condition. Mcm4/6/7 was also pulled down by the biotin DNA under the same condition, and this binding was significantly stimulated by the presence of Cdt1 protein (supplemental Fig. S1, compare lanes 2 versus 3, 5 versus 6, 8 versus 9, and 11 versus 12). Cdt1-mediated stimulation of DNA binding of Mcm was more prominent in the absence of a nucleotide than in its presence. Strikingly, the addition of ATPγS or ADP decreased DNA binding of Mcm4/6/7 in the presence of Cdt1, whereas addition of ATP did not significantly affect it under the same condition. DNA binding assays using the glycerol gradient fractions of Mcm2/3/4/5/6/7+Cdt1 or Mcm4/6/7+Cdt1 also indicated increased formation of a complex of slower mobility compared with the Mcm complexes alone (data not shown). The above results on Cdt1-mediated promotion of DNA binding of Mcm may reflect an aspect of the Mcm-chromatin loading activity of Cdt1 protein.
Cdt1 Stimulates DNA Helicase Activity of Mcm—Because Cdt1 protein forms a complex with Mcm4/6/7 and increases its DNA binding activity, we examined whether Cdt1 protein can stimulate the helicase activity of Mcm4/6/7. The same reaction mixtures for DNA binding assays (Fig. 2A) were examined for DNA helicase activities (Fig. 2B). Addition of increasing amounts of Cdt1 significantly stimulated the helicase activity of Mcm4/6/7, whereas that of T-antigen helicase was not affected under the same condition (Fig. 2B, lower). Cdt1 stimulated the helicase activity of Mcm4/6/7 as much as 5-fold (lanes 7 or 8 in Fig. 2B). The isolated Mcm4/6/7-Cdt1 complex exhibited more efficient DNA binding and helicase activities than Cdt1-free Mcm4/6/7 complex (supplemental Fig. S2). However, addition of excess amount of Cdt1 led to inhibition of the helicase activity (Fig. 2D). Titration of Cdt1 indicated that addition of more than 18 molecules of the Cdt1 monomer per Mcm4/6/7 hexamer resulted in inhibition. Excess Cdt1 may compete with Mcm for binding to the substrate DNA and titrate out the effective Mcm4/6/7 helicase. These behaviors of Cdt1 are similar to the known effect of DnaC on DnaB helicase activity (21, 32, 33).
Geminin binds to Cdt1 and inhibits its activity. We purified the wild-type geminin and a mutant geminin lacking the coiled-coiled domain. This mutant geminin does not interact with Cdt1 protein, as judged from immunoprecipitation assays (data not shown). Fig. 3A shows that addition of the wild-type geminin to the helicase reaction in the presence of Cdt1 resulted in strong inhibition of the helicase activity, whereas the mutant geminin did not affect the increase of helicase activity by Cdt1. Because geminin alone did not affect the helicase action of Mcm4/6/7 at the same concentrations (data not shown), almost complete inhibition by geminin (lanes 6 and 7) may be due to the inhibitory effect of the geminin-Cdt1 complex on Mcm helicase. In DNA binding assays, the wild-type geminin, when added to the Cdt1-DNA complex, formed a new complex migrating at a distinct position which represents the Cdt1-geminin-DNA ternary complex (Fig. 3B, lanes 5 and 6). When added to the Mcm-Cdt1-DNA complex, it disrupted the slow-migrating complex, regenerating the Mcm-DNA complex (Fig. 3B, lanes 9 and 10). In contrast, the mutant geminin did not affect the Mcm-Cdt1-DNA complex even at a high concentration (Fig. 3B, lanes 11 and 12), in keeping with the preservation of helicase activity under the same conditions. These results clearly indicate that the stimulation of the helicase activity was due to the action of Cdt1 protein. Although Cdt1 stimulated DNA binding activity of Mcm2/3/4/5/6/7 hexamer on single-stranded DNA, the Mcm heterohexamer did not show any DNA helicase activity even in the presence of Cdt1 protein (data not shown).
FIGURE 3.
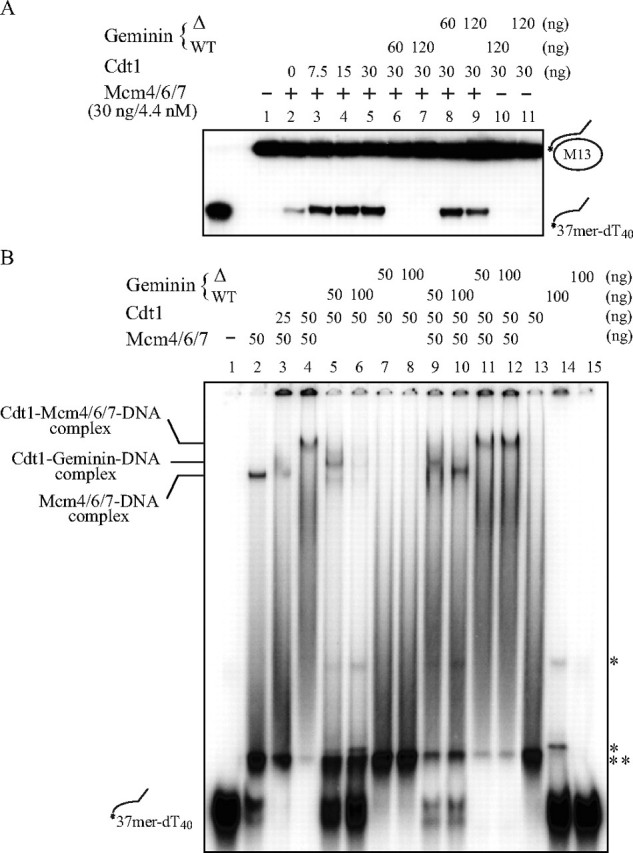
Effect of geminin on helicase and DNA binding activities of Mcm4/6/7 in the presence of Cdt1. The helicase (A) and gel shift (B) assays were carried out as described under “Experimental Procedures.” Amounts of proteins used are shown above each panel. Geminin WT and Δ represent, respectively, the wild-type protein and a mutant protein lacking the Cdt1-interacting domain. The wild-type geminin but not the mutant strongly inhibited the helicase activity of Mcm4/6/7 in the presence of Cdt1 protein and disrupted the Cdt1-Mcm4/6/7-DNA complex, regenerating the Mcm4/6/7-DNA complex. * indicates weak shifted bands generated by the wild-type geminin preparation. The band indicated by ** appears only when Tris and Hepes buffers derived from the reaction mix and protein preparations, respectively, are mixed, and could represent the substrate DNA adopting a different conformation.
The above results point to functional similarity between Cdt1 and DnaC proteins, the latter of which binds to DnaB helicase and stimulates its helicase activity. In light of critical roles of ATP in regulation of the DnaC-DnaB complex, we then examined the effect of ATP concentration on the DNA binding and helicase activities of Mcm4/6/7 in the absence and presence of Cdt1 protein (Fig. 4A). The optimum ATP concentration for the helicase reaction in the presence of Cdt1 was 10 mm, but DNA binding of Mcm4/6/7 did not require ATP either in the presence or absence of Cdt1 (Fig. 4A, lower). In contrast, DNA binding was inhibited, as more ATP was added (Fig. 4B).
FIGURE 4.
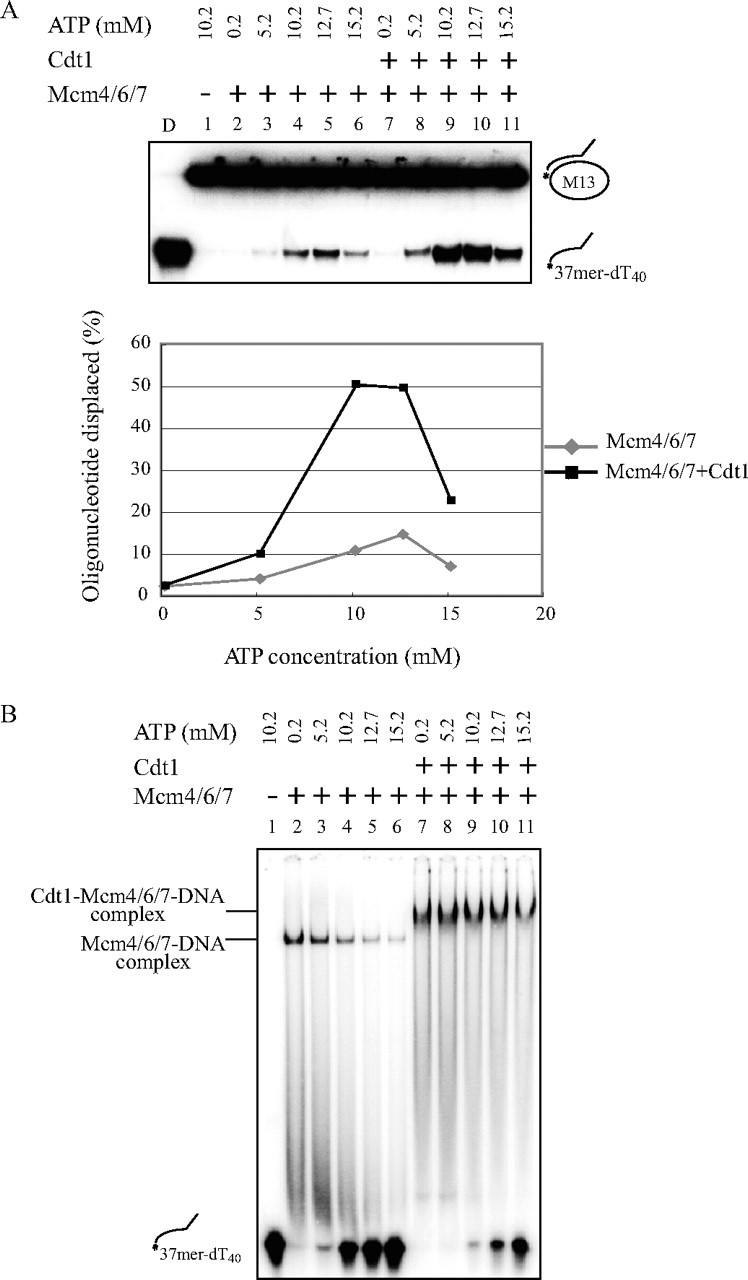
Effect of ATP concentration on DNA binding and helicase activities of Mcm4/6/7 in the absence and presence of Cdt1. DNA helicase (A) and DNA binding (B) activities of Mcm4/6/7 (40 ng) in the absence (lanes 2–6) or presence (lanes 7–11) of Cdt1 (30 ng) were examined with increasing concentrations of ATP. The level of DNA helicase activity, calculated as percent oligonucleotide displaced, is presented at various ATP concentrations. The stimulation of helicase activity by Cdt1 was more prominent at 10–13 mm ATP.
Mcm4/6/7-Cdt1 Complex Formation in the Presence of ATP—We then examined whether the presence of ATP affects the nature of the Mcm4/6/7-Cdt1 complexes. When Mcm4/6/7 and Cdt1 were mixed and were subjected to glycerol gradient centrifugation in the presence of ATP, most of the Cdt1 protein cosedimented with the Mcm4/6/7 complex (Fig. 5A). The peak fractions corresponded to the protein marker thyroglobulin with an estimated molecular mass of about 650 kDa. This value is larger than the Mcm4/6/7 protein alone in the presence of ATP, which sediments at ∼450 kDa (Fig. 5E). We previously reported that the Mcm4/6/7 in the absence of ATP sedimented at ∼350 kDa in glycerol gradient and migrates at 550 kDa in native acrylamide gel (22, 23). This was confirmed in Fig. 5F. Meanwhile, Cdt1 protein alone peaked at 70 kDa (fraction 12) as expected for a monomer under this condition (Fig. 5G). In the absence of ATP, on the other hand, the Mcm4/6/7-Cdt1 complex sedimented in a rather broad peak at around 350 kDa, smaller than the Mcm-Cdt1 complex in the presence of ATP (Fig. 5B). The Mcm4/6/7-Cdt1 complex formation was confirmed also in the regular native gel or blue native polyacrylamide gel (BN-PAGE, data not shown).
FIGURE 5.

Cdt1 forms high molecular weight complexes with Mcm4/6/7 in the presence of ATP. Purified Mcm4/6/7 with or without Cdt1 protein (purified from insect cells; A and B, wild-type; C and D, KKAA mutant) were fractionated by glycerol gradient (15–30%). The buffer used in the gradient was 50 mm sodium acetate, 50 mm Hepes-Na, pH 7.5, 0.5 mm EDTA, 1 mm DTT, and 0.01% Triton X-100, and 2 mm Mg-acetate, with (A, C, E, and G) or without (B, D, and F) 1 mm ATP. Centrifugation was at 38,000 rpm for 17 h. Each fraction was run on 4–20% SDS-PAGE, and proteins were stained with silver. The positions of molecular weight markers are indicated along with their molecular sizes. H, Cdt1 KKAA mutant protein (purified from insect cells) fractionated by glycerol gradient (15–30%) at 45,000 rpm for 20 h.
The ratio of the protein amount of Mcm4/6/7 and Cdt1 was 3:1 in the peak fractions (fraction 2) of the glycerol gradient in Fig. 5A. Taking the molecular masses of Cdt1 (70 kDa) and Mcm4/6/7 (300 kDa) into account, a molar ratio of Cdt1 to Mcm is estimated to be ∼3:2. Thus, in the presence of ATP, about three Cdt1 molecules may bind to the two Mcm4/6/7 trimers (hexamers). In contrast, the Mcm4/6/7-Cdt1 complexes in the absence of ATP was more heterogeneous in size, and the amount of Cdt1 associated with Mcm4/6/7 was lower than the complex in the presence of ATP (Fig. 5B). These data suggest that the Mcm4/6/7-Cdt1 complex exists as a mixture of different forms with different numbers of bound Cdt1 molecules, and that a large complex containing three Cdt1 molecules may be preferentially generated in the presence of ATP.
A Mutant Cdt1 Protein Fails to Activate Mcm4/6/7 Helicase—To identify residues critical for stimulation of MCM helicase, we generated a mutant Cdt1 (KKAA) in which two lysine residues in the 441KXXXK445 were change to alanine (Fig. 6A). The Lys441 residue is conserved from fission yeast to human. We purified the mutant protein after overexpression in insect cells or in E. coli.
FIGURE 6.

Biochemical characterization of Cdt1 (KKAA) mutant protein. A, schematic drawing of domain structures of Cdt1, and the mutagenized amino acids. B, using the identical amount of wild-type or KKAA Cdt1 protein, the effect on DNA binding (lower panel) and helicase (upper panel) activities of Mcm4/6/7 were examined. The proteins used were; 30 ng of Mcm4/6/7 and 10, 20, 40, and 80 ng of Cdt1. C, kinetics of helicase reactions using 30 ng of Mcm4/6/7 and 15 ng of Cdt1 (wild type or KKAA). D, pull-down assay was carried out with 50 ng of Cdt1 (wild type or KKAA) and 50 pmol of biotinylated oligonucleotide in the presence or absence of ATP. The bound (left half) and unbound (right half) proteins are shown. * indicates nonspecific bands. E, purified Mcm4/6/7 (200 ng) was mixed with the same amount (200 ng) of wild-type or KKAA T7-tagged Cdt1, and immunoprecipitation experiments were performed using T7-antibody beads as described under “Experimental Procedures,” except that beads were further washed twice with T7-wash buffer (4.29 mm Na2HPO4, 1.47 mm KH2PO4, 2.7 mm KCl, 137 mm NaCl, and 0.1% Tween-20, pH 7.3). F, purified geminin (200 ng) was mixed with same amount (200 ng) of wild-type or KKAA T7-tagged Cdt1, and immunoprecipitation experiments were performed using T7-antibody beads as described under “Experimental Procedures.” In D, E, and F, the samples were run on 4–20% SDS-PAGE, and the proteins were detected by silver staining. In E and F: I, input; U, unbound; B, bound. Cdt1 was overexpressed and purified from insect cells (B and C) or from E. coli (D, E, and F). The KKAA mutant did not promote the high molecular weight complex formation with Mcm at a higher concentration and did not stimulate the helicase activity of Mcm4/6/7. The interaction of the mutant Cdt1 with Mcm was significantly reduced, although it interacted with DNA and with geminin as efficiently as wild-type.
We next examined the activity of the mutant protein in DNA binding and helicase assays of Mcm4/6/7. The KKAA mutant showed very little stimulatory effect on the helicase activity of Mcm4/6/7 and only slightly stimulated its DNA binding activity (Fig. 6B). The slow-migrating bands representing the Mcm-Cdt1-DNA complex were not detected and the smeared bands representing Cdt1-DNA increased, suggesting that the KKAA mutant Cdt1 fails to generate a complex with Mcm (Fig. 6B, lower, lane 10). Kinetics of helicase assays with the identical amounts of the wild-type or mutant Cdt1 protein also indicated very little or no stimulatory effect of the mutant protein on the Mcm helicase activity (Fig. 6C). We have also conducted helicase and DNA binding assays with the wild-type and mutant recombinant Cdt1 protein generated in E. coli, and obtained similar results (supplemental Fig. S3). This indicates that the reduced activity of the mutant protein is not due to differential modification (such as phosphorylation).
To clarify the nature of the mutant protein, we examined DNA binding activity and protein-protein interactions. DNA binding activity of the mutant protein in a pull-down assay with biotinylated DNA was comparable to that of the wild-type protein (Fig. 6D). Interaction between T7-tagged Cdt1 and Mcm4/6/7 complex was examined with immunoprecipitation with anti-T7-tag antibody. The mutant Cdt1 was found to interact with Mcm4/6/7 with reduced affinity (Fig. 6E), consistent with the results of gel shift assays (Fig. 6B). In the control, binding of the mutant Cdt1 protein to geminin was not affected (Fig. 6F). These results indicate that the KKAA mutant interacts with Mcm with reduced affinity and fails to stimulate DNA binding and helicase activities of Mcm4/6/7. The mutant Cdt1 did not generate the high molecular weight complex as efficiently as the wild-type did even in the presence of ATP (Fig. 5, C and D), and the amount of the cosedimenting Cdt1 polypeptide was significantly reduced, reflecting its reduced affinity to Mcm. The mutant Cdt1 alone behaved as a monomer, as the wild-type did (Fig. 5H). We also detected the Mcm-Cdt1 complex in native polyacrylamide gel electrophoresis, but this complex was not detected with the KKAA mutant protein (data not shown). These results strongly indicate that the mutant Cdt1 fails to generate a helicase-active high molecular weight Mcm-Cdt1 complex because of its reduced affinity to Mcm.
DISCUSSION
DNA helicase plays a central role during the course of DNA replication and loading of the replicative helicase onto a template DNA is a crucial event for initiation of DNA replication. In E. coli, DnaB is the replicative helicase, and DnaC forms a heterohexamer with DnaB (DnaC6-DnaB6) to deliver the DnaB helicase onto the unwound single-stranded DNA at the origin (17, 34). Formation of the pre-RC in eukaryotes is carried out by assembly of proteins such as the ORC, Cdc6, Cdt1, and the Mcm protein complex at a replication origin. Mcm is the central component for the replicative helicase, and Cdc6 and Cdt1 have been shown to play roles in delivering Mcm onto replication origins. Among them, Cdt1 is known to interact with Mcm and is critical for loading of Mcm onto the ORC-Cdc6-DNA complex. In this report, we have conducted detailed analyses of physical and functional interactions of mammalian Cdt1 with Mcm. The results reported here indicate functional similarity between Cdt1 and DnaC.
Mechanisms of Stimulation of Mcm4/6/7 Helicase by Cdt1— Highly purified Cdt1 protein forms complexes with various Mcm assembly and stimulates DNA helicase activity of Mcm4/6/7 at an optimal concentration. Coimmunoprecipitation assays indicate that Cdt1 interacts with Mcm2 and Mcm4 –6 proteins. Furthermore, Cdt1 stimulates DNA binding activity of Mcm4/6/7 in gel-shift and biotin-DNA pull-down assays. Cdt1 forms a high molecular weight complex in the presence of ATP with Mcm4/6/7, which may contain three molecules of Cdt1 protein per Mcm4/6/7 dimer, and generation of this form may be correlated with high DNA helicase activity. The KKAA mutant is not capable of activating the Mcm4/6/7 helicase and also is reduced in its ability to interact with MCM. We previously reported that the C-terminal region (residues 407– 477) of Cdt1 is responsible for binding to Mcm4/6/7 (31), suggesting a possibility that the mutated lysine residues in the KKAA mutant (positions 441 and 445) may be involved in interaction with Mcm. In contrast, the mutant protein interacts with DNA as efficiently as wild-type, indicating that the ability of Cdt1 to interact with DNA is not sufficient for activation of Mcm helicase. However, an N-terminal truncation mutant of Cdt1, which is capable of interacting with Mcm but not with DNA also has lost the ability to stimulate the Mcm helicase (data not shown), suggesting that both Mcm and DNA-interacting domains of Cdt1 are required for stimulation of Mcm helicase. Because Cdt1 dissociates from chromatin and gets degraded after initiation of S phase, it is unlikely that it acts as a part of the helicase complex at the fork. Instead, Cdt1-assisted Mcm complex may facilitate the initial opening of the duplex DNA (35).
On the other hand, Mcm4/6/7 helicase was inhibited in the presence of excess amount of Cdt1. Careful titration indicated that the presence of more than 18-fold molar excess Cdt1 molecule per one Mcm4/6/7 hexamer leads to inhibition. Results from glycerol gradient analyses were also consistent with the formation of (Mcm4/6/7)2Cdt13 complex in the presence of ATP. At the fork, the Mcm4/6/7 protein forms a double hexamer (27, 29), and a dimer of (Mcm4/6/7)2Cdt13 complex may be generated. Excess free Cdt1 may inhibit the helicase reaction by directly binding to substrate DNA, thus competing out Mcm4/6/7. Consistent with the inhibitory effect of Cdt1 on duplex unwinding, Cdt1 is rapidly released from chromatin once S phase is initiated (36).
Similarity between Cdt1 and DnaC—DnaC of E. coli is a DnaB-loading protein and utilizes ATP binding to assemble the DnaB helicase at the origin. In the presence of ATP, a maximum of six DnaC monomers bind cooperatively to the DnaB hexamer to generate a DnaC6-DnaB6 heterohexamer (16, 17, 19). Once DnaB is delivered to the origin, DnaC is released from both DnaB and the origin (34, 38). DnaC stimulates DnaB helicase activity, and excess DnaC inhibits DnaB helicase (21, 37). Together with the fact that ATP can be UV-cross-linked to Cdt1 protein in vitro,3 these point to a significant similarity between the Cdt1 and DnaC proteins. On the other hand, there is very little similarity on the amino acid level between the two proteins. In E. coli, DNA-dependent ATPase activity of DnaB was reported to be unaffected by dnaC (16), but more recently was reported to be activated by DnaC (37). DNA-dependent ATPase activity of Mcm4/6/7 protein was not affected by Cdt1 protein, and Cdt1 alone exhibited very little or no ATPase activity (data not shown).
Another protein, Cdc6, an ATP-binding protein, is also required for assembly of the Mcm helicase at the origin in vivo. Cdc6 is a member of the AAA+ family of ATPases, shows sequence similarity to DnaC and was thought to be analogous to the DnaC helicase loader (5, 39). It is known that both Cdc6 and Cdt1 proteins are required for association of Mcm with replication origins in vivo (5). Cdt1 interacts with Cdc6 protein in vitro (40).3 Thus, it is possible that the combined actions of Cdt1 and Cdc6 are required for loading of Mcm, as in the case of Bacillus subtilis DnaB and DnaI, which are a pair of helicase loaders required for the loading of the Bacillus replicative helicase DnaC (41). However, Cdc6 shares strong sequence similarity with Orc1 (42, 43), suggesting that Cdc6 may be a part of the ORC initiator assembly on which pre-RC would be assembled. In fact, Stillman's group recently showed in a single-particle reconstruction of electron microscopic images that the ORC-Cdc6 complex forms a ring-shaped structure with dimensions similar to those of the ring-shaped MCM helicase. Thus, Cdc6 could be regarded as an additional subunit of the ORC complex (43, 44). In contrast, the Cdt1 forms a stable complex with Mcm proteins in vivo (30, 45) and in vitro (this study). Taken together, we would like to propose the possibility that Cdt1 may be the functional homolog of DnaC protein in E. coli. Future studies on the structure of the Mcm-Cdt1 complex as well as its interaction with ATP would be needed to test this hypothesis.
Supplementary Material
Acknowledgments
We thank Dr. Hideo Nishitani for his generous gifts of various reagents (antibodies, plasmid DNA etc.) and for very helpful discussions. We also thank Dr. Masatoshi Fujita for his gifts of plasmids and proteins, and Dr. Haruhiko Takizawa and Kouji Ohide for collaboration on experiments using Xenopus egg extracts. We also thank Dr. Yukio Ishimi for his critical reading of the manuscript, Dr. Seiji Matsumoto and Naoko Kakusho for their experiments, and other members of the laboratory for useful discussions.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
Footnotes
The abbreviations used are: pre-RC, pre-replicative complex; DTT, dithiothreitol; NTA, nitrilotriacetic acid; ssDNA, single-stranded DNA; ATPγS, adenosine 5′-O-(thiotriphosphate); ORC, origin recognition complex; Mcm, minichromosome maintenance.
Z. You and H. Masai, unpublished data.
References
- 1.Cocker, J. H., Piatti, S., Santocanale, C., Nasmyth, K., and Diffley, J. F. (1996) Nature 379180 –182 [DOI] [PubMed] [Google Scholar]
- 2.Maiorano, D., Lemaitre, J. M., and Mechali, M. (2000) J. Biol. Chem. 2758426 –8431 [DOI] [PubMed] [Google Scholar]
- 3.Nishitani, H., Lygerou, Z., Nishimoto, T., and Nurse, P. (2000) Nature 404 625– 628 [DOI] [PubMed] [Google Scholar]
- 4.Wohlschlegel, J. A., Dwyer, B. T., Dhar, S. K., Cvetic, C., Walter, J. C., and Dutta, A. (2000) Science 2902309 –2312 [DOI] [PubMed] [Google Scholar]
- 5.Bell, S. P., and Dutta, A. (2002) Annu. Rev. Biochem. 71333 –374 [DOI] [PubMed] [Google Scholar]
- 6.Mendez, J., and Stillman, B. (2003) Bioessays 251158 –1167 [DOI] [PubMed] [Google Scholar]
- 7.Koonin, E. V. (1993) Nucleic Acids Res. 212541 –2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blow, J. J., and Dutta, A. (2005) Nat. Rev. Mol. Cell Biol. 6476 –486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li, S. S., Xue, W. C., Khoo, U. S., Ngan, H. Y., Chan, K. Y., Tam, I. Y., Chiu, P. M., Ip, P. P., Tam, K. F., and Cheung, A. N. (2005) Histopathology 46307 –313 [DOI] [PubMed] [Google Scholar]
- 10.Lutzmann, M., Maiorano, D., and Mechali, M. (2006) EMBO J. 255764 –5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machida, Y. J., Hamlin, J. L., and Dutta, A. (2005) Cell 12313 –24 [DOI] [PubMed] [Google Scholar]
- 12.Ballabeni, A., Melixetian, M., Zamponi, R., Masiero, L., Marinoni, F., and Helin, K. (2004) EMBO J. 233122 –3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihaylov, I. S., Kondo, T., Jones, L., Ryzhikov, S., Tanaka, J., Zheng, J., Higa, L. A., Minamino, N., Cooley, L., and Zhang, H. (2002) Mol. Cell Biol. 221868 –1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wuarin, J., Buck, V., Nurse, P., and Millar, J. B. (2002) Cell 111419 –431 [DOI] [PubMed] [Google Scholar]
- 15.Xouri, G., Squire, A., Dimaki, M., Geverts, B., Verveer, P. J., Taraviras, S., Nishitani, H., Houtsmuller, A. B., Bastiaens, P. I., and Lygerou, Z. (2007) EMBO J. 261303 –1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobori, J. A., and Kornberg, A. (1982) J. Biol. Chem. 25713770 –13775 [PubMed] [Google Scholar]
- 17.Wickner, S., and Hurwitz, J. (1975) Proc. Natl. Acad. Sci. U. S. A. 72921 –925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanka, E., and Schuster, H. (1983) Nucleic Acids Res. 11987 –997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galletto, R., Jezewska, M. J., and Bujalowski, W. (2003) J. Mol. Biol. 329 441– 465 [DOI] [PubMed] [Google Scholar]
- 20.Barcena, M., Ruiz, T., Donate, L. E., Brown, S. E., Dixon, N. E., Radermacher, M., and Carazo, J. M. (2001) EMBO J. 201462 –1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davey, M. J., Fang, L., McInerney, P., Georgescu, R. E., and O'Donnell, M. (2002) EMBO J. 213148 –3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishimi, Y. (1997) J. Biol. Chem. 27224508 –24513 [DOI] [PubMed] [Google Scholar]
- 23.You, Z., Komamura, Y., and Ishimi, Y. (1999) Mol. Cell Biol. 198003 – 8015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tye, B. K. (1999) Annu. Rev. Biochem. 68649 –686 [DOI] [PubMed] [Google Scholar]
- 25.Labib, K., Tercero, J. A., and Diffley, J. F. (2000) Science 2881643 –1647 [DOI] [PubMed] [Google Scholar]
- 26.You, Z., Ishimi, Y., Masai, H., and Hanaoka, F. (2002) J. Biol. Chem. 27742471 – 42479 [DOI] [PubMed] [Google Scholar]
- 27.You, Z., Ishimi, Y., Mizuno, T., Sugasawa, K., Hanaoka, F., and Masai, H. (2003) EMBO J. 226148 –6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You, Z., and Masai, H. (2005) Nucleic Acids Res. 333033 –3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, J. K., and Hurwitz, J. (2001) Proc. Natl. Acad. Sci. U. S. A. 9854 –59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka, S., and Diffley, J. F. (2002) Nat. Cell Biol. 4198 –207 [DOI] [PubMed] [Google Scholar]
- 31.Yanagi, K., Mizuno, T., You, Z., and Hanaoka, F. (2002) J. Biol. Chem. 277 40871– 40880 [DOI] [PubMed] [Google Scholar]
- 32.Wahle, E., Lasken, R. S., and Kornberg, A. (1989) J. Biol. Chem. 2642463 –2468 [PubMed] [Google Scholar]
- 33.Walter, J., Sun, L., and Newport, J. (1998) Mol. Cell 1519 –529 [DOI] [PubMed] [Google Scholar]
- 34.Bramhill, D., and Kornberg, A. (1988) Cell 52743 –755 [DOI] [PubMed] [Google Scholar]
- 35.Costa, A., and Onesti, S. (2008) Biochem. Soc. Trans. 36136 –140 [DOI] [PubMed] [Google Scholar]
- 36.Nishitani, H., Taraviras, S., Lygerou, Z., and Nishimoto, T. (2001) J. Biol. Chem. 276 44905– 44911 [DOI] [PubMed] [Google Scholar]
- 37.Wahle, E., Lasken, R. S., and Kornberg, A. (1989) J. Biol. Chem. 2642469 –2475 [PubMed] [Google Scholar]
- 38.Seufert, W., and Messer, W. (1987) Cell 4873 –78 [DOI] [PubMed] [Google Scholar]
- 39.Lee, D. G., and Bell, S. P. (2000) Curr. Opin. Cell Biol. 12280 –285 [DOI] [PubMed] [Google Scholar]
- 40.Cook, J. G., Chasse, D. A., and Nevins, J. R. (2004) J. Biol. Chem. 2799625 –9633 [DOI] [PubMed] [Google Scholar]
- 41.Velten, M., McGovern, S., Marsin, S., Ehrlich, S. D., Noirot, P., and Polard, P. (2003) Mol. Cell 111009 –1020 [DOI] [PubMed] [Google Scholar]
- 42.Bell, S. P., Mitchell, J., Leber, J., Kobayashi, R., and Stillman, B. (1995) Cell 83563 –568 [DOI] [PubMed] [Google Scholar]
- 43.Speck, C., Chen, Z., Li, H., and Stillman, B. (2005) Nat. Struct. Mol. Biol. 12965 –971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speck, C., and Stillman, B. (2007) J. Biol. Chem. [DOI] [PMC free article] [PubMed]
- 45.Chen, S., de Vries, M. A., and Bell, S. P. (2007) Genes Dev. 212897 –2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.