

Abstract
GABAB receptors mediate slow synaptic inhibition in the central nervous system and are important for synaptic plasticity as well as being implicated in disease. Located at pre- and postsynaptic sites, GABAB receptors will influence cell excitability, but their effectiveness in doing so will be dependent, in part, on their trafficking to, and stability on, the cell surface membrane. To examine the dynamic behavior of GABAB receptors in GIRK cells and neurons, we have devised a method that is based on tagging the receptor with the binding site components for the neurotoxin, α-bungarotoxin. By using the α-bungarotoxin binding site-tagged GABAB R1a subunit (R1aBBS), co-expressed with the R2 subunit, we can track receptor mobility using the small reporter, α-bungarotoxin-conjugated rhodamine. In this way, the rates of internalization and membrane insertion for these receptors could be measured with fixed and live cells. The results indicate that GABAB receptors rapidly turnover in the cell membrane, with the rate of internalization affected by the state of receptor activation. The bungarotoxin-based method of receptor-tagging seems ideally suited to follow the dynamic regulation of other G-protein-coupled receptors.
γ-Aminobutyric acid (GABA)2 is the major inhibitory neurotransmitter in the central nervous system (CNS) activating ionotropic GABAA/C, as well as the metabotropic GABAB receptor. GABAB receptors are expressed in all major brain structures (1–3) and are important for synaptic plasticity as well as having therapeutic implications for epilepsy, pain, spasticity, drug addiction, schizophrenia, depression, and anxiety (4).
The trafficking and cell surface mobility of ligand-gated GABAA receptors has been studied using reporter tags with electrophysiological (5) or imaging approaches (6, 7). However, the mobility and trafficking of extrasynaptic GABAB receptors has provided diverse results (8–11). The GABAB receptor is a heterodimeric G-protein-coupled receptor (GPCR), requiring R1 and R2 subunits to co-assemble before trafficking to the cell surface to form functional receptors. The R1 subunit possesses an ER retention motif that is masked by binding to the R2 subunit (12–14). Although, generally, GPCRs are readily internalized from the cell surface following agonist activation and receptor phosphorylation (15–17); the GABAB receptor was thought to behave differently, being relatively stable in the cell membrane (8, 9). However, other reports indicate that agonist activation of GABAB receptors may promote internalization and/or rapid recycling (10, 11, 18). To address the topic of GABAB receptor trafficking, prior studies have used various techniques to monitor receptor movement, including: receptor biotinylation (8, 9); antibody labeling of extracellular GABAB receptor epitopes on live cells (9); as well as fluorescence recovery after photobleaching (FRAP) (10). These methods have therefore relied on the use of relatively large reporter molecules, such as antibodies. Although such studies have revealed some aspects of trafficking behavior for GABAB receptors, there is still uncertainty regarding: how stable GABAB receptors are in the surface membrane; over what time scale they are likely to traffic; and whether trafficking observed in secondary cell lines is relevant to the movements of GABAB receptors in neurons.
To address these questions, we adopted a different strategy, based on incorporating a minimal-size epitope into the R1a subunit of the GABAB receptor. This comprised a 13-amino acid α-bungarotoxin binding site (BBS) motif, which retains high affinity for its ligand (19). The mobility of GABAB receptors can then be tracked, in real time, using fluorescent derivatives of α-bungarotoxin (BTX), which is a small reporter molecule. This high affinity site has been previously incorporated into ligand-gated ion channels, including AMPA (20) and GABAA (6) receptors, to monitor their trafficking, but its use in GPCRs is unexplored.
Here, we report that in HEK-293 cells, stably expressing inwardly rectifying Kir3.1 and 3.2 potassium channels (GIRK cells), and in hippocampal neurons expressing R1aBBSR2 subunits, the GABAB receptor undergoes quite rapid endocytosis and exocytosis. This indicates that the levels of this GPCR in the cell surface membrane are dynamically regulated, with implications for inhibitory synaptic plasticity.
EXPERIMENTAL PROCEDURES
GABAB Receptor Containing the BTX-binding Site—Complementary DNA fragments for the 13 amino acid BBS (WRYYESSLEPYPD;(19)) were synthesized with the nucleotide sequences: CTAGCTGGAGATACTACGAGAGCTCCCTGGAGCCCTACCCTGACG (sense) and CTAGCGTCAGGGTAGGGCTCCAGGGAGCTCTCGTAGTATCTCCAG (anti-sense). BBS fragments were annealed and phosphorylated then subcloned into a NheI site that was introduced six amino acids after the start of the mature R1a subunit (Fig. 1A). cDNAs for the R1a and R2 subunits (21) were mutated to include epitope tags for Myc (R1a) or FLAG (R2) inserted four amino acids from the start of the mature proteins. cDNAs were subcloned into the pRK5 vector, sequenced, and analyzed using Sequencher 3.1. The α7/5HT3a chimeric receptor was provided by N. Millar (UCL).
FIGURE 1.

BBS tag inserted into the R1a subunit is able to bind BTX without altering receptor pharmacology. A, schematic of R1a subunit showing relative positions for the myc tag (blue) and the BBS (red) and the primary sequence. B, images of fixed GIRK cells transfected with R1aBBSR2 + GFP, α7/5HT3a + GFP and GFP alone after treatment with 3 μg/ml BTX-Rhd. GFP (FITC) and rhodamine channels are shown separately and then merged. C, concentration response curves from GIRK cells transfected with either R1aR2 (▪; n = 8–11) or R1aBBSR2 with (▴; n = 5) or without (•; n = 5) an application of 3 μg/ml BTX-Rhd. D, CGP55845 inhibition curves for R1aR2 (▪) and R1aBBSR2 (•) using an EC50 GABA concentration (n = 5). E, time course of responses to submaximal GABA concentration (10 μm) for R1aR2 (▪; n = 5–8), and R1aBBSR2 with either BTX-Rhd (3 μg/ml) pre-applied (•; n = 6–7) or applied after the first and second GABA applications (○; n = 5–6).
Cell Culture and Transfection—GIRK cells (22) were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with penicillin-G/streptomycin (100 units/100 μg/ml; Invitrogen), 2 mm glutamine, 10%v/v fetal calf serum, and geneticin (0.5 mg/ml). These cells were transfected using a calcium phosphate method (23) with cDNAs (1 mg/ml) mixed in the following ratios: R1 (or R1BBS)/R2/EGFP reporter, 1:5:1, or for α7/5HT3a/EGFP reporter, 1:1. Primary hippocampal neurons were prepared from postnatal day 4 (P4) rat brains and plated onto poly-d-lysine-treated 22 mm glass coverslips (Assistence (5)). Neurons were transfected at 6–8 DIV using a calcium phosphate method (Clontech).
Patch Clamp Electrophysiology—Membrane currents were recorded, using whole cell patch clamp, from single GIRK cells as described previously (24). Patch pipettes (3–5 MΩ) were filled with a solution containing (mm): 120 KCl, 2 MgCl2, 11 EGTA, 30 KOH, 10 HEPES, 1 CaCl2, 1 GTP, 2 ATP, 14 creatine phosphate, pH 7.0. The cells were continuously perfused with Krebs solution containing (mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 11 glucose, and 5 HEPES, pH 7.4. To increase the size of the GABAB receptor-activated K+ currents and convert them to inward currents, prior to GABA application, the Krebs concentration of KCl was increased to 25 mm and that of NaCl reduced to 120 mm. This change altered EK from approximately -90 mV to -47mV. Peak amplitude GABA-activated K+ currents, were recorded from cells 48–72 h after transfection, at -70 mV holding potential and filtered at 5 kHz (-3dB, 6th pole Bessel, 36 dB/octave) before storage on a Dell Pentium III computer for analysis with Clampex 8. Changes >10% in the membrane input conductance or series resistance resulted in the recording being discarded.
To construct GABA concentration-response relationships, the current (I) was measured in the presence of each concentration of GABA applied at 3-min intervals as described previously (24). The currents were normalized to the maximum GABA response (Imax) and the concentration response relationship fitted with the Hill equation (24). Drugs and solutions were rapidly applied to the cells using a modified Y-tube, positioned ∼200–300 μm from the recorded cell. Inhibition concentration response curves, for the antagonist CGP55845, were fitted to Equation 1,
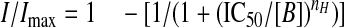 |
(Eq. 1) |
where the IC50 is the antagonist concentration (B) eliciting half-maximal inhibition of the GABA-activated potassium current.
For the analysis of GABA-activated potassium current run-down, current amplitudes were measured at 3-min intervals, in the absence or presence of 3 μg/ml BTX-Rhd (Molecular Probes), and the resulting time stability relationships were fitted with a single exponential function as described previously (24). The best fits to the data were determined using a Marquardt nonlinear least squares routine (Origin 6).
Immunocytochemistry—Transfected GIRK cells were fixed 48 h after transfection. Briefly, cells were washed twice with a phosphate-buffered saline (PBS, Sigma). BTX-Rhd was applied for different times and/or at different concentrations, in the absence or presence of unlabeled BTX. The cells were then fixed with 4% PFA for 15 min and quenched with NH4Cl (50 mm; VWR), for 10 min, prior to adhering the coverslips to slides using glycerol/gelatin (Sigma). When antibodies were added, cells were permeabilized with 0.1% v/v Triton X in 10% v/v blocking serum (see below). Cells were then incubated in 10% blocking serum (5% v/v horse (Invitrogen) and 5% v/v Donkey (Sigma) serum in PBS) for 25 min. Primary and secondary antibodies were diluted in 1.5% or 3% blocking serum, respectively. Antibodies against the C-terminal of the GABAB receptor R2 (guinea pig; 1:1000; Chemicon) were used in conjunction with a secondary Cy5 antibody (Donkey anti Guinea pig; Chemicon). Hippocampal neurons were transfected at 6–8DIV and fixed, as above, 6–8 days after transfection. We found that available N-terminal antibodies performed poorly in our immunoassays (see also Ref. 25).
Confocal Imaging—To image the fixed cells, a Zeiss Axioskop LSM 510 Meta Confocal microscope was used with a ×40/1.3 oil DIC objective and the following laser settings: FITC, 543 nm, 5% of maximum; TRITC, 633 nm, 80%; Cy5, 488 nm, 30%. The top and bottom of the cell was determined using a rapid z-stack scan, and a mid image of each cell was optimized and acquired with a mean of 8 scans, and stored for further analysis. Analysis of the confocal images was performed using Image J, where 3 regions of interest (ROI) were identified per cell (the cell surface membrane, intracellular compartment, and total cell fluorescence; see Fig. 2A, inset). Each pixel in the ROI was graded on a scale of 0 to 255 (max) and a mean fluorescence value was determined for the specific area (μm2) of interest. The background, a region where no cells were present, was subtracted from the ROI of each cell to give a normalized mean.
FIGURE 2.

BBS-tagged R1a subunit retains high affinity for BTX. A, concentration-fluorescence curves for cells expressing α7/5HT3a (□; n = 6–10) and R1aBBSR2 (•; n = 8–12) receptors, and GFP only (⋄; n = 8–10). B, whole cell radioligand binding experiments for the R1aBBSR2 receptor (n = 6).
To image live transfected neurons, PBS with 1 mm d-tubocurarine (d-TC) was applied for 2 min to block endogenous nicotinic ACh α7 subunit-containing receptors, and then with d-TC (1 mm) + BTX-Rhd (3 μg/ml) for 5 min to allow BTX-Rhd to bind to R1aBBSR2 receptors. Cells were washed (2×) with PBS, to remove any excess BTX-Rhd or d-TC, and superfused with Krebs, in a recording chamber, at either 37, 15, or at room temperature, 23 °C, by using a Peltier device. Transfected neurons were identified by the expression of GFP and images were scanned at specified time points using a Achroplan ×40/0.8 water DIC objective.
Radioactive α-Bungarotoxin Binding Assay—To ascertain the apparent affinity of BTX for its binding site in the GABAB R1aBBSR2 receptor, binding studies with 125I-BTX (200Ci/mmol; Amersham Biosciences) to cell surface receptors were performed with intact cells. Transfected GIRK cells were washed twice in PBS and re-suspended in Hanks buffered salt solution (GIBCO) containing 0.5% bovine serum albumin (Sigma). Cells were incubated in radioligand for 60 min at room temperature, with gentle agitation, in a total volume of 150 μl. Nonspecific binding was determined by the addition of a 1000-fold higher concentration of unlabeled BTX (Molecular Probes). Radioligand binding was assayed by filtration onto 0.5% polyethylenimine pre-soaked Whatman GF/A filters, followed by rapid washing with PBS using a Brandel cell harvester. Filters were assayed in a Wallac 1261 gamma counter. Scatchard analysis with non linear regression was used to obtain Bmax and Kd values (Origin 6) with Equation 2.
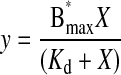 |
(Eq. 2) |
For comparison, the same analysis was used for GIRK cells expressing a chimeric α7/5HT3a receptor which is known to retain a high affinity BTX binding site (26).
RESULTS
Engineering the BBS into the GABAB Receptor—Incorporating a high affinity binding site for BTX into ligand-gated ion channels has demonstrated its usefulness in allowing the tracking of receptor movements into and out of the cell membrane (6, 7). To explore whether a similar strategy could be used to monitor GPCR trafficking, with their quite different transmembrane topologies, we engineered the N terminus of the R1a subunit of the GABAB receptor, to include a BBS (R1aBBS). To empirically maximize access for BTX to its binding site, the BBS was inserted just after the start of the mature protein (Fig. 1A), a position noted for GABAA receptors to be silent in terms of its impact on receptor structure and function (27). This region of the GABAB receptor was also deemed suitable since inserting a Myc epitope, just 4 amino acids from the start of the protein (upstream of the BBS), did not affect receptor function (21). The R1a subunit used here contained both Myc and BBS epitopes.
To determine if the R1aBBS subunit could bind α-bungarotoxin-rhodamine (BTX-Rhd) in vitro, GIRK cells were transfected with cDNAs encoding for: R1aBBS/R2/GFP; or the nicotinic/5-HT3a receptor chimera, α7/5HT3a/GFP (control for BTX binding); or GFP (negative control). The chimera was used to enhance the surface expression of bungarotoxin-binding α7 receptors, which is quite poor otherwise in HEK cells. After allowing receptor expression for 48 h, exposure to 3 μg/ml BTX-Rhd for 10 min, revealed prominent cell surface immunoreactivity for both the R1aBBSR2 and α7/5HT3a receptors. By contrast, no surface-specific BTX-Rhd immunoreactivity was observed on GFP-only transfected cells (Fig. 1B).
Activation of Kir3.1 and 3.2 by GABAB R1aBBSR2 Receptors—The effect of the BBS on the GABAB receptor functional properties was studied using GIRK cells and patch clamp electrophysiology. The activation of Kir 3.1 and 3.2 channels by GABA was used to construct concentration response curves for wild-type (R1aR2) and mutant R1aBBSR2 GABAB receptors in the presence and absence of 3 μg/ml BTX-Rhd (Fig. 1C). There was no significant shift in the concentration response curves, or the potency of GABA, determined from the EC50 values for the R1aBBSR2 receptor in the presence (0.48 ± 0.06 μm) or absence (0.36 ± 0.05 μm) of BTX-Rhd, compared with the wild-type receptor (0.43 ± 0.05 μm; n = 5–11; p > 0.05; Fig. 1C). In addition, antagonism by the competitive GABAB antagonist CGP55845 at R1aBBSR2 was minimally affected by the BBS (Fig. 1D) with only a small increase in the IC50 at R1aBBSR2 (118 ± 14 nm) compared with wild type (50 ± 6 nm).
The time-dependent stability of GABA-activated potassium currents was assessed by sequential applications of GABA, at 3-min intervals. By comparison with wild-type receptors, the effect of the BBS was assessed in R1aBBSR2 receptors by either using a 10-min pretreatment with BTX-Rhd or by applying BTX-Rhd for 3 min following the first two sequential GABA applications. With each protocol, the BBS appeared silent as there was no significant difference in the run-down profiles of either of the BTX-Rhd treated receptors compared with the wild-type receptors (Fig. 1E). The run-down of currents could be fitted with a single exponential providing time constants of: 2.9 ± 0.8 min (wild-type); 4.2 ± 0.7 min (R1aBBSR2 + 10 min BTX-Rhd pretreatment); and 2.3 ± 0.3 min (R1aBBSR2 + BTX-Rhd treatment after the first two sequential GABA applications).
These results suggested that the incorporation of the BBS into the N terminus of the GABAB R1a subunit, when expressed with the R2 subunit, did not alter the activation or pharmacological profiles of this receptor. Similarly, the addition of BTX-Rhd, which binds to the BBS on the N terminus of the R1aBBS subunit, did not impact on the function of the receptor. Overall, this indicates that the inserted BBS has the capability to operate as a functionally silent reporter of GABAB receptor trafficking.
BTX Binding to the R1aBBS Subunit—To determine the apparent affinity of BTX-Rhd for the BBS in R1a subunits, GIRK cells were transfected with cDNAs encoding for: R1aBBSR2/GFP; or α7/5HT3a/GFP; or just GFP. Cells were exposed for 10 min to increasing concentrations of BTX-Rhd before fixation with PFA. BTX-Rhd concentration cell surface fluorescence curves were constructed to deduce the apparent affinities of BTX-Rhd for these receptors, as well as for GFP (negative control; Fig. 2A). Receptors on or near the cell surface were determined by having an ROI, which incorporated just the surface fluorescence of the cell thus, providing mean fluorescence values that accounted for cell size (Fig. 2A, inset).
The BTX-Rhd concentration-fluorescence curves, using mean cell surface fluorescence, were normalized to the mean maximum fluorescence obtained from the α7/5HT3a positive controls exposed to 300 nm BTX-Rhd. The EC50 for BTX on the R1aBBSR2 receptor (52 ± 18 nm) was ∼80-fold greater when compared with that for the α7/5HT3a receptor (0.8 ± 0.1 nm; n = 8–12), indicating that the affinity of BTX for the BBS in GABAB receptors was lower than that for the site in α7/5HT3a receptor. The GFP-only expressing GIRK cells did not exhibit any concentration-dependent specific cell surface binding of the BTX-Rhd (Fig. 2A).
To further evaluate the affinity of BTX for the R1aBBSR2 receptor, radioligand binding studies were performed with 125I-BTX. Increasing concentrations of 125I-BTX were applied to GIRK cells, expressing either R1aBBSR2 or α7/5HT3a receptors, for 1 h at room temperature. These cells were then washed, harvested, and placed in the gamma counter. The GABAB R1aBBSR2 containing receptors clearly bound 125I-BTX in a concentration-dependent manner (Fig. 2B). Using a Scatchard analysis, the Kd for BTX binding was 9.8 ± 2.6 nm (n = 6) for the R1aBBSR2 receptor, which was 11-fold lower than the Kd for BTX binding to the α7/5HT3a receptor (0.86 ± 0.04 nm; n = 3).
Constitutive Internalization and Membrane Insertion of GABAB Receptors—The rate at which GABAB R1aBBSR2 receptors were internalized from the cell surface membrane was examined by exposing GIRK cells expressing R1aBBSR2 to 3 μg/ml BTX-Rhd for 10 min at room temperature. The cells were washed free of unbound BTX-Rhd and then kept at either 37 or 18 °C, until fixed at selected time points after the initial exposure to BTX (Fig. 3A). At 18 °C, a temperature that inhibits endocytosis (28), there was little change in the mean cell surface fluorescence over a 300-min period, demonstrating the stability of BTX-Rhd binding to the BBS on the GABAB receptor. However, over a similar duration at 37 °C, the mean cell surface fluorescence was rapidly reduced, according to a single exponential process with a time constant of 39.6 ± 4 min (Fig. 3, A and B). During this period, there was little change (∼10% reduction) in intracellular fluorescence and only a slight reduction in total fluorescence over 300 min at 37 °C (data not shown). To ascertain that there was no significant dissociation of BTX-Rhd from the BSS at 37 °C, 125I-BTX was incubated with cells expressing R1aBBSR2 for 1 h at room temperature and then in the presence of excess unlabeled-BTX for up to 120 min at 37 °C. No decrease in 125I-BTX binding was observed over this period (97 ± 14% of control bound at t = 120 min; n = 6), confirming that BTX-Rhd binds with high affinity to the BBS. Taken overall, these data suggested that recombinant GABAB receptors in GIRK cells are subjected to substantive and constitutive internalization.
FIGURE 3.

Constitutive turnover of cell surface GABAB receptors. A and B, images and time course relationship for surface fluorescence ROI for the R1aBBSR2 receptor at 37 °C (•) and 18 °C (○) (n = 7–22). Scale bars, 10 μm. C and D, images and time course relationships from fixed GIRK cells showing the appearance of surface fluorescence, over time, in the absence (▪; n = 7–22) and presence (□; n = 7–22) of unlabeled (UL) BTX for the R1aBBSR2 receptor at 37 °C. Scale bars, 10 μm.
To identify insertion of the GABAB R1aBBSR2 receptor into the plasma membrane and determine its rate, GIRK cells expressing R1aBBSR2 were exposed to unlabeled BTX (20 μg/ml), to block all the existing GABAB receptors on the cell surface, at room temperature for 5 min. After removing the excess unlabeled BTX, by washing, 3 μg/ml BTX-Rhd was applied for different times at 37 °C. The cells were then fixed prior to measuring levels of surface fluorescence. Within the first minute, there was little evidence of receptor insertion indicating that all pre-existing surface receptors were saturated and bound by unlabeled BTX. However, by 5–10 min, cell surface fluorescence appeared, indicating the membrane insertion of new R1aBBSR2 receptors, and by 20–25 min this had reached a steady state (Fig. 3, C and D). The rate for receptor insertion was best described by a single exponential process with a time constant of 7.8 ± 1.6 min (Fig. 3D). After longer incubation times (70 min), BTX-Rhd labeling appeared in intracellular compartments in accord with these newly inserted receptors being subject to internalization (Fig. 3C).
These results indicate that there are GABAB receptors, forming part of an intracellular pool, which are ready for rapid insertion, and these receptors are also subject to internalization as part of a dynamic cycling/rapid turnover of receptors at the cell surface of GIRK cells.
GABAB Receptor Activation and the Rate of Internalization—In accord with the process of internalization for other GPCRs, it might be expected that the state of GABAB receptor activation would be similarly influential. To investigate this, we examined whether the endocytosis of the R1aBBSR2 receptor, in GIRK cells, was influenced by receptor activation or inhibition at 37 °C. Cells expressing R1aBBSR2 were exposed to BTX-Rhd in the presence of either GABA (EC50, 0.3 μm), or the competitive inhibitor CGP55845 (IC50, 175 nm). Activation by GABA significantly increased the rate of internalization of GABAB receptors (τ = 18 ± 4 min) when compared with similar receptors expressed in untreated cells at 37 °C (39.6 ± 4 min; n = 5–12; p < 0.05; Fig. 4). By contrast, inhibiting GABAB receptors with CGP55845 did not change the rate of internalization compared with untreated controls (τ = 30 ± 7 min; n = 12; p > 0.05; Fig. 4).
FIGURE 4.

Activation of R1aBBSR2 receptors increases the rate of endocytosis. Time courses for the surface fluorescence of fixed GIRK cells expressing R1aBBSR2 receptors after application of either 0.3 μm GABA (•; n = 5–15) or 175 nm CGP55845 (▴; n = 10–12) at 37 °C. Control shown as a dotted line taken from Fig. 3B.
Tracking GABAB Receptors in Live Hippocampal Neurons using BTX—Although GABAB receptors can dynamically traffic in GIRK cells, it is unclear whether such processes occur in neurons, and if they do, whether BTX can track receptor mobility. R1aBBSR2 receptors were expressed in hippocampal neurons, exposed to BTX-Rhd and then fixed with PFA, permeabilized and a C terminus R2 antibody, plus Cy-5 secondary, were used to identify co-localization of BTX-Rhd tagged R1aBBS and R2 subunits (Fig. 5A). To track the R1aBBSR2/GFP receptors in hippocampal neurons, the nicotinic acetylcholine (nACh) antagonist, d-tubocurarine (1 mm) was applied for 2 min prior to BTX-Rhd application, to selectively block BTX binding to native hippocampal α7 nACh receptors (20); this concentration of d-tubocurarine did not affect the binding of BTX-Rhd to GABAB R1aBBSR2 expressed in GIRK cells, (data not shown). Transfected neurons were then exposed to 3 μg/ml BTX-Rhd, plus 1 mm d-tubocurarine, for 5 min at room temperature, washed twice with PBS, and placed in the recording chamber with heated, cooled, or room temperature Krebs. As an additional control, we confirmed that the application of 1 mm d-TC failed to affect the internalization of R1aBBSR2 GABAB receptors in GIRK cells at 37 °C (τ = 46 ± 8 min; n = 6–12; p > 0.05 compared with controls in the absence of d-TC). By distinguishing the neuronal somatic membrane from the intracellular compartment using separate ROIs, cell membrane fluorescence was monitored in real time. At 37 °C BTX-labeled GABAB receptors were rapidly internalized within 5–10 min (Fig. 5B) with the process being slowed, but not entirely prevented, at 15 °C (Fig. 5B). Internalization was correlated with the Krebs temperature and was accounted for by a single exponential that decreased with an increase in temperature (15 °C, τ = 20 ± 2 min; 22 °C, τ = 11 ± 1 min; 37 °C, τ = 7.5 ± 1 min (n = 4–6; Fig. 5C; p < 0.05 comparing data at 15 °C with 22 °C or 37 °C).
FIGURE 5.

Rate of endocytosis for R1aBBSR2 receptors can be monitored in hippocampal neurons. A, image of a hippocampal neuron transfected at 7DIV with R1aBBS, R2, and GFP cDNAs (FITC, green). At 14DIV, 3 μg/ml BTX-Rhd was applied (rhodamine, red) prior to being fixed and permeabilized. Additionally, an R2 C-terminal antibody with Cy5 (blue) secondary was applied. Scale bars, 10 μm. Merged image (without FITC) is shown. B, images of live neurons expressing R1aBBSR2 subunits at 15 °C (upper panel) and 37 °C (lower panel). C, time course of surface fluorescence for live neurons expressing R1aBBSR2 subunits at 37 °C (•), 22 °C (□), and 15 °C (○); n = 4–6.
Modulating GABAB Receptor Trafficking in Live Hippocampal Neurons—Applying the GABAB receptor agonist, baclofen (EC50; 3 μm), did not change the rate (τ = 13 ± 2 min) or the steady-state (ss) level of endocytosis (ss = 41 ± 3%) for R1aBBSR2 receptors expressed in hippocampal neurons compared with untreated controls at room temperature (Fig. 6A; τ = 12 ± 1 min; ss = 46 ± 1%). However, the application of the GABAB receptor antagonist, CGP55845 (EC50′ 500 nm), significantly reduced the steady-state level (ss = 59 ± 2%), but not the rate of GABAB receptor endocytosis (τ = 11 ± 1 min) (Fig. 6A). These results suggest that in neurons, there is a basal rate of endocytosis that can be modulated by GABAB receptor inhibition.
FIGURE 6.

Modulation of GABAB receptor endocytosis in hippocampal neurons. Time courses for the surface fluorescence of live neurons, expressing R1aBBSR2 receptors, with the application of either A, 3 μm Baclofen (•; n = 5) or 500 nm CGP55845 (▵; n = 5) or B, 3 μm glutamate (▴; n = 5), 30 μm glutamate (▵; n = 5), or 1 mm kynurenic acid (•; n = 5) at room temperature. Control rates of endocytosis are shown as dotted lines and are taken from Fig. 5. Solid lines indicates the duration of drug application.
As postsynaptic GABAB receptors reside in close proximity to excitatory terminals, it was conceivable that the activation or inhibition of excitatory receptors might influence the trafficking of GABAB receptors. This was examined by chronically applying glutamate, for up to 60 min, at both low (3 μm) and high (30 μm) concentrations (Fig. 6B). At 3 μm, there was no change in the rate (τ = 14 ± 2 min) or the steady-state level of endocytosis (ss = 47 ± 2%) of GABAB receptors compared with untreated controls at room temperature. However, at 30 μm, the rate of GABAB receptor endocytosis was slowed (τ = 25 ± 1 min), and the steady-state level of endocytosis reduced (ss = 33 ± 1%) compared with untreated controls at room temperature (Fig. 6B). The non-selective excitatory receptor antagonist, kynurenic acid (1 mm), neither affected the rate (τ = 10 ± 1 min) nor the steady state (44 ± 1%) of GABAB receptor endocytosis (Fig. 6B). These results suggest that the endocytosis of GABAB receptors in hippocampal neurons can be influenced by the activation of excitatory receptors with glutamate, or the inhibition of GABAB receptors.
DISCUSSION
This study presents a BBS tagging method for tracking the mobility of GABAB receptors in real time using secondary cell lines and live neurones. This method also appears eminently suitable for studying the trafficking of other GPCRs. Our results indicate, by using electrophysiological techniques, that there are no untoward functional consequences of tagging GABAB receptors with the BBS, and even when bound by BTX, activation of the receptor by GABA and its inhibition by CGP55845 are largely unaffected. Thus, the BBS insert appears to be functionally silent, fulfilling the most important criterion of a reporter tag.
By attaching BTX-Rhd to the R1aBBS subunit, it was clear, using confocal microscopy, that GABAB receptors constitutively internalize in a matter of minutes and that replenishment of cell surface receptors occurred over a similar time scale. Interestingly, activation by the natural transmitter, GABA, increased the rate of internalization in GIRK cells, but this rate was unaffected by a competitive antagonist.
GABAB receptors are key components for slow synaptic inhibition in the CNS and they are purported to have a role in neuropsychiatric and neurological disorders (13) making them potential drug targets for several disease states. The desensitization and endocytosis of the GABAB receptor, either constitutively or after receptor activation, is important because of the potential implications for synaptic plasticity and regulation of neuronal inhibition.
G-protein-coupled receptors often exhibit a generic mechanism for endocytosis, after receptor activation, which usually involves rapid phosphorylation by G-protein receptor kinases (GRKs), followed by arrestin and dynamin-stimulated internalization via clathrin-coated pits (15, 29, 30). However, the desensitization and endocytosis of GABAB receptors does not conform to this pattern. Notably, using patch clamp recording, phosphorylation of the R2 subunit, on Ser-892 by PKA (21), or on Ser-783 by 5′-AMP-dependent protein kinase (AMPK) (24), appeared to stabilize GABAB receptors on the cell surface. By contrast, although GABAB receptors seem not to be phosphorylated by GRKs, in cerebellar granule neurons, GRK4 does mediate baclofen-induced desensitization and endocytosis, but probably not via phosphorylation of either R1 or R2 subunits (31).
Previous studies have provided differing results regarding GABAB receptor trafficking, ranging from demonstrations of highly stable GABAB receptors in the cell membrane of HEK cells (8), to receptors that undergo constitutive endocytosis in CHO-K1 cells (18). Recently, there is increasing evidence in support of rapid GABAB receptor recycling (11), constitutive endocytosis (9), and lateral diffusion of these receptors in the cell membrane (10). Why these studies reach different conclusions is unclear. Some variation may result from differences in the techniques used to monitor GABAB receptor mobility or in the cell types used for study. One specific advantage of using the BTX-Rhd tag is that it has a significantly smaller volume compared with antisera, which are used in more traditional receptor labeling approaches. It is therefore less likely to interfere with the innate trafficking of the GABAB receptor. Indeed, antibodies may interfere with aspects of GABAB receptor trafficking, since high concentrations of antisera, used to monitor the trafficking of the R2 subunit, can reduce internalization (9), which may not be surprising given their ability to promote clustering or capping of cell surface antigens. Significantly, the incorporation of the high affinity BBS into the R1a subunit offers an important technical advance, allowing us to track GABAB receptor mobility in real time using live neurons. We could not use antibodies for live trafficking studies since those available and capable of recognizing external N-terminal receptor epitopes performed poorly (25).
The affinity of the BBS for BTX in the R1aBBSR2 receptor (Kd = 9.8 ± 2.6 nm), estimated from radioligand binding, was similar to that reported previously for AMPA or GABAA receptors with Kd values of 7.85 ± 2.4 and 13.6 nm, respectively (6, 20). In comparison, BTX affinity for R1aBBSR2 was 11-fold lower than for the α7/5HT3a chimeric receptor (Kd = 0.86 ± 0.04 nm), suggesting that the protein folding and juxtaposed residues of the BBS in the R1a subunit effectively, but not completely, reproduce that of the chimeric α7/5HT3a receptor. However, this proved to be sufficient for BTX to act as a sensitive reporter of GABAB receptor trafficking. Of importance, electrophysiological studies demonstrated that there was no change in GABAB receptor pharmacology after insertion of the BBS, even with bound BTX-Rhd, confirming the suitability of the method for tracking these receptors in real time.
The analysis of fluorescence, in specific ROIs, enabled the rate of internalization of surface receptors into intracellular compartments to be quantified. The potential issue of BTX-Rhd dissociation from the BBS, which would confound its use as a reporter for receptor trafficking, was addressed in two ways: by a time course study with BTX-Rhd at 18 °C, a temperature sufficient to inhibit the endocytosis of membrane proteins (28); and by a radioligand binding experiment with 125I-BTX at 37 °C, in the presence of excess unlabeled BTX that would replace any dissociated 125I-BTX. Under either of these conditions, BTX did not dissociate from the receptor during the time courses of the experiments (5 h and 2 h, respectively), suggesting that the internalization of the R1aBBS subunit was a real phenomenon, with a monoexponential rate of internalization (τ = 39.6 ± 4 min), and an even faster rate of insertion (τ = 7.8 ± 2 min). This strongly indicated that the GABAB receptor constitutively traffics into and out of the cell membrane. This constant turnover of surface GABAB receptors might explain why previous studies reported relative stability (8) as the steady-state number of surface receptors are unlikely to change much over time.
After the application of BTX-Rhd, clusters of fluorescence formed in intracellular compartments, which are, most likely, BTX-labeled R1aBBS subunits that have trafficked into endosomes via clathrin-coated pits. These can be identified by co-staining with transferrin (data not shown) and is seemingly a favored mechanism of GABAB receptor endocytosis (9, 11, 18).
Agonist activation of GPCRs generally increases the rate of endocytosis (29); however, for the GABAB receptor, the consequences of receptor activation are unclear. Prior reports suggest that the GABAB receptor undergoes no (8, 9) or increased (18) agonist-induced endocytosis. Furthermore, GABAB receptors have been observed to undergo clathrin-dependent internalization and rapid recycling to the plasma membrane, following activation with baclofen, in dorsal root ganglionic and spinal cord neurons, in addition to evidence of dissociation and trafficking of the GABAB receptor heterodimer upon chronic stimulation with capsaicin (11). Using BTX-Rhd, the rate of internalization of GABAB receptors, in GIRK cells, was increased by the addition of an EC50 concentration of GABA, which may have been resolved by this method due to its higher time resolution. The increased rate of endocytosis, with agonist activation, could be an important regulatory function of GABAB receptors that might prevent excessive perisynaptic inhibition.
Although the BBS with BTX-Rhd enabled the trafficking of GABAB receptors in GIRK cells to be followed, it was important to demonstrate a similar utility for this method in neurons, providing an essential step in understanding the regulatory mechanisms of endocytic pathways for these receptors. Using live transfected primary hippocampal cultures expressing the R1aBBS and R2 subunits, along with GFP, it was possible to monitor the rate of endocytosis of these receptors in real time. We used d-TC to block the binding of BTX to endogenous neuronal nACh receptors; however, these were quite sparse in our hippocampal cultures. Nevertheless, d-TC did not have any effect on binding of BTX-Rhd to the BBS on the GABAB receptor, or on the trafficking of GABAB receptors in GIRK cells. This is not surprising since a prior study found that d-TC did not affect GABAB receptor mediated outward K+ currents in substantia nigra dopaminergic neurons (32). The rate of endocytosis of GABAB receptors in neurons was more rapid than that observed in fixed GIRK cells. Reducing the temperature to 15 °C slowed the rate of endocytosis in neurons, but did not cause complete inhibition. This may indicate that other pathways can regulate the trafficking of GABAB receptors (33), which may not be surprising, given the number of associating/scaffolding proteins, kinases and phosphatases that are likely to be present in neuronal cells. It was also conceivable that manipulating the activity of excitatory and inhibitory receptors could play an important role in the synaptic plasticity of GABAB receptors. With GABA increasing the rate of endocytosis in GIRK cells, it was surprising that baclofen did not have a similar effect on the rate or steady-state level of endocytosis in neurons. This lack of effect could be due to basal levels of GABA tonically activating the GABAB receptors, which would explain why endocytosis was reduced by the GABAB receptor inhibitor, CGP55845. With regard to excitatory receptors, addition of a low concentration of glutamate or the non-selective inhibitor, kynurenic acid, had no effect on the rate or steady-state of GABAB receptor endocytosis. Only when the glutamate concentration was increased to 30 μm was there a decrease in the rate of GABAB receptor endocytosis, which may reflect a counteraction to excessive overexcitation of the neuron.
Overall, this study demonstrates that GABAB receptors undergo constitutive turnover at the cell membrane in recombinant cells and neurons, which can be modulated by receptor activation/inhibition. Using the BBS-epitope tag technique, the identification of mechanisms that control GABAB receptor turnover can now be investigated in live neuronal cells. Furthermore, we predict that changes in GABAB receptor turnover are likely to have implications for slow synaptic inhibition in the CNS, not only under physiological conditions, but also in diseased states.
Acknowledgments
We thank Stuart Lansdell for technical help with the radioligand binding experiments.
This work was supported by the MRC and the Wellcome Trust. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: GABA, γ-amino butyric acid; GPCR, G-protein-coupled receptor; PBS, phosphate-buffered saline; BBS, α-bungarotoxin binding site; BTX, α-bungarotoxin; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine isothiocyanate; ROI, region of interest; GFP, green fluorescent protein.
References
- 1.Benke, D., Honer, M., Michel, C., Bettler, B., and Mohler, H. (1999) J. Biol. Chem. 274 27323-27330 [DOI] [PubMed] [Google Scholar]
- 2.Fritschy, J. M., Meskenaite, V., Weinmann, O., Honer, M., Benke, D., and Mohler, H. (1999) Eur. J. Neurosci. 11 761-768 [DOI] [PubMed] [Google Scholar]
- 3.Fritschy, J. M., Sidler, C., Parpan, F., Gassmann, M., Kaupmann, K., Bettler, B., and Benke, D. (2004) J Comp Neurol. 477 235-252 [DOI] [PubMed] [Google Scholar]
- 4.Enna, S. J., and Bowery, N. G. (2004) Biochem. Pharmacol 68 1541-1548 [DOI] [PubMed] [Google Scholar]
- 5.Thomas, P., Mortensen, M., Hosie, A. M., and Smart, T. G. (2005) Nat. Neurosci. 8 889-897 [DOI] [PubMed] [Google Scholar]
- 6.Bogdanov, Y., Michels, G., Armstrong-Gold, C., Haydon, P. G., Lindstrom, J., Pangalos, M., and Moss, S. J. (2006) EMBO J. 25 4381-4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luscher, B., and Keller, C. A. (2004) Pharmacol. Ther. 102 195-221 [DOI] [PubMed] [Google Scholar]
- 8.Fairfax, B. P., Pitcher, J. A., Scott, M. G., Calver, A. R., Pangalos, M. N., Moss, S. J., and Couve, A. (2004) J. Biol. Chem. 279 12565-12573 [DOI] [PubMed] [Google Scholar]
- 9.Grampp, T., Sauter, K., Markovic, B., and Benke, D. (2007) J. Biol. Chem. 282 24157-24165 [DOI] [PubMed] [Google Scholar]
- 10.Pooler, A. M., and McIlhinney, R. A. J. (2007) J. Biol. Chem. 282 25349-25356 [DOI] [PubMed] [Google Scholar]
- 11.Laffray, S., Tan, K., Dulluc, J., Bouali-Benazzouz, R., Calver, A. R., Nagy, F., and Landry, M. (2007) Eur. J Neurosci 25 1402-1416 [DOI] [PubMed] [Google Scholar]
- 12.Margeta-Mitrovic, M., Jan, Y. N., and Jan, L. Y. (2000) Neuron 27 97-106 [DOI] [PubMed] [Google Scholar]
- 13.Bettler, B., Kaupmann, K., Mosbacher, J., and Gassmann, M. (2004) Physiol. Rev. 84 835-867 [DOI] [PubMed] [Google Scholar]
- 14.Couve, A., Filippov, A. K., Connolly, C. N., Bettler, B., Brown, D. A., and Moss, S. J. (1998) J. Biol. Chem. 273 26361-26367 [DOI] [PubMed] [Google Scholar]
- 15.Marchese, A., Paing, M. M., Temple, B. R., and Trejo, J. (2008) Annu. Rev. Pharmacol. Toxicol. 48 601-629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore, C. A., Milano, S. K., and Benovic, J. L. (2007) Annu. Rev. Physiol. 69 451-482 [DOI] [PubMed] [Google Scholar]
- 17.Hanyaloglu, A. C., and Zastrow, M. v. (2008) Annu. Rev. Pharmacol. Toxicol. 48 537-568 [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Maeso, J., Wise, A., Green, A., and Koenig, J. A. (2003) Eur. J. Pharmacol. 481 15-23 [DOI] [PubMed] [Google Scholar]
- 19.Harel, M., Kasher, R., Nicolas, A., Guss, J. M., Balass, M., Fridkin, M., Smit, A. B., Brejc, K., Sixma, T. K., Katchalski-Katzir, E., Sussman, J. L., and Fuchs, S. (2001) Neuron 32 265-275 [DOI] [PubMed] [Google Scholar]
- 20.Sekine-Aizawa, Y., and Huganir, R. L. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 17114-17119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couve, A., Thomas, P., Calver, A. R., Hirst, W. D., Pangalos, M. N., Walsh, F. S., Smart, T. G., and Moss, S. J. (2002) Nat. Neurosci. 5 415-424 [DOI] [PubMed] [Google Scholar]
- 22.Leaney, J. L., Milligan, G., and Tinker, A. (2000) J. Biol. Chem. 275 921-929 [DOI] [PubMed] [Google Scholar]
- 23.Wilkins, M. E., Hosie, A. M., and Smart, T. G. (2005) J. Physiol. 567 365-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuramoto, N., Wilkins, M. E., Fairfax, B. P., Revilla-Sanchez, R., Terunuma, M., Tamaki, K., Iemata, M., Warren, N., Couve, A., Calver, A., Horvath, Z., Freeman, K., Carling, D., Huang, L., Gonzales, C., Cooper, E., Smart, T. G., Pangalos, M. N., and Moss, S. J. (2007) Neuron 53 233-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Correa, S. A., Munton, R., Nishimune, A., Fitzjohn, S., and Henley, J. M. (2004) Neuropharmacology 47 475-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisele, J. L., Bertrand, S., Galzi, J. L., Devillers-Thiery, A., Changeux, J. P., and Bertrand, D. (1993) Nature 366 479-483 [DOI] [PubMed] [Google Scholar]
- 27.Connolly, C. N., Krishek, B. J., McDonald, B. J., Smart, T. G., and Moss, S. J. (1996) J. Biol. Chem. 271 89-96 [DOI] [PubMed] [Google Scholar]
- 28.Connolly, C. N., Kittler, J. T., Thomas, P., Uren, J. M., Brandon, N. J., Smart, T. G., and Moss, S. J. (1999) J. Biol. Chem. 274 36565-36572 [DOI] [PubMed] [Google Scholar]
- 29.Bunemann, M., and Hosey, M. M. (1999) J. Physiol. 517 5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couve, A., Calver, A. R., Fairfax, B., Moss, S. J., and Pangalos, M. N. (2004) Biochem. Pharmacol. 68 1527-1536 [DOI] [PubMed] [Google Scholar]
- 31.Perroy, J., Adam, L., Qanbar, R., Chenier, S., and Bouvier, M. (2003) EMBO J. 22 3816-3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caputi, L., Bengtson, C. P., Guatteo, E., Bernardi, G., and Mercuri, N. B. (2003) Synapse 47 236-239 [DOI] [PubMed] [Google Scholar]
- 33.Mayor, S., and Pagano, R. E. (2007) Nat. Rev. Mol. Cell. Biol. 8 603-612 [DOI] [PubMed] [Google Scholar]