

Abstract
An immunodetection study of protein tyrosine phosphatase 1B (PTP-1B), SHP-2, and Src in isolated mitochondria from different rat tissues (brain, muscle, heart, liver, and kidney) revealed their exclusive localization in the brain. Given this result, we sought whether mitochondria respond to ATP and to the general tyrosine phosphatase inhibitor orthovanadate and found little or no change in the tyrosine phosphorylation profile of mitochondria from muscle, heart, liver, and kidney. In contrast, ATP induced an enhancement in the tyrosine-phosphorylated protein profile of brain mitochondria, which was further greatly enhanced with orthovanadate and which disappeared when Src was inhibited with two inhibitors: PP2 and PP1. Importantly, we found that in brain mitochondria, ATP addition induced Src autophosphorylation at Tyr-416 in its catalytic site, leading to its activation, whereas the regulatory Tyr-527 site remained unphosphorylated. Functional implications were addressed by measurements of the enzymatic activity of each of the oxidative phosphorylation complexes in brain mitochondria in the presence of ATP. We found an increase in complex I, III, and IV activity and a decrease in complex V activity, partially reversed by Src inhibition, demonstrating that the complexes are Src substrates. These results complemented and reinforced our initial study showing that respiration of brain mitochondria was partially dependent on tyrosine phosphorylation. Therefore, the present data suggest a possible control point in the regulation of respiration by tyrosine phosphorylation of the complexes mediated by Src auto-activation.
Mitochondria provide the energy necessary for cell growth and biological activities through oxidative phosphorylation (OxPhos).4 This relies on electron transfer from oxidative substrates to oxygen, via a series of redox reactions, to generate water. In this process, protons are pumped from the matrix across the mitochondrial inner membrane, via respiratory complexes I, III, and IV. When protons return to the mitochondrial matrix, ATP is synthesized via complex V.
As the energy demand of a cell depends on its function and activity, energy production is adjusted and controlled by different mechanisms (1–3). One major regulatory system is protein phosphorylation/dephosphorylation (4). Evidence indicates that mitochondrial proteins undergo posttranslational phosphorylation (2, 5, 6), and reports have unambiguously revealed the existence of several kinases within the mitochondria, such as cAMP-dependent protein kinase (7) and members of the Src kinase family (8). Protein phosphatases have also been described, such as Ser/Thr phosphatases, PP2C-γ and PP2A (7), and tyrosine phosphatases, SHP-2 (9), PTP-1B (10), and PTPMT1, a dual-specific phosphatase (11), the latter found exclusively in mitochondria but not in the cytosol.
As indicated by earlier reports, mitochondrial signaling enzyme distribution and stimulation may vary according to the tissue. In a preceding report, which also explored the presence of PTP-1B in three other tissues, muscle, heart, and liver, we showed that it was detected exclusively in rat brain mitochondria (10). An unidentified tyrosine kinase was shown to basically phosphorylate subunit I of complex IV in cow heart mitochondria, but high cAMP levels were required to phosphorylate this subunit in mitochondria from cow liver (12). Low Src levels or compensation by other tyrosine kinases in mitochondria from mouse muscle were deduced from studies showing no change in complex IV enzymatic activity in Src–/– mice, whereas activity was reduced in liver and kidney mitochondria (13). Other reports evoked the possibility that Src translocated within mitochondria in a human kidney cell line (14, 15). Therefore, this tissue-specific distribution of mitochondrial kinases and phosphatases could be correlated to functional tissue differences. Prompted by this earlier research, the aim of this report was to analyze the variability in signaling enzyme expression in mitochondria from different rat tissues to highlight the role of Src in tyrosine phosphorylation and to study some functional consequences.
In agreement with earlier studies (8–10), we confirmed that brain mitochondria expressed Src, PTP-1B, and SHP-2. However, we did not detect these enzymes in the other tissues. Accordingly, stimulation or inhibition of tyrosine kinase and phosphatase activities induced corresponding changes in the tyrosine-phosphorylated protein status of brain mitochondria, whereas little or no change was observed in mitochondria from other tissues. Assuming that Src was responsible for increased tyrosine phosphorylation in brain mitochondria incubated with ATP, we analyzed the phosphorylation status of Src and found that it autophosphorylated in its active Tyr-416 residue, resulting in its activation. We also showed that the OxPhos complex activities were differently modulated by ATP. Although critical questions remain to be solved, our data indicated that Src played a role in brain mitochondrial functions by self-activation within mitochondria without any extracellular triggers.
EXPERIMENTAL PROCEDURES
Chemicals—Micro-BC assay protein quantification kit was from Pierce. n-Dodecyl-b-d-maltoside was from Calbiochem. The mouse monoclonal antibody to phosphotyrosine (PY20) directly coupled to horseradish peroxidase and the mouse monoclonal antibody to Src were from Santa Cruz Biotechnology (Santa Cruz, CA), together with the rabbit polyclonal antibodies to manganese superoxide dismutase, GRP78, SHP-2, and Src. The Src antibody sampler kit (containing phospho-Tyr-416-Src, phospho-Tyr-527-Src, non-phospho-Tyr-416-Src, non-phospho-Tyr-527-Src, and Src antibodies) was from Cell Signaling. The mouse monoclonal antibody to complex IV subunit I was from Mitosciences (Eugene, OR). The monoclonal antibody to PTP-1B and the Src kinase inhibitors, PP2 and PP1, were from Calbiochem. All other chemicals were from Sigma.
Preparing Mitochondria from Different Rat Tissues—Male Wistar rats were sacrificed by cervical shock and decapitation. Brain mitochondria were isolated from whole brain, as described by Clark and Nicklas (16).
Muscle and heart mitochondria were isolated as described by Morgan-Hughes et al. (17), and liver and kidney mitochondria were isolated as described by Jumelle-Laclau et al. (18) and Johnson and Lardy (19), respectively. Cytosolic contamination detected by lactate dehydrogenase activity never exceeded 0.5%. After isolation, 1-mg mitochondrial fractions were immediately stored at –80 °C.
In Vitro ATP Addition Studies of Tyrosine Phosphorylation—Mitochondria from the various tissues were thawed and preincubated in parallel at 30 °C for 10 min in 10 mm Tris-HCl, pH 7.5, 8 mm MgCl2 (kinase buffer) containing 20 μm oligomycin and 30 μg/ml rotenone (10), together with 10 μm PP2, 10 μm PP1, or 10 mm NaF plus 2 mm orthovanadate, when indicated. Then 1 mm ATP was added, except in the control samples, and incubation continued for another 10 min. The reaction was stopped by adding 5× Laemmli buffer.
Gel Electrophoresis and Western Blotting—Samples were subjected to SDS-PAGE (10% or 12%), and separated proteins were transferred onto a nitrocellulose membrane (10). Tyrosine-phosphorylated proteins were labeled with a specific mouse monoclonal antibody to phosphotyrosine directly coupled to horseradish peroxidase. Other primary antibodies were revealed with horseradish-peroxidase-conjugated secondary antibody (F(ab′)2 fragments of anti-mouse or anti-rabbit, F(ab′)2 fragment-specific, Jackson ImmunoResearch Laboratories, West Grove, PA).
Immunocytochemistry for Electron Microscopy—Freshly isolated brain or muscle mitochondria were fixed in 4% paraformaldehyde and 0.2% glutaraldehyde, rinsed in distilled water, and then embedded in 1% agarose in distilled water at 45 °C. Small pieces (1.0 mm3) were cut under the stereomicroscope and embedded at –35 °C in hydrophobic Lowicryl® HM20 resin (Electron Microscopy Sciences) by the progressive lowering temperature technique, using the Leica AFS system according to the instruction manual. Ultrathin sections from Lowicryl blocks were mounted on Butvar-coated single nickel grids and submitted to the post-embedding immunogold procedure to detect PTP-1B. The grids were floated twice for 5 min on drops of 100 mm glycine in TBS and twice for 10 min on drops of TBS supplemented with 1% (w/v) bovine serum albumin to block nonspecific labeling. The grids were then transferred to drops of the primary antibody (1/50 or 1/500, depending on the size of the gold particles: 15 or 10 nm, respectively), diluted in TBS supplemented with 0.1% (w/v) bovine serum albumin, and incubated in a moist chamber at room temperature overnight. After rinsing several times on large drops of TBS supplemented with 0.1% (w/v) bovine serum albumin, the grids were incubated at room temperature for 1 h with goat anti-mouse colloidal gold conjugate diluted 1/30 (15 or 10 nm, EMS or Aurion, respectively). Samples were rinsed once with TBS supplemented with 0.1% (w/v) bovine serum albumin, once with TBS, and twice with distilled water (5 min each) and post-fixed in 1% glutaraldehyde for 1 min. After rinsing several times on large drops of distilled water, sections were counterstained with 2% uranyl acetate in distilled water for 15 min and lead citrate for 2 min. The grids were finally observed under a FEI Tecnai 12 electron microscope. Control sections were incubated without the primary antibody.
Measuring Enzymatic Activity of the Respiratory Complexes and ATP Synthase—Thawed mitochondria (1 mg/ml) were preincubated at 30 °C for 10 min in the kinase buffer with 20 μm oligomycin (except for ATPase activity), 30 μg/ml rotenone, 1 μm cAMP-dependent protein kinase inhibitor (to inhibit mitochondrial cAMP-dependent protein kinase), and 10 μm PP2 or PP1, when indicated. Then 1 mm ATP was added, and incubation continued for 10 min. A control sample was prepared under the same conditions without PP2, PP1, and ATP. Spectrophotometry standard methods were used, consisting of measuring the enzymatic activity of a complex by providing the required substrate while inhibiting the others. The ATPase activity was determined with an ATP-regenerating system (20–22). The concentrations of mitochondrial protein were 40, 3, 5, and 30 μg, respectively, for complexes I, III, IV, and V.
RESULTS
PTP-1B, SHP-2, and Src Found Exclusively in Brain Mitochondria—Fig. 1 shows immunodetection of PTP-1B, SHP-2, and Src in mitochondria from different rat tissues. As positive controls, we used total lysate of human blood platelets, which contain large amounts of these enzymes. Contamination by the endoplasmic reticulum in our mitochondrial preparations was minimal, as shown by labeling with an antibody to GRP78, an endoplasmic reticulum marker (Fig. 1, bottom). Labeling with an antibody to manganese superoxide dismutase was used as a loading control (Fig. 1, bottom).
FIGURE 1.

PTP-1B, SHP-2, and Src expression in mitochondria from different rat tissues and human platelets. The protein content in purified mitochondria from the different tissues was assayed by the Micro-BCA method, and the same amount of protein lysate was loaded per lane: 100 μg for immunodetection of PTP-1B, SHP-2, and GRP78 and 30 μg for Src and manganese superoxide dismutase (MnSOD). For platelets, 10 μg of proteins were used.
As expected, PTP-1B, SHP-2, and Src were strongly labeled in brain mitochondria, in confirmation of previous studies (8–10), and in platelets (Fig. 1). In contrast, we did not find any labeling for these enzymes in mitochondria from other tissues.
SHP-2 and Src were already localized within brain mitochondria by immunogold EM staining (8, 9). By subfractionment of brain mitochondria, we have immunodetected PTP-1B in inner membranes and matrix (10). To further confirm this PTP-1B localization, we performed immunogold staining. Ultrathin sections of brain mitochondria were probed with a mouse anti-PTP-1B primary antibody and gold particle-conjugated goat anti-mouse secondary antibody. As shown in Fig. 2A, gold particles were observed mainly in the mitochondria with little background staining. Higher magnification clearly showed intense labeling, mostly in the electron-dense areas of the mitochondria, associated with the cristae, and also in the white regions corresponding to matrix. The outer membrane was not labeled. No background staining was seen with the second antibody alone (result not shown), thus confirming the specificity of the labeling.
FIGURE 2.
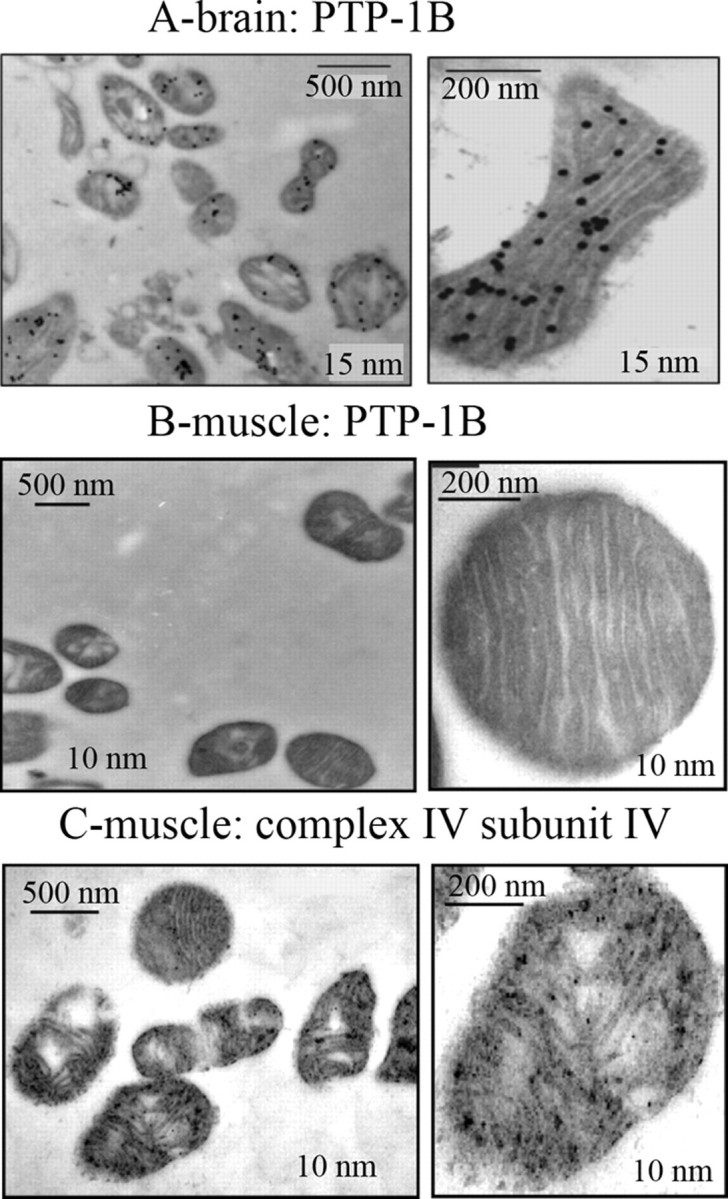
Immunogold labeling of PTP-1B in isolated brain mitochondria. An overall view and a mitochondrium was shown, in which PTP-1B (A and B) and subunit I of complex IV (C) were immunodetected with 15- or 10-nm gold-labeled secondary antibody.
In comparison, muscle mitochondria were not labeled with the anti-PTP-1B antibody (Fig. 2B), confirming the Western blot experiment (Fig. 1). As a positive control, muscle mitochondria were treated with an antibody to subunit I of complex IV, and results showed, as expected, positive labeling of mitochondria (Fig. 2C).
Tyrosine Phosphorylation Profiles of Mitochondria from the Different Tissues—To further investigate the effect of the presence or absence of these signaling enzymes in mitochondria, we compared the tyrosine phosphorylation profiles of mitochondrial proteins from the various tissues in response to ATP, added alone or with the Src kinase inhibitor PP2 (8, 23), or orthovanadate, a generic tyrosine phosphatase inhibitor. We reasoned that if tyrosine kinases were present and active, adding ATP would increase tyrosine phosphorylation. A decrease in labeling in the presence of PP2 would indicate the presence of Src kinases. On the contrary, if tyrosine phosphatases were present, their inhibition would increase tyrosine phosphorylation.
As shown in Fig. 3, none of the treatments (lanes 2–4) induced any significant changes in tyrosine phosphorylation levels for muscle, heart, and kidney mitochondria when compared with control (lane 1), which is consistent with the fact that Src, SHP-2, and PTP-1B were not detected in these tissues (Fig. 1). In liver mitochondria, the presence of ATP induced a small but significant increase in the tyrosine phosphorylation labeling of virtually all the bands (lane 2). This increase was not affected by PP2 (lane 3), reflecting the absence of Src in this tissue, as shown in Fig. 1. A further increase was observed in the presence of orthovanadate (lane 4), indicating inhibition of tyrosine phosphatases, which are not SHP-2 or PTP-1B, as shown in Fig. 1.
FIGURE 3.

Changes in tyrosine phosphorylation of mitochondrial proteins from liver, kidney, heart, and muscle. Mitochondria (20 μg) were untreated (lane 1) or treated at 30 °C for 10 min with 1 mm ATP alone (lane 2) or after preincubation at 30 °C for 10 min with 10 μm PP2 (lane 3) or 2 mm orthovanadate plus 10 mm NaF (lane 4). The reaction was stopped by the addition of 5× Laemmli buffer. Phosphorylated proteins on tyrosine residues were detected with an antibody to phosphotyrosine directly labeled with horseradish peroxidase. The membranes were stripped and reprobed with an antibody to manganese superoxide dismutase (MnSOD) as a loading control (below). Data are representative of at last three experiments.
In contrast, brain mitochondria exhibited a robust increased tyrosine phosphorylation labeling in the presence of ATP (Fig. 4, left, lane 2) when compared with control (lane 1), showing active tyrosine kinases, including some Src kinases, as labeling decreased in the presence of PP2 (lane 3). Labeling also increased strongly in the presence of orthovanadate (lane 4), indicating the presence of tyrosine phosphatases.
FIGURE 4.

Changes in tyrosine phosphorylation of mitochondrial proteins from brain. Left, see the legend for Fig. 3. Right, similar to left, except that 10 and 100 μm PP1 were used. MnSOD, manganese superoxide dismutase.
To confirm this result, we performed experiments with PP1, another well known Src inhibitor (23). Results showed that tyrosine phosphorylation, which increased in the presence of ATP (Fig. 4, right, lane 2), decreased in the presence of 10 μm PP1 and returned to control level at 100 μm PP1 (Fig. 4, right, lanes 3 and 4). Therefore, the different responses of mitochondria from the different tissues to direct exposure to ATP, PP2, PP1, and orthovanadate confirmed the presence or absence of these signaling enzymes.
Src Activation as Detected by Study of Its Tyrosine Phosphorylation Status—Given that our experiments clearly showed that Src was active in the presence of ATP, we examined in detail the phosphorylation status of Src. Src contains a regulatory Tyr-527 residue in the C terminus, phosphorylated by Csk, resulting in inhibition, and a Tyr-416 residue in the catalytic domain, the autophosphorylation of which promotes kinase activity (24). Using phospho-specific antibodies to Tyr-416 and Tyr-527 of Src, we showed that mitochondria pretreated with ATP were labeled with the anti-pY416-Src antibody (Fig. 5, left, top, lane 2) but not when untreated (lane 1), whereas Tyr-527 residue was not phosphorylated under both conditions (Fig. 5, right, top, lanes 1 and 2). This latter result was confirmed with the antibody to non-phosphorylated Tyr-527, which labeled a band of similar intensity in the absence and presence of ATP (Fig. 5, right, middle). The antibody to non-phosphorylated Tyr-416 detected a band in mitochondria in the absence of ATP (Fig. 5, left, middle, lane 1). This antibody also detected a band in mitochondria incubated with ATP (Fig. 5, left, middle, lane 2) because proteins are usually partially phosphorylated. Therefore, our data support a direct autophosphorylation of Src with added ATP promoting its activity toward mitochondrial proteins.
FIGURE 5.

Phosphorylation status of Src in brain mitochondria. Left, Src in untreated or ATP-treated mitochondria (20 μg) was immunodetected with the anti pY416-Src antibody. The membrane was stripped and reprobed with the anti non-pY416-Src antibody, stripped again, and reprobed with an anti Src antibody. Right, a similar procedure was used to study Src phosphorylation at Tyr-527 residue.
Effect of ATP on the Activities of the OxPhos Complexes in Brain Mitochondria—The above results showed that brain mitochondria contained Src and responded to ATP addition by increased protein tyrosine phosphorylation due, at last in part, to Src autophosphorylation and activation. Thus the possibility that tyrosine phosphorylation influenced the function of the oxidative phosphorylation complexes in brain mitochondria was studied by measuring the effect of ATP and the Src inhibitors PP2 and PP1 on their enzymatic activities. The assays were performed in the presence of cAMP-dependent protein kinase inhibitor to prevent any participation of c-AMP-dependent kinase.
Results (Table 1) showed that ATP stimulated the specific activity of complexes I (about 20%, p < 0.1), III (20%, p < 0.001), and IV (about 30%, p < 0.1) and inhibited the ATPase activity of complex V (about 38%, p < 0.001). The presence of PP2 and PP1 partially but significantly restored this activity to control levels, except for complex I.
TABLE 1.
Effect of ATP and the Src inhibitors PP2 and PP1 on the enzymatic activities of the oxidative phosphorylation complexes
Thawed mitochondria were pre-incubated at 30 °C for 10 min in kinase buffer with 1 μm cAMP-dependent protein kinase inhibitor and 20 μg/ml oligomycin (except for ATPase activity of complex V), together with 10 μm PP2 or PP1, when indicated. Then 1 mm ATP was added, and incubation continued for 10 min. Control experiments were performed without ATP and PP2 or PP1. Enzymatic assays were performed in 50 mm K2HPO4, pH = 7.2 by standard methods (see “Experimental Procedures”). Results are expressed as mean values of at least five different experiments ± standard error. p < 0.1 is considered statistically significant.
| CONTROL | ATP | ATP + PP2 | ATP + PP1 | |
|---|---|---|---|---|
| mm/min/mg | ||||
| Complex I | 0.117 ± 0.002 | 0.135 ± 0.007 | 0.136 ± 0.008 | 0.126 ± 0.012 |
| p < 0.1 | p > 0.1 | p > 0.1 | ||
| Complex III | 2.260 ± 0.032 | 2.669 ± 0.012 | 2.577 ± 0.013 | 2.295 ± 0.041 |
| p < 0.001 | p < 0.01 | p < 0.01 | ||
| Complex IV | 1.585 ± 0.014 | 2.101 ± 0.025 | 1.801 ± 0.066 | 1.859 ± 0.012 |
| p < 0.01 | p < 0.01 | p < 0.01 | ||
| Complex V | 0.412 ± 0.034 | 0.254 ± 0.011 | 0.293 ± 0.013 | 0.305 ± 0.021 |
| p < 0.001 | p < 0.01 | p < 0.01 | ||
DISCUSSION
Mitochondria are involved in crucial cellular processes, including ATP production, and a role of reversible phosphorylation in the regulation of mitochondrial functions remains poorly understood. Therefore, the discovery of mitochondrial kinases and phosphatases regulating mitochondrial physiology is thus of critical importance.
Although the distribution patterns of several tyrosine kinases and phosphatases are known to range from ubiquitous to tissue-specific (25), one of the novel features of this report is the immunodetection of Src, PTP-1B, and SHP-2 in an organelle, mitochondria, and exclusively in brain. Moreover, immunogold labeling showed that these enzymes reside in inner membrane and matrix (8, 9, 13) (Fig. 2). Therefore, the slight labeling of SHP-2 in liver and kidney mitochondria (Fig. 1) could result from some contamination from cytosol and endoplasmic reticulum.
Preliminary studies revealed that mitochondrial Src distribution and effect varied according to the tissue. As reported in the Introduction, complex IV activity in muscle mitochondria from Src-deficient mice was not different from normal mice, indicating low expression of Src in muscle mitochondria (13). This is consistent with our results showing neither Src labeling (Fig. 1) nor change in tyrosine phosphorylation in muscle mitochondria following ATP treatment (Fig. 4C).
In bovine liver mitochondria, experiments demonstrated that subunit I of complex IV was tyrosine-phosphorylated in a cAMP-dependent manner by an unknown tyrosine kinase (12). Our results ruled out the possibility that this could be due to Src. On the other hand, we did not detect SHP-2 or PTP-1B, but tyrosine phosphorylation of liver mitochondrial proteins increased on application of orthovanadate (Fig. 3B), suggesting the presence of another tyrosine phosphatase. Notably, a dual-specific protein tyrosine phosphatase PTPMT1 was detected in the inner mitochondrial membrane of rat liver (11).
The absence of Src, SHP-2, or PTP-1B labeling in muscle, heart, liver, or kidney mitochondria is puzzling. Their intramitochondrial localization (Refs. 8, 9, 13 and the present work) ruled out the possibility that they were washed out. However, recent observations indicate that the protein kinase A anchor protein AKAP121 associates with Src and another tyrosine phosphatase, PTP-1D, on the outer membrane of mitochondria in a human embryonic kidney cell line, HEK293, in response to epidermal growth factor (14, 15). The authors proposed that Src translocated inside the mitochondria to regulate tyrosine phosphorylation. As AKAP121 is ubiquitously expressed, this mechanism may transport Src into mitochondria in mammalian cells, in response to extracellular stimulation. Such changes in the subcellular localization of signaling proteins are now well documented (4).
Several groups reported that the addition of radiolabeled ATP to purified mitochondria led to the formation of numerous phosphoproteins, which increases in number with the addition of phosphatases inhibitors (26–28). Nevertheless, the majority of studies investigated the effect of the cAMP-dependent pathway catalyzed by protein kinase A. A recent report has used exogenously added kinases of the Src family to identify tyrosine phosphorylated proteins in brain mitochondria (29). In our work, we focused on tyrosine phosphorylation and showed no response for muscle, heart, and kidney mitochondria and a small response for liver mitochondria to ATP, PP2, and orthovanadate in their phosphorylation status on tyrosine residues (Fig. 3), in confirmation with their content in tyrosine kinases and phosphatases (Fig. 1). In contrast, we showed strong responses for brain mitochondria (Fig. 4), which occurred with endogenous Src, which was activated by autophosphorylation at Tyr-417 residue, whereas the regulatory Tyr-527 residue remains unphosphorylated (Fig. 5), despite the presence in brain mitochondria of Csk (8). Interestingly, autophosphorylation of Src has been shown to protect its inactivation by Csk (30).
The responsiveness of brain mitochondria to ATP and to PP2 and PP1, together with the finding that they contain signaling enzymes, suggested that mitochondrial energy production depends on tyrosine phosphorylation. However, the complexity of the OxPhos system implies that the influence of tyrosine phosphorylation on the activity of the individual complex be studied as a first step before interpretation of more integrative studies. We found enzymatic activities for controls (Table 1) in the range described in the literature (31, 32). In the presence of ATP, our finding that the enzymatic activity of complex IV was greatly stimulated and partially returned to control levels in the presence of PP2 and PP1 was consistent with previous results showing increased activity in osteoclasts due to Src phosphorylation of subunit II (13). Complex III activity also increased in the presence of ATP and decreased in the presence of Src inhibitors, suggesting that Src was involved. For complex I, the fact that the ATP-induced increase in activity was not sensitive to Src inhibitors suggested that it was not due to Src phosphorylation. Finally, the ATPase activity of complex V decreased in the presence of ATP and was partially Src-dependent (Table 1). However, it is difficult to link the extent of tyrosine phosphorylation of enzymatic complexes to their effect on respiratory rates because it is well established that in living cells, the OxPhos system is not functioning to its maximal activity. Furthermore, the existence of a reserve of enzymes not initially involved in the respiration rate, and which can be mobilized upon energy demand, has been demonstrated (1–3). Finally, control of mitochondrial metabolic fluxes is shared among the different complexes (33).
In a previous report, we have already shown that ATP, which is produced from ADP during oxidative phosphorylations in an oxygraphic cuvette during state 3 respiration in brain mitochondria, could activate tyrosine phosphorylation (10). We have also found a significant decrease in state 3 respiration (88% of control values, p < 0.05) at the same time as tyrosine phosphorylation was reversed, when state 3 respiration was measured in the presence of 10 μm PP2. Therefore, these previous data showed that mitochondrial tyrosine kinases can sense and use ATP synthesized in situ by mitochondria to phosphorylate proteins. They also showed that tyrosine phosphorylation is involved in state 3 respiration. The present study showed that this occurred by means of differential regulation of the activities of the enzymes of the respiratory chain by tyrosine phosphorylation. It also suggested that Src can autophosphorylate at the Tyr-416 residue to active enzyme with ATP synthesized in situ, whereas the regulatory Tyr-517 remained unphosphorylated. Therefore, these results raise the issue that activation of signaling pathways can happen within mitochondria without the need for an extracellular triggering effector.
We are aware that further studies are required to link ATP stimulation or inhibition of enzymatic activity to tyrosine phosphorylation of a defined subunit on a complex. At the present time, except subunits I, IV, and II for complex IV (12, 13), and recently, subunits e and γ for complex V (29, 34), no phosphoproteomic study has unambiguously identified tyrosine-phosphorylated subunit for the other complexes.
It is noteworthy that both PTP-1B and SHP-2 can dephosphorylate Src (24), suggesting their possible involvement in Src regulation and activity in brain mitochondria. However, it is too early to assume that expression of PTP-1B, SHP-2, and Src could be related to a specific function of mitochondria in brain.
This work was supported in part by grants from INSERM, the Université Victor Segalen-Bordeaux 2, and the Fondation de France. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: OxPhos, oxidative phosphorylation; GRP78, glucose regulated protein 78; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine; PTP-1B, protein tyrosine phosphatase 1B; PTP-1D, protein tyrosine phosphatase 1D; PTPMT1, protein tyrosine phosphatase mitochondrial T1; TBS, Tris-buffered saline.
References
- 1.Gnaiger, E., Lassnig, B., Kuznetsov, A., Rieger, G., and Margreiter, R. (1998) J. Exp. Biol. 201 1129–1139 [DOI] [PubMed] [Google Scholar]
- 2.Devin, A., and Rigoulet, M. (2007) Am. J. Physiol. 292 C52–C58 [DOI] [PubMed] [Google Scholar]
- 3.Benard, G., Faustin, B., Galinier, A., Rocher, C., Bellance, N., Smolkova, K., Casteilla, L., Rossignol, R., and Letellier, T. (2008) Int. J. Biochem. Cell Biol. 40 1545–1554 [DOI] [PubMed] [Google Scholar]
- 4.Hüttemann, M., Lee, I., Samavati, L., Yu, H., and Doan, J. W. (2007) Biochim. Biophys. Acta 1773 1701–1720 [DOI] [PubMed] [Google Scholar]
- 5.Pagliarini, D. J., and Dixon, J. E. (2006) Trends Biochem. Sci. 31 26–34 [DOI] [PubMed] [Google Scholar]
- 6.Salvi, M., Brunati, A. M., and Toninello, A. (2005) Free Radic. Biol. Med. 38 1267–1277 [DOI] [PubMed] [Google Scholar]
- 7.Papa, S, Scacco, S., Sardanelli, A. M., Petruzzella, V., Vergari, R., Signorile, A., and Technikova Dobrova, Z. (2002) Biosci. Report 22 3–16 [DOI] [PubMed] [Google Scholar]
- 8.Salvi, M., Brunati, A. M., Bordin, L., La Rocca, N., Clari, G., and Toninello, A. (2002) Biochim. Biophys. Acta 1589 181–195 [DOI] [PubMed] [Google Scholar]
- 9.Salvi, M., Stringaro, A., Brunati, A. M., Agostinelli, E,. Arancia, G., Clari, G., and Toninello, A. (2004) CMLS Cell Mol. Life Sci. 61 2393–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augereau, O., Claverol, S., Boudes, N., Basurko, M. J., Bonneu, M., Rossignol, R., Mazat, J. P., Letellier, T., and Dachary-Prigent, J. (2005) CMLS Cell Mol. Life Sci. 62 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagliarini, D. J., Wiley, S. E., Kimple, M. E., Dixon, J. R., Kelly, P., Worby, C. A., Casey, P. J., and Dixon, J. E. (2005) Mol. Cell 19 197–207 [DOI] [PubMed] [Google Scholar]
- 12.Lee, I., Salomon, A. R., Ficarro, S., Mathes, I., Lottspeich, F., Grossman, L. I., and Huttemann, M. (2005) J. Biol. Chem. 280 6094–6100 [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki, T., Neff, L., Tanaka, S., Horne, W. C., and Baron, R. (2003) J. Cell Biol. 160 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardone, L., Carlucci, A., Affaitati, A., Livigni, A., DeCristofaro, T., Garbi, C., Varrone, S., Ullrich, A., Gottesman, M. E., Avvedimento, E. V., and Feliciello, A. (2004) Mol. Cell Biol. 24 4613 4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livigni, A., Scorziello, A., Agnese, S., Adornetto, A., Carlucci, A., Garbi, C., Castaldo, I., Annunziato, L., Avvedimento, E. V., and Feliciello, A. (2006) Mol. Biol. Cell 17 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark, J. B., and Nicklas, W. J. (1970) J. Biol. Chem. 245 4724–4731 [PubMed] [Google Scholar]
- 17.Morgan-Hughes, J. A., Schapira, A. H., Cooper, J. M., Holt, I. J., Harding, A. E., and Clark, J. B. (1990) Biochim. Biophys. Acta 1018 217–222 [DOI] [PubMed] [Google Scholar]
- 18.Jumelle-Laclau, M., Rigoulet, M., Averet, N., Leverve, X., Dubourg, L., Carbonneau, A., Clerc, M., and Guérin, B. (1993) Biochim. Biophys. Acta 1144 90–94 [DOI] [PubMed] [Google Scholar]
- 19.Johnson, D., and Lardy, H. (1958) Nature 181 701–702 [DOI] [PubMed] [Google Scholar]
- 20.Solaini, G., Baracca, A., Parenti-Castelli, G., and Strambini, G. B. (1993) Eur. J. Biochem. 214 729 734 [DOI] [PubMed] [Google Scholar]
- 21.Birch-Machin, M. A., Shepherd, I. M., Watmough, N. J., Sherratt, H. S., Bartlett, K., Darley Usmar, V. M., Milligan, D. W., Welch, R. J., Aynsley-Green, A., and Turnbull, D. M. (1989) Pediatr. Res. 25 553–559 [DOI] [PubMed] [Google Scholar]
- 22.Rimoldi, M., Bottacchi, E., Rossi, L., Cornelio, F., Uziel, G., and Di Donato, S. (1982) J. Neurol. 227 201–207 [DOI] [PubMed] [Google Scholar]
- 23.Bain, J., McLauchlan, H., Elliott, M., and Cohen, P. (2003) Biochem. J. 371 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roskoski, R., Jr. (2004) Biochem. Biophys. Res. Commun. 324 1155–1164 [DOI] [PubMed] [Google Scholar]
- 25.Alonso, A., Sasin, J., Bottini, N., Friedberg, I., Osterman, A., Godzik, A., Hunter, T., Dixon, J., and Mustelin, T. (2004) Cell 117 699–711 [DOI] [PubMed] [Google Scholar]
- 26.Technikova-Dobrova, Z., Sardanelli, A. M., Stanca, M. R., and Papa, S. (1994) FEBS Lett. 350 187 191 [DOI] [PubMed] [Google Scholar]
- 27.Sardanelli, A. M., Technikova-Dobrova, Z., Scacco, S. C., Speranza, F., and Papa, S. (1995) FEBS Lett. 377 470–474 [DOI] [PubMed] [Google Scholar]
- 28.Chen, R., Fearnley, I. M., Peak-Chew, S. Y., and Walker, J. E. (2004) J. Biol. Chem. 279 26036–26045 [DOI] [PubMed] [Google Scholar]
- 29.Lewandrowski, U., Sickmann, A., Cesaro, L., Brunati, A. M., Toninello, A., and Salvi, M. (2008) FEBS Lett. 582 1104–1110 [DOI] [PubMed] [Google Scholar]
- 30.Sun, G., Sharma, A. K., and Budde, R. J. (1998) Oncogene 17 1587–1595 [DOI] [PubMed] [Google Scholar]
- 31.Kipp, J. L., and Ramirez, V. D. (2001) Endocrine 15 165–175 [DOI] [PubMed] [Google Scholar]
- 32.Benard, G., Faustin, B., Passerieux, E., Galinier, A., Rocher, C., Bellance, N., Delage, J.-P., Casteilla, L., Letellier, T., and Rossignol, R. (2006) Am. J. Physiol. 29 C1172–C1182 [DOI] [PubMed] [Google Scholar]
- 33.Rossignol, R., Letellier, T., Malgat, M., Rocher, C., and Mazat, J. P. (2000) Biochem. J. 347 45–53 [PMC free article] [PubMed] [Google Scholar]
- 34.Di Pancrazio, F., Bisetto, E., Alverdi, V., Mavelli, I., Esposito, G., and Lippe, G. (2006) Proteomics 6 921–926 [DOI] [PubMed] [Google Scholar]