

Abstract
Conditions of stress, such as myocardial infarction, stimulate up-regulation of heme oxygenase (HO-1) to provide cardioprotection. Here, we show that CO, a product of heme catabolism by HO-1, directly inhibits native rat cardiomyocyte L-type Ca2+ currents and the recombinant α1C subunit of the human cardiac L-type Ca2+ channel. CO (applied via a recognized CO donor molecule or as the dissolved gas) caused reversible, voltage-independent channel inhibition, which was dependent on the presence of a spliced insert in the cytoplasmic C-terminal region of the channel. Sequential molecular dissection and point mutagenesis identified three key cysteine residues within the proximal 31 amino acids of the splice insert required for CO sensitivity. CO-mediated inhibition was independent of nitric oxide and protein kinase G but was prevented by antioxidants and the reducing agent, dithiothreitol. Inhibition of NADPH oxidase and xanthine oxidase did not affect the inhibitory actions of CO. Instead, inhibitors of complex III (but not complex I) of the mitochondrial electron transport chain and a mitochondrially targeted antioxidant (Mito Q) fully prevented the effects of CO. Our data indicate that the cardioprotective effects of HO-1 activity may be attributable to an inhibitory action of CO on cardiac L-type Ca2+ channels. Inhibition arises from the ability of CO to promote generation of reactive oxygen species from complex III of mitochondria. This in turn leads to redox modulation of any or all of three critical cysteine residues in the channel's cytoplasmic C-terminal tail, resulting in channel inhibition.
CO is an established and important signaling molecule in both the heart and vasculature as well as other tissues (1, 2). Cardiac atrial and ventricular myocytes express heme oxygenases HO-14 and HO-2, which generate CO along with biliverdin and free Fe2+ by heme catabolism, and HO-1 levels can be increased by various stress factors (3), including myocardial infarction (4). CO limits the cellular damage of ischemia/reperfusion injury in the heart (5). Indeed, greater cardiac damage is seen following ischemia/reperfusion injury in HO-1 knock-out mice (6). Conversely, HO-1 overexpression in the heart reduces infarct size and other markers of damage following ischemia/reperfusion injury (7). CO also improves cardiac blood supply through coronary vessel dilation (8, 9) and reduces cardiac contractility (9). However, the mechanisms underlying this cardioprotective effect of CO are not understood.
In the vasculature, CO also exerts numerous beneficial effects. Its ability to dilate blood vessels is long established (9–11) and endothelium-independent (12) and not due to development of hypoxia through displacement of O2 (see Ref. 13). CO has clear, protective effects in various vascular diseases, such as systemic and pulmonary hypertension, development of atherosclerosis, and neointimal hyperplasia due to proliferation of vascular smooth muscle cells following vascular injury (all reviewed in Refs. 2, 13, and 14). Importantly, up-regulation of HO-1 in a model of hypertension can provide significant vascular protection by suppressing the effects of constricting agents (15). This beneficial effect is extended to the pulmonary circulation (16) and is supported fully by studies in transgenic HO-1 knock-out mice (17).
Although the involvement of CO in various intracellular signaling pathways is established (1), the mechanism(s) by which CO exerts protective effects in the cardiovascular system remains to be determined. We reasoned that the L-type Ca2+ channel, the major route of Ca2+ entry into cardiac myocytes (18) and vascular smooth muscle cells (19), may be a target site of action for CO; although Ca2+ overload-mediated cardiac cell death may not primarily involve L-type Ca2+ channels (20), their inhibition is protective in this respect (21), and such drugs are of clinical importance (21, 22). The role of these channels in the control of vascular tone is fundamental (19). Our findings indicate that L-type Ca2+ channels are indeed a site of action for CO.
EXPERIMENTAL PROCEDURES
Reagents—The following compounds were used as described under “Results”: PKG inhibitor, PET-cGMPS (100 nm), BIOLOG (Bremen, Germany); NO donors, SIN-1 (10 μm) and S-nitrosoglutathione (2 mm), both soluble in DMSO and used in the presence of 50 units/ml superoxide dismutase; NO scavenger, carboxy-PTIO (1 mm); nitric-oxide synthase inhibitor, NG-nitro-l-arginine methyl ester (1 mm); antioxidant, Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP) (100 μm) (Calbiochem); reducing agent, dithiothreitol (2 mm); xanthine oxidase inhibitor, allopurinol (1 μm); NADPH oxidase inhibitors, apocynin (30 μm) and diphenyleneiodonium (DPI; 3 μm); mitochondrial antioxidant and control compound, MitoQ10 methanesulfonate (MitoQ) and decyl-TPP bromide (TPP) (both 250 nm) (Antipodean Pharmaceuticals, Auckland, New Zealand); mitochondrial complex inhibitors, rotenone (2 μm), stigmatellin (1 μm), and antimycin A (3 μm). All other chemicals were obtained from Sigma unless otherwise stated.
Application of CO—For these experiments, we employed either CO dissolved in perfusate or the well established CO-releasing molecule, CORM-2 ([Ru(CO3)Cl2]2, tricarbonyldichlororuthenium(II) dimer) (23). To prepare CO-containing solutions, extracellular perfusate was bubbled for at least 2 h under positive pressure with CO. This solution was assumed to be saturated with CO and then diluted 1:5 before application to cells. This dilution was selected because it would result in the pO2 of the perfusate only falling to a potential minimum of ∼120 mm Hg, which is not sufficiently hypoxic to alter Ca2+ channel function through lack of available O2 (24). CORM-2 was purchased from Sigma, freshly prepared in DMSO, and diluted to the required concentration in extracellular recording solution for the experiments described. Also prepared in DMSO, dichlorotetrakis (dimethylsulfoxide) ruthenium(II), the breakdown product (RuCl2(DMSO)4) of CORM-2 kindly synthesized by the Department of Chemistry and referred to herein as inactive CORM-2 (iCORM-2), was used as a control. In all experiments, the concentration of DMSO used did not exceed 0.1% (i.e. 1:1000 dilution).
Cardiac Myocyte Isolation—Rats were killed by an intraperitoneal overdose of sodium pentobarbitone, and the hearts were rapidly excised into oxygenated physiological saline solution. Hearts were mounted on a Langendorff apparatus and perfused at 5 ml/min at 37 °C with a series of solutions based on an “isolation solution” of the following composition: 130 mm NaCl, 5.4 mm KCl, 1.4 mm MgCl2, 0.4 mm NaH2PO4, 5 mm Hepes, 10 mm glucose, 20 mm taurine, 10 mm creatine, pH 7.3. The first solution, containing 750 μm CaCl2, was perfused for 4 min. The heart was then perfused for 4 min with Ca2+-free isolation solution containing 100 μm Na2EGTA. Finally, perfusion was switched to the isolation solution containing 200 μm CaCl2 and collagenase (Worthington type 2; 0.1 mg/ml) for 9–12 min. The left ventricle was dissected and finely chopped in an enzyme-containing solution with 1% bovine serum albumin and gently agitated in a water bath at 37 °C. Aliquots of the cell suspension were examined every 5 min until a >80% yield of rod-shaped cells with a clear striation pattern was obtained. Myocytes were collected by filtration through nylon gauze and gentle centrifugation.
Cell Culture—HEK293 cells were cultured in growth medium comprising MEM with Earle's salts and l-glutamine, supplemented with 9% (v/v) fetal calf serum (Globepharm, Esher, Surrey, UK), 1% (v/v) nonessential amino acids, 50 μg/ml gentamicin, 100 units/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin in a humidified atmosphere of air/CO2 (19:1) at 37 °C. All culture reagents were purchased from Invitrogen unless otherwise stated.
Mutagenesis—Full-length hHT and rHT α1C cDNAs, previously described and deposited in GenBank™ (accession numbers are L04569 and L29536 for hHT and rHT, respectively), were ligated into the pcDNA3.1(+) mammalian expression vector (Invitrogen) and used for transfection of HEK293 cells as described below. Deletion of segments within the 71-amino acid insert of hHT (shown in Fig. 3) was performed by introducing two unique NruI restriction sites flanking the desired stretch using the MegaPrimer PCR strategy, cutting with NruI, and then religating the α1C(hHT) cDNA. Procedures have been described in depth previously (25). Individual Cys to Ser substitutions were similarly generated using the MegaPrimer PCR strategy.
FIGURE 3.

CO sensing requires a C-tail splice insert. A, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz) in cells expressing the hHT channel isoform (top, as in Fig. 2) or the rHT isoform (bottom), which lacks a C-tail insert. Scale bars, 100 pA (vertical) and 50 ms (horizontal). B, schematic of the splice insert found in hHT but not rHT channel variants, along with illustration of deletion mutations. The open bars indicate regions remaining, and horizontal lines indicate regions deleted. Only mutants containing a region that spans the shaded area displayed sensitivity to CO. C, bar graph illustrating mean ± S.E. (n = 9–20) inhibitory effect of CORM-2 (30 μm) on the various mutants tested. Dotted line, inhibitory effect of iCORM-2 on hHT activity (from Fig. 2B). ***, p < 0.001 versus the response of hHT to iCORM-2 (unpaired t test).
Cell Transfections—HEK293 cells were transfected with the appropriate pcDNA3.1/α1C construct using the PolyFect reagent (Qiagen, Hybaid Ltd., Teddington, UK) according to the manufacturer's instructions. Stably transfected cell lines were selected with G-418 antibiotic (1 mg/ml; Invitrogen) added 3 days after transfection. Selection was applied for 4 weeks (media changed every 4–5 days), colonies were then picked, grown to confluence, and screened electrophysiologically after plating onto coverslips for 24–48 h. G418 selection was maintained throughout the cloning process at 1 mg/ml and then reduced to 200 μg/ml in all subsequent passages once stable clones had been positively identified.
Electrophysiology—Cardiac myocytes were placed in the recording chamber and allowed to settle before perfusion was applied. Patch pipettes (1–3 megaohms resistance) were filled with 115 mm CsCl, 10 mm HEPES, 10 mm EGTA, 20 mm tetraethylammonium chloride, 5 mm MgATP, 0.1 mm Tris-GTP, 1 mm CaCl2 (pH adjusted to 7.0 with CsOH). Once the whole cell configuration was established, perfusion was activated with a solution of composition 140 mm NaCl, 5.4 mm CsCl, 2.5 mm CaCl2, 0.5 mm MgCl2, 5.5 mm HEPES, and 11 mm glucose (pH adjusted to 7.4 with NaOH). Cells were voltage-clamped at –80 mV, and to inactivate Na+ and T-type Ca2+ channels, a 50-ms prepulse to –30 mV was applied immediately before each test pulse. L-type currents were then activated by successive depolarizations to +10 mV (75-ms duration, 0.1 Hz).
HEK 293 cells attached to coverslip fragments were placed in a perfused (2–4 ml/min) chamber, and whole cell patch clamp recordings were made as previously described (26). Perfusate contained 95 mm NaCl, 5 mm CsCl, 0.6 mm MgCl2, 20 mm BaCl2, 5 mm HEPES, 10 mmd-glucose, 20 mm tetraethylammonium chloride (pH 7.4, 21–24 °C). Patch electrodes (resistance 4–7 megaohms) contained 120 mm CsCl, 20 mm tetraethylammonium chloride, 2 mm MgCl2, 10 mm EGTA, 10 mm HEPES, 2 mm ATP (pH adjusted to 7.2 with CsOH). Cells were clamped at –80 mV, and whole cell capacitance was determined from analogue compensation. Series resistance compensation of 70–90% was applied. Whole cell currents were evoked by 100-ms step depolarizations to various test potentials (0.1 Hz), and leak subtraction was applied as previously described (26). Holding current in any recording was <50 pA, and any recordings in which this fluctuated by more than 10% were discarded. Evoked currents were filtered at 1 kHz and digitized at 2 kHz, and current amplitudes were measured over the last 10–15 ms of each step depolarization. Since Ba2+ was used as the charge carrier, they displayed little or no inactivation during step depolarizations. Currents showing notable run down before CORM-2 application were discarded.
All voltage clamp and analysis protocols were performed with the use of an Axopatch 200A amplifier/Digidata 1200 interface controlled by Clampex 9.0 software (Molecular Devices, Foster City, CA). Offline analysis was performed using the data analysis package Clampfit 9.0 (Molecular Devices). Results are presented as means ± S.E., and statistical analysis was performed using unpaired Student's t tests, where p < 0.05 was considered statistically significant.
Generation of ρ0 Cells—Cells lacking mitochondrial DNA (ρ0 cells) were generated by incubation in medium containing EtBr (50 ng/ml), sodium pyruvate (1 mm), and uridine (50 μg/ml) (27). Verification of mitochondrial DNA loss was performed following RNA extraction using the Aurum total RNA minikit (Bio-Rad) according to the manufacturer's instructions. cDNA was synthesized from eluted RNA samples using the iScript™ Select cDNA synthesis kit (Bio-Rad), and real time RT-PCR was carried out using the Applied Biosystems (ABI) 7500 real time PCR system and Taqman® probes (ABI). β-Actin and 28 S ribosomal RNA were used as endogenous controls (ABI). Cytochrome b and COX2 (cytochrome oxidase subunit II) were the mitochondrial target genes (ABI). Data were analyzed using the 7500 software (ABI), and relative gene expression was calculated using the 2–ΔΔCT method using either β actin or 28 S as an endogenous control and cells cultured in the absence of EtBr as a calibrator.
RESULTS
CO Inhibits Cardiac L-type Ca2+ Channels—Ca2+ channel currents evoked in rat ventricular cardiac myocytes by successive depolarizations from –30 to +10 mV were reversibly inhibited by exposure to the CO donor CORM-2 (30 μm) in the perfusate (Fig. 1, A, D, and E). Effects were maximal within 2 min but required much longer to recover (e.g. Fig. 2E). CORM-2 is an established CO donor molecule (28–30), and its effects can be attributed to released CO, since the inactive form (iCORM-2) of the donor was without significant effect (Fig. 1, B and D). Furthermore, exposure of cells to dissolved CO (see “Experimental Procedures”) similarly inhibited currents (Fig. 1, C and D). Cardiac myocyte Ca2+ currents commonly run down with time during prolonged recordings. We observed a small degree of run down (Fig. 1E), but this clearly does not account for the slowly reversible inhibitory effects of CO or CORM-2 (Fig. 1E).
FIGURE 1.
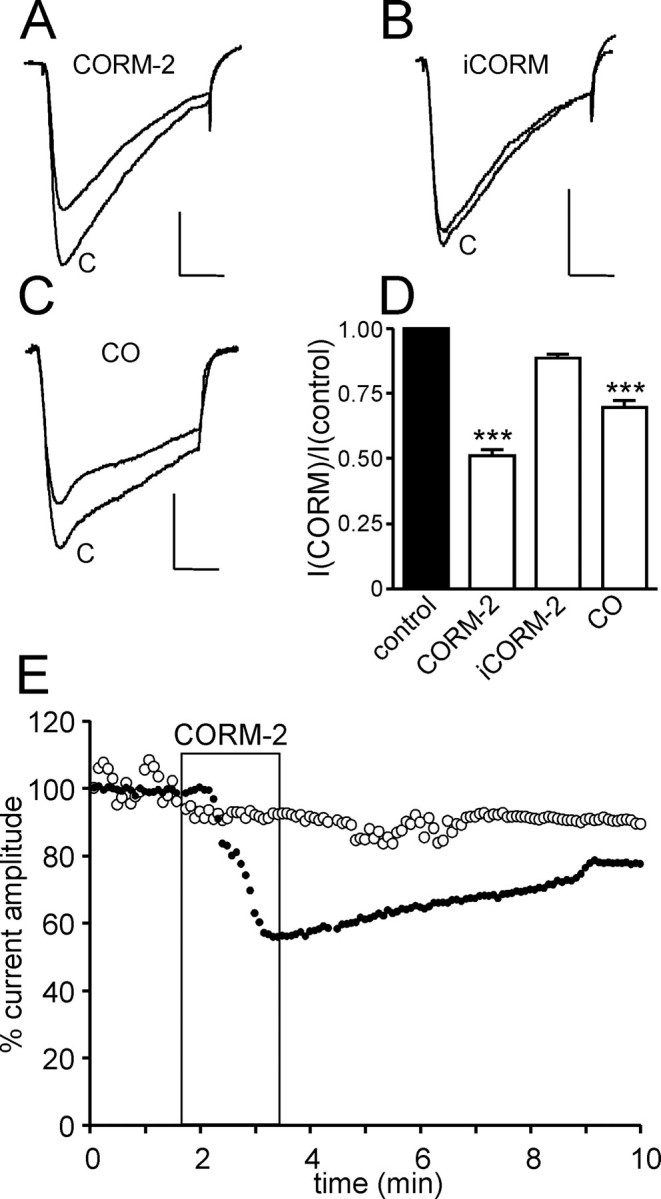
CO inhibits L-type Ca2+ currents in rat cardiomyocytes. Traces A–C show currents evoked in isolated rat cardiomyocytes before (control (C)) and during exposure to the CO donor CORM-2 (30 μm)(trace A) or its inactive form (iCORM-2; trace B) or dissolved CO (trace C), as indicated. Scale bars, 200 pA (vertical) and 20 ms (horizontal) in each case. Currents were evoked by step depolarizations from –30 to +10 mV. D, bar graph illustrating mean ± S.E. (n = 8–10) inhibitory effect of CORM-2 and iCORM-2 (30 μm) as well as dissolved CO. ***, p < 0.001 (unpaired t test). E, example time course traces showing either modest run down in the absence of drug application (open circles) or the slowly reversible inhibitory action of 30 μm CORM-2 (applied for the period indicated by the shaded region). Each point plotted is the current amplitude evoked by a step depolarization from –30 to +10 mV (75 ms, 0.1 Hz).
FIGURE 2.
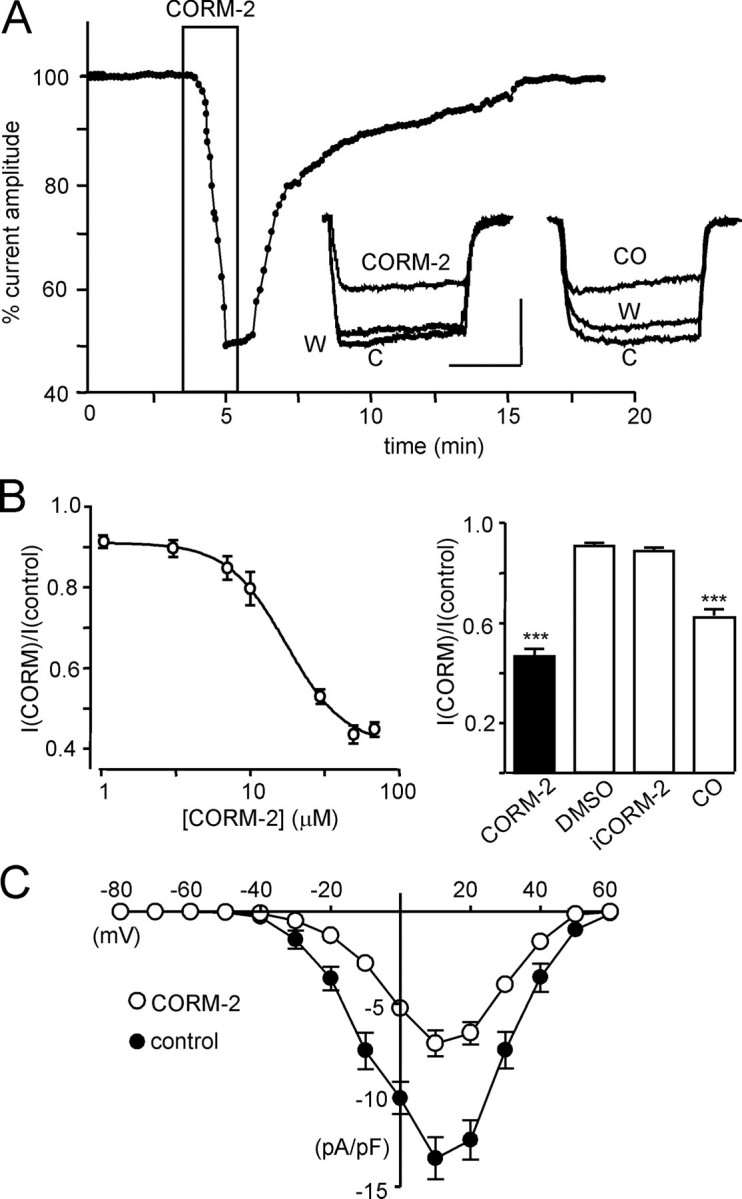
CO inhibits the human L-type Ca2+ channel α1C subunit. A, example time course of the reversible inhibitory effect of CO on whole cell Ca2+ channel currents in HEK293 cells expressing the full-length hHT isoform of the α1C subunit before, during, and after exposure to CORM-2 (30 μm; applied for period indicated by the boxed region). Each point plotted is the current amplitude evoked by a step depolarization from –80 to +10 mV (100 ms, 0.1 Hz). Inset, left, superimposed example currents obtained under control conditions (C), during exposure to CORM-2, and after washout (W). Also shown (right) are example traces evoked under identical conditions before (C), during (CO), and after (W) application of dissolved CO. Scale bars, 100 pA (vertical) and 50 ms (horizontal). B, left, concentration-response relationship; each plotted point is mean ± S.E. (n = 5–9 cells) inhibitory effect of CORM-2. Right, bar graph illustrating mean ± S.E. (n = 9–24) inhibitory effect of CORM-2 (30 μm), DMSO (0.1%), iCORM-2 (30 μm), and dissolved CO. ***, p < 0.001 versus the response to iCORM-2 (unpaired t test). C, mean ± S.E. (n = 10) current density versus voltage relationships taken before (solid circles) and during (open circles) exposure to 30 μm CORM-2 (n = 10).
Exposure of HEK293 cells expressing the α1C subunit of the human cardiac L-type Ca2+ channel (Cav1.2) to the CO donor CORM-2 caused marked inhibition of currents, as exemplified in Fig. 2A. As in cardiac myocytes, effects were largely but slowly reversible and not associated with any marked changes in current kinetics (Fig. 2A). The effects of CORM-2 were concentration-dependent (Fig. 2B) with a calculated IC50 of 14.8 ± 0.9 μm. Importantly, currents were largely unaffected (∼10% inhibition) either by the vehicle, DMSO, or by the inactive breakdown product, iCORM-2 (Fig. 2B), indicating that the effects of CORM-2 were largely due to its release of CO, as has previously been established (29–31). To confirm this, we exposed cells to dissolved CO and observed a similar degree of reversible inhibition (Fig. 2, A and B). Construction of current-voltage relationships indicated that the inhibitory effects of CO were apparent at all activating test potentials (Fig. 2C), indicating a lack of voltage dependence. Thus, the ability of CO to inhibit L-type Ca2+ currents in myocytes was fully reproducible in cells expressing only the pore-forming α1C subunit of the human channel. To investigate the structural requirements for CO inhibition of the channel and to probe candidate mechanisms of inhibition, we subsequently employed the recombinant α1C subunit and examined its responses to CORM-2.
Splice Insert Requirement for Ca2+ Channel CO Sensitivity—We have previously shown that splice variation in the cytoplasmic C-tail region of the α1C channel protein is a key determinant of some of the physiological responses of this channel (26). Fig. 3A indicates that this 71-amino acid (aa) region is also an absolute requirement for inhibition by CO. Thus, the full-length hHT variant is CO-sensitive, but the rHT variant, lacking the C-tail insert, was insensitive to CO. To investigate which region of this insert is required for CO sensitivity, we generated deletion mutants according to the schematic of Fig. 3B, and responses to 30 μm CORM-2 are summarized in Fig. 3C. We found that only those mutants with an intact region spanning residues 1787–1818 (i.e. mutants Δ4 and Δ7) demonstrated CO sensitivity. Importantly, their responses were quantitatively similar to the full-length hHT variant, suggesting that this region of the channel was necessary and sufficient for CO sensitivity.
CO Acts Independently of NO and Protein Kinase G—CO is an established activator of NO production, possibly by activating nitric-oxide synthase (1, 32). To investigate any role of NO, we first tested the effects of an NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO), and the nitric-oxide synthase inhibitor, NG-nitro-l-arginine methyl ester (both applied at 1 mm for 20 min prior to and during recordings). Neither affected the ability of CO (applied as CORM-2; 30 μm) to inhibit Ca2+ currents (Fig. 4, A and B). We also found that two distinct NO donors, SIN-1 (10 μm) and S-nitrosoglutathione (2 mm), failed to mimic the inhibitory actions of CO and did not significantly alter Ca2+ currents (Fig. 4, A and B). Finally, the protein kinase G inhibitor PET-cGMPS (100 nm, 2-h preincubation) was also without effect on the ability of CO to inhibit Ca2+ currents. All of these NO-related compounds were functionally active, as determined by their ability to modulate the activity of the recombinant leak K+ channel hTREK-1 (33). Thus, the inhibitory actions of CO on L-type Ca2+ currents were independent of the NO/PKG pathway.
FIGURE 4.

Inhibition of hHT Ca2+ currents by CO does not involve NO or protein kinase G. A, upper traces, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz) in hHT-expressing cells indicating the effects of CO applied via 30 μm CORM-2 in the absence of other drugs (upper left) and in the presence of a NO scavenger (carboxy-PTIO (c-PTIO); 1 mm) and a nitric-oxide synthase inhibitor (NG-nitro-l-arginine methyl ester (L-NAME); 1 mm). The lower traces show the lack of effect of two NO donors, SIN-1 (10 μm) and S-nitrosoglutathione (GSNO; 2 mm), and the lack of effect of the PKG inhibitor PET-cGMPS (100 nm) on the CO-mediated inhibition of currents, as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). B, bar graph illustrating mean ± S.E. (n = 5–10) effect of CO in the presence or absence (solid column) of other drugs examined, as illustrated in A. Dotted line, inhibitory effect of iCORM-2 on hHT activity (from Fig. 2B). ***, p < 0.001 versus the response of hHT to iCORM-2 (unpaired t test).
CO Inhibition of Ca2+ Currents Requires Reactive Oxygen Species (ROS)—Several reports indicate that CO can increase intracellular ROS levels via multiple potential mechanisms (34, 35). To investigate their potential involvement, we examined the ability of the antioxidant MnTMPyP (100 μm, 30-min preincubation) to interfere with the effects of CORM-2. As illustrated in Fig. 5A, MnTMPyP fully prevented the inhibitory actions of CORM-2 (30 μm). Similarly, the CO donor was ineffective during a simultaneous exposure to the reducing agent dithiothreitol (2 mm; Fig. 5A). These data suggested a possible involvement of ROS in CO-mediated inhibition of L-type Ca2+ channels. We therefore next investigated possible sources of intracellular ROS. As shown in Fig. 5B, exposure of cells to the xanthine oxidase inhibitor allopurinol (1 μm, 30-min preincubation) did not alter the ability of CO to inhibit Ca2+ currents. We also examined the effects of apocynin (APO; 30 μm) and (3 μm), known (though not highly selective) inhibitors of NADPH oxidase. Both agents themselves had modest but significant inhibitory effects (Fig. 5B), but in their continued presence, the degree of inhibition caused by CORM-2 was not significantly different from that observed in the absence of these compounds. Thus, although ROS appear to mediate the effects of CO, they do not appear to be derived from either xanthine oxidase or NADPH oxidase.
FIGURE 5.

Inhibition of hHT Ca2+ channels by CO involves ROS but not derived from NADPH or xanthine oxidase. A, left, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz) in hHT-expressing cells indicating the effects of CO applied via 30 μm CORM-2 in the absence or presence of MnTMPyP (100 μm) or dithiothreitol (DTT; 2 mm), as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). Right, bar graph illustrating mean ± S.E. (n = 7–10) inhibitory effect of CO in the absence (solid column) or presence of MnTMPyP or dithiothreitol, as indicated. B, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz) in hHT-expressing cells illustrating the effects of CO applied via 30 μm CORM-2 in the additional presence of allopurinol (allo.; 1 μm), DPI (3 μm), or APO (30 μm), as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). Below, bar graph illustrating mean ± S.E. (n = 6–9) effect of CO in the presence or absence (solid column) of other compounds as well as the effects of DPI and APO themselves. Brackets highlight the percentage inhibition caused by CO above that caused by either DPI or APO itself. Dotted line, inhibitory effect of iCORM-2 on hHT activity (from Fig. 2B). ***, p < 0.001 versus the response of hHT to iCORM-2 (unpaired t test). †††, p < 0.001 versus the response to DPI alone (unpaired t test). ‡‡‡, p < 0.001 versus the response to APO alone (unpaired t test).
Mitochondrial ROS Mediate CO Inhibition of Ca2+ Channels—Recent studies have implicated mitochondria as a source of CO-evoked rises in ROS (34, 35). To investigate their role in CO-mediated L-type Ca2+ channel inhibition, we first examined the effects of MitoQ, a mitochondria-targeted antioxidant (36). MitoQ (250 nm, 1-h preincubation) fully prevented the inhibition of Ca2+ currents by CO, whereas its control compound (TPP; 250 nm, 1-h preincubation, mitochondria-targeted but without antioxidant properties) was without effect (Fig. 6A). Thus, the source of ROS appeared to be mitochondrial. To test this idea further, we generated ρ0 cells lacking functional mitochondria (see “Experimental Procedures”). Real time reverse transcription-PCR measurements indicated >99% reduction in mitochondrial DNA for cytochrome b and COX2 in ρ0 cells, expression levels being 0.006 ± 0.003% (n = 3) and 0.004 ± 0.002% (n = 3) of that observed in control (untreated) cells. In these cells, CO was again without effect (Fig. 6A). ROS can be produced by the electron transport chain through electron leak at complexes I and III (37). The complex I inhibitor rotenone (2 μm) did not alter the ability of CO to inhibit Ca2+ currents (Fig. 6B). However, this effect of CO was fully prevented by either stigmatellin (1 μm, 1-h preincubation) or antimycin A (3 μm), two inhibitors of complex III (Fig. 6B). These data therefore suggest that CO inhibition of L-type Ca2+ currents is mediated by ROS generated at complex III of the mitochondrial electron transport chain.
FIGURE 6.
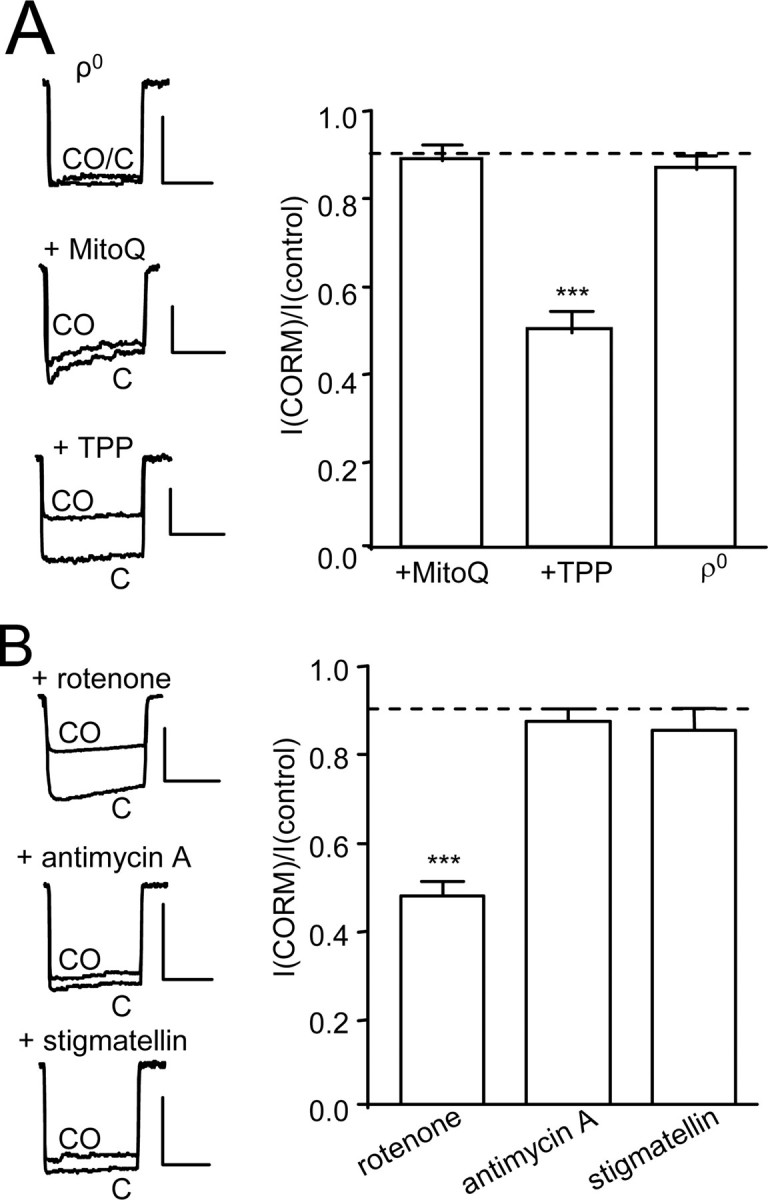
ROS from complex III of mitochondria mediate CO inhibition of hHT Ca2+ currents. A, left, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz), indicating the effects of CO applied via 30 μm CORM-2 in ρ0 cells or in control cells following a 1-h preexposure to either MitoQ (250 nm) or its control compound, TPP (250 nm), as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). Right, bar graph illustrating mean ± S.E. (n = 6–8) inhibitory effect of CO in ρ0 cells or in control cells following a 1-h preexposure to either MitoQ or TPP as indicated. B, left, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz) in hHT-expressing cells, indicating the effects of CO applied via 30 μm CORM-2 in the presence of rotenone (2 μm), antimycin A (3 μm), or stigmatellin (1 μm) as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). Right, bar graph illustrating mean ± S.E. (n = 5–6) inhibitory effect of CO in the presence of the three mitochondrial inhibitors as indicated. Dotted line, inhibitory effect of iCORM-2 on hHT activity (from Fig. 2B). ***, p < 0.001 versus the response of hHT to iCORM-2 (unpaired t test).
Cysteine Residues Are Essential for CO Modulation of Ca2+ Channels—Given that ROS clearly mediate the effects of CO on L-type Ca2+ channels, we investigated the potential involvement of redox-sensitive cysteine residues. The C-tail splice insert contains seven cysteine residues as shown in the schematic of Fig. 7. Of these, three (Fig. 7, shaded area) lie in the region previously determined to be required for CO sensitivity (aa 1787–1818; Fig. 3). We mutated each in turn to a serine residue (neutral and of a similar size). Remarkably, each point mutation rendered the channels completely insensitive to CO, as illustrated and quantified in Fig. 7. This indicated that all three cysteine residues were required for CO sensing by the L-type Ca2+ channel.
FIGURE 7.

CO inhibition of hHT Ca2+ channels is dependent on three cysteine residues. The upper schematic shows the position of cysteine residues within the hHT splice insert. The shaded region indicates the region required for CO sensitivity (aa 1787–1818). Bottom, left, example currents (evoked by step depolarizations from –80 to +10 mV, 100 ms, 0.1 Hz), indicating the effects of CO applied via 30μm CORM-2 on currents evoked in cells expressing point mutations for each of the three cysteine residues, as indicated. Scale bars, 100 pA (vertical) and 50 ms (horizontal). Right, bar graph illustrating mean ± S.E. (n = 6–12) inhibitory effect of CO on the wild type (WT) hHT channel and on each of the cysteine to serine mutant channels. Dotted line, inhibitory effect of iCORM-2 on hHT activity (from Fig. 2B). ***, p < 0.001 versus the response of hHT to iCORM-2 (unpaired t test).
Mitochondrial ROS Mediate CO Inhibition of Native Cardiac L-type Ca2+ Channels—In order to investigate whether mitochondrial ROS also mediated CO inhibition of native L-type Ca2+ channels, we exposed cardiac myocytes to MitoQ. Myocytes appeared less tolerant to this agent than HEK293 cells, since 250 nm MitoQ frequently caused spontaneous contraction (not shown). However, when exposed to 100 nm MitoQ for 30 min prior to application of 30 μm CORM-2, the inhibitory effect of the CO donor was not significantly different from that produced by vehicle alone (Fig. 8). By contrast, identical treatment of cells with the control compound TPP did not alter the inhibition of native Ca2+ currents by CORM-2 (Fig. 8). Thus, the mechanism of CO-mediated channel inhibition determined in HEK 293 cells also appears to account for the effects of CO in cardiac myocytes.
FIGURE 8.

Mitochondrial ROS mediate CO inhibition of Ca2+ channels in cardiac myocytes. Left-hand traces show currents evoked in isolated rat cardiomyocytes before (C) and during exposure to the CO donor CORM-2 (30 μm; CO), as indicated. Cells were previously incubated for 30 min with either MitoQ (100 nm; upper traces) or TPP (100 nm; lower traces). Scale bars, 100 pA (vertical) and 50 ms (horizontal) in each case. Right, bar graph illustrating mean ± S.E. (n = 6–8) inhibitory effect of CORM-2 (30 μm) on cardiac L-type Ca2+ currents in the absence of other pretreatment and following 30-min exposure to either MitoQ (100 nm) or TPP (100 nm), as indicated. ***, p < 0.001 versus the response of myocyte currents to iCORM-2 (unpaired t test). n.s., not significant.
DISCUSSION
This study reveals several novel physiological aspects of the gasotransmitter CO, which is being increasingly appreciated as an important messenger molecule (1, 2). Most fundamentally, we report that CO, applied either as the dissolved gas or from the donor molecule CORM-2, inhibits both native (rat) and recombinant (human) cardiac L-type Ca2+ channels (Figs. 1 and 2). The inhibition by CO is due to modulation of the α1C subunit, since no auxiliary subunits were co-expressed in our recombinant experiments, suggesting that auxiliary subunits are not required for channel modulation by CO. Our results support a previous brief report indicating that CO at saturating concentration reversibly inhibited L-type Ca2+ currents in the rat embryonic cardiac cell line H9c2 in order to afford protection against ischemic cell death (38). Surprisingly, given the cardioprotective properties of CO, this is the only other such study directly examining the effects of CO on cardiac L-type (or other) Ca2+ channel activity.
We also found that channel inhibition by CO requires a naturally occurring splice insert in the cytoplasmic C-tail portion of the channel (aa 1787–1857 of the hHT variant), a feature that probably affords variation in sensitivity to CO through differential expression of splice variants (39). When these splice variants were first reported, they were not believed to be of functional significance (39). However, we subsequently found that the splice insert required for CO sensing as shown in the present study is also necessary for O2 sensing (26), indicating that this region is likely to be of physiological importance for channel regulation, at least by some (CO and O2) but not all (NO; Fig. 4) physiologically active gases.
Of the numerous signaling pathways now known to be modulated by CO (1, 29), we found that inhibition of both recombinant and native L-type channels occurs via an increase in ROS production specifically from mitochondria. Cellular ROS production can occur at numerous sites, including NADPH oxidases and xanthine oxidase, and indeed CO can modulate NADPH oxidase (34). However, the present study suggests that mitochondria are the source of ROS increases evoked by CO. Thus, both a general antioxidant (MnTMPyP) and a mitochondrial targeted one (MitoQ), but not inhibition of NADPH or xanthine oxidases, prevented the actions of CO. This is in accordance with previous studies (34, 35, 40). CO is known to bind to complex IV (cytochrome c oxidase) of the electron transport chain (see Ref. 40), thereby presumably inhibiting its acceptance of electrons from complex III and so allowing more to leak from the chain and form ROS. In support of this, the effect of CO was prevented by complex III (but not complex I) inhibition (Fig. 6; see also Ref. 35).
Given that effects of CO were ROS-mediated, we probed the possible involvement of cysteine residues, since these have previously been indirectly implicated in O2 sensing by this channel (41), and maintaining a reduced environment with dithiothreitol also prevented the effects of CO (Fig. 5). We found that the three cysteine residues present in the CO-sensitive splice insert region (aa 1787–1818) were all necessary for sensitivity to CO. This is intriguing, given that two are adjacent and the third is separated by 10 other residues. However, we cannot speculate as to their possible interactions (e.g. through formation of disulfide bridges) until the structure of this region is better understood. The present study, however, reveals that CO may exert some of its important cardioprotective properties by suppression of Ca2+ influx through L-type Ca2+ channels. This occurs through increased formation of ROS at complex III of the mitochondrial electron transport chain, which alters channel function through redox modulation of three critical cysteine residues in the cytoplasmic C-tail region of this channel.
Acknowledgments
We are grateful to Dr. J. Bould (Department of Chemistry, University of Leeds) for the synthesis of iCORM-2, to Dr. M. P. Murphy (Cambridge) for the kind gift of MitoQ, and to Dr. D. S. Steele and Dr. Z. Yang (University of Leeds) for the supply of cardiac myocytes.
This work was supported by the British Heart Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HO, heme oxygenase; PET-cGMPS, β-phenyl-1,N2-etheno-8-bromoguanosine-3′,5′-cyclic monophosphorothioate; SIN-1, 3-morpholinosydnonimine hydrochloride; MnTMPyP, Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride; MitoQ, MitoQ10 methanesulfonate; TPP, decyl-TPP bromide; aa, amino acid(s); ROS, reactive oxygen species; APO, apocynin; DPI, diphenyleneiodonium.
References
- 1.Kim, H. P., Ryter, S. W., and Choi, A. M. (2006) Annu. Rev. Pharmacol. Toxicol. 46 411–449 [DOI] [PubMed] [Google Scholar]
- 2.Ryter, S. W., Alam, J., and Choi, A. M. (2006) Physiol. Rev. 86 583–650 [DOI] [PubMed] [Google Scholar]
- 3.Ewing, J. F., Raju, V. S., and Maines, M. D. (1994) J. Pharmacol. Exp. Ther. 271 408–414 [PubMed] [Google Scholar]
- 4.Lakkisto, P., Palojoki, E., Backlund, T., Saraste, A., Tikkanen, I., Voipio-Pulkki, L. M., and Pulkki, K. (2002) J. Mol. Cell Cardiol. 34 1357–1365 [DOI] [PubMed] [Google Scholar]
- 5.Clark, J. E., Naughton, P., Shurey, S., Green, C. J., Johnson, T. R., Mann, B. E., Foresti, R., and Motterlini, R. (2003) Circ. Res. 93 e2–e8 [DOI] [PubMed] [Google Scholar]
- 6.Yet, S. F., Perrella, M. A., Layne, M. D., Hsieh, C. M., Maemura, K., Kobzik, L., Wiesel, P., Christou, H., Kourembanas, S., and Lee, M. E. (1999) J. Clin. Invest. 103 R23–R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yet, S. F., Tian, R., Layne, M. D., Wang, Z. Y., Maemura, K., Solovyeva, M., Ith, B., Melo, L. G., Zhang, L., Ingwall, J. S., Dzau, V. J., Lee, M. E., and Perrella, M. A. (2001) Circ. Res. 89 168–173 [DOI] [PubMed] [Google Scholar]
- 8.Grilli, A., De Lutiis, M. A., Patruno, A., Speranza, L., Gizzi, F., Taccardi, A. A., Di, N. P., De, C. R., Conti, P., and Felaco, M. (2003) Ann. Clin. Lab. Sci. 33 208–215 [PubMed] [Google Scholar]
- 9.McGrath, J. J. (1984) Pharmacol. Biochem. Behav. 21 Suppl. 1, 99–102 [DOI] [PubMed] [Google Scholar]
- 10.Sylvester, J. T., and McGowan, C. (1978) Circ. Res. 43 429–437 [DOI] [PubMed] [Google Scholar]
- 11.McGrath, J. J., and Smith, D. L. (1984) Proc. Soc. Exp. Biol. Med. 177 132–136 [DOI] [PubMed] [Google Scholar]
- 12.McFaul, S. J., and McGrath, J. J. (1987) Toxicol. Appl. Pharmacol. 87 464–473 [DOI] [PubMed] [Google Scholar]
- 13.Durante, W., Johnson, F. K., and Johnson, R. A. (2006) J. Cell Mol. Med. 10 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durante, W. (2002) Vasc. Med. 7 195–202 [DOI] [PubMed] [Google Scholar]
- 15.Yang, L., Quan, S., Nasjletti, A., Laniado-Schwartzman, M., and Abraham, N. G. (2004) Hypertension 43 1221–1226 [DOI] [PubMed] [Google Scholar]
- 16.Christou, H., Morita, T., Hsieh, C. M., Koike, H., Arkonac, B., Perrella, M. A., and Kourembanas, S. (2000) Circ. Res. 86 1224–1229 [DOI] [PubMed] [Google Scholar]
- 17.Wiesel, P., Patel, A. P., Carvajal, I. M., Wang, Z. Y., Pellacani, A., Maemura, K., DiFonzo, N., Rennke, H. G., Layne, M. D., Yet, S. F., Lee, M. E., and Perrella, M. A. (2001) Circ. Res. 88 1088–1094 [DOI] [PubMed] [Google Scholar]
- 18.Bodi, I., Mikala, G., Koch, S. E., Akhter, S. A., and Schwartz, A. (2005) J. Clin. Invest. 115 3306–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis, M. J., and Hill, M. A. (1999) Physiol. Rev. 79 387–423 [DOI] [PubMed] [Google Scholar]
- 20.Moens, A. L., Claeys, M. J., Timmermans, J. P., and Vrints, C. J. (2005) Int. J. Cardiol. 100 179–190 [DOI] [PubMed] [Google Scholar]
- 21.Ravens, U., and Himmel, H. M. (1999) Pharmacol. Res. 39 167–174 [DOI] [PubMed] [Google Scholar]
- 22.Pepine, C. J. (1995) Eur. Heart J. 16 Suppl. H, 19–24 [DOI] [PubMed] [Google Scholar]
- 23.Motterlini, R., Clark, J. E., Foresti, R., Sarathchandra, P., Mann, B. E., and Green, C. J. (2002) Circ. Res. 90 E17–E24 [DOI] [PubMed] [Google Scholar]
- 24.Fearon, I. M., Palmer, A. C. V., Balmforth, A. J., Ball, S. G., Mikala, G., Schwartz, A., and Peers, C. (1997) J. Physiol. 500 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scragg, J. L., Fearon, I. M., Ball, S. G., Schwartz, A., Varadi, G., and Peers, C. (2004) Methods Enzymol. 381 290–302 [DOI] [PubMed] [Google Scholar]
- 26.Fearon, I. M., Varadi, G., Koch, S., Isaacsohn, I., Ball, S. G., and Peers, C. (2000) Circ. Res. 87 537–539 [DOI] [PubMed] [Google Scholar]
- 27.King, M. P., and Attardi, G. (1989) Science 246 500–503 [DOI] [PubMed] [Google Scholar]
- 28.Wu, L., and Wang, R. (2005) Pharmacol. Rev. 57 585–630 [DOI] [PubMed] [Google Scholar]
- 29.Boczkowski, J., Poderoso, J. J., and Motterlini, R. (2006) Trends Biochem. Sci. 31 614–621 [DOI] [PubMed] [Google Scholar]
- 30.Chatterjee, P. K. (2007) Br. J. Pharmacol. 150 961–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams, S. E., Wootton, P., Mason, H. S., Bould, J., Iles, D. E., Riccardi, D., Peers, C., and Kemp, P. J. (2004) Science 306 2093–2097 [DOI] [PubMed] [Google Scholar]
- 32.Lim, I., Gibbons, S. J., Lyford, G. L., Miller, S. M., Strege, P. R., Sarr, M. G., Chatterjee, S., Szurszewski, J. H., Shah, V. H., and Farrugia, G. (2005) Am. J. Physiol. 288 G7–G14 [DOI] [PubMed] [Google Scholar]
- 33.Dallas, M., Scragg, J. L., and Peers, C. (2008) Neuroreport 19 345–348 [DOI] [PubMed] [Google Scholar]
- 34.Taille, C., El-Benna, J., Lanone, S., Boczkowski, J., and Motterlini, R. (2005) J. Biol. Chem. 280 25350–25360 [DOI] [PubMed] [Google Scholar]
- 35.Zuckerbraun, B. S., Chin, B. Y., Bilban, M., de Costa, D. J., Rao, J., Billiar, T. R., and Otterbein, L. E. (2007) FASEB J. 21 1099–1106 [DOI] [PubMed] [Google Scholar]
- 36.Jauslin, M. L., Meier, T., Smith, R. A., and Murphy, M. P. (2003) FASEB J. 17 1972–1974 [DOI] [PubMed] [Google Scholar]
- 37.Turrens, J. F. (2003) J. Physiol. 552 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uemura, K., Adachi-Akahane, S., Shintani-Ishida, K., and Yoshida, K. (2005) Biochem. Biophys. Res. Commun. 334 661–668 [DOI] [PubMed] [Google Scholar]
- 39.Klockner, U., Mikala, G., Eisfeld, J., Iles, D. E., Strobeck, M., Mershon, J. L., Schwartz, A., and Varadi, G. (1997) Am. J. Physiol. 272 H1372–H1381 [DOI] [PubMed] [Google Scholar]
- 40.D'Amico, G., Lam, F., Hagen, T., and Moncada, S. (2006) J. Cell Sci. 119 2291–2298 [DOI] [PubMed] [Google Scholar]
- 41.Fearon, I. M., Palmer, A. C. V., Balmforth, A. J., Ball, S. G., Varadi, G., and Peers, C. (1999) J. Physiol. 514 629–637 [DOI] [PMC free article] [PubMed] [Google Scholar]