

Abstract
Protein S-nitrosylation mediated by cellular nitric oxide (NO) plays a primary role in executing biological functions in cGMP-independent NO signaling. Although S-nitrosylation appears similar to Cys oxidation induced by reactive oxygen species, the molecular mechanism and biological consequence remain unclear. We investigated the structural process of S-nitrosylation of protein-tyrosine phosphatase 1B (PTP1B). We treated PTP1B with various NO donors, including S-nitrosothiol reagents and compound-releasing NO radicals, to produce site-specific Cys S-nitrosylation identified using advanced mass spectrometry (MS) techniques. Quantitative MS showed that the active site Cys-215 was the primary residue susceptible to S-nitrosylation. The crystal structure of NO donor-reacted PTP1B at 2.6 Å resolution revealed that the S-NO state at Cys-215 had no discernible irreversibly oxidized forms, whereas other Cys residues remained in their free thiol states. We further demonstrated that S-nitrosylation of the Cys-215 residue protected PTP1B from subsequent H2O2-induced irreversible oxidation. Increasing the level of cellular NO by pretreating cells with an NO donor or by activating ectopically expressed NO synthase inhibited reactive oxygen species-induced irreversible oxidation of endogenous PTP1B. These findings suggest that S-nitrosylation might prevent PTPs from permanent inactivation caused by oxidative stress.
Two major groups of free radicals have been identified as second messengers
in signal transduction. Superoxide
(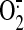 ), generated by cellular
NADPH oxidases, and its derivative hydrogen peroxide
(H2O2) are members of the reactive oxygen species
(ROS)3
(1). Nitric oxide (NO) and its
derivatives, which make up the reactive nitrogen species (RNS), are the
products of NO synthases (NOSs)
(2).
), generated by cellular
NADPH oxidases, and its derivative hydrogen peroxide
(H2O2) are members of the reactive oxygen species
(ROS)3
(1). Nitric oxide (NO) and its
derivatives, which make up the reactive nitrogen species (RNS), are the
products of NO synthases (NOSs)
(2).
Both NADPH oxidases and NOSs can be activated in signaling response to a number of extracellular stimuli (3-6). ROS and RNS play a critical role in regulating important signaling events, such as tyrosine phosphorylation-dependent signal transduction (7, 8). In addition, ROS (9) and RNS (10) may function as typical second messengers to transiently control the activity of signaling modulators when they are generated at physiological concentrations. Under certain pathological conditions, however, the role of ROS and RNS becomes more complicated. For example, when ischemia/reperfusion (I/R) occurs in heart and liver, an increased RNS level in ischemic tissues is critical in the protection of cells against subsequent damage due to a ROS burst upon reperfusion (11, 12). Therefore, although both ROS and RNS are classified as free radical-based second messengers, they may regulate cell signaling differently. The molecular details of their activities are largely unexplored.
ROS and RNS regulate the activity of signaling modulators through post-translational modification, which is how they perform their cellular functions (9, 10). One of them is the regulation of protein-tyrosine phosphatase (PTP) activities through Cys oxidation (13-15). Studies have described how ROS is involved in regulating cell signaling through oxidation-dependent, reversible inactivation of PTPs in response to various physiological stimuli (16). It has been proposed that PTPs are reversibly inactivated by the molecular modification of the active site from cysteine to sulfenic acid (7, 9, 17) or cyclized sulfenyl-amide derivative (13, 14). The ROS-mediated reversible inhibition of PTP activity is considered a critical step in facilitating phosphotyrosine signaling in response to a variety of physiological stimuli (7, 9). Recent studies have demonstrated that the same active site Cys residue is irreversibly oxidized to sulfinic (Cys-SO2H) or sulfonic (Cys-SO3H) acid states (17, 18), thus rendering PTPs permanently inactive under certain pathological conditions (e.g. when certain cancer cells produce high levels of ROS) (17).
Because of the low pKa (between 4.5 and 5.5) (19) at the active site, the Cys residue in PTPs may also be susceptible to S-nitrosylation mediated by RNS. Some researchers have suggested that PTP inactivation induced by RNS is one of the regulatory mechanisms of cell signaling independent of cGMP (20-22). Although S-nitrosylation has been shown to control the activity of important signaling modulators, such as caspases (10, 23) and plasma membrane-associated cation channels (24), the RNS-mediated redox switch for PTP activity has not been well understood. Recent studies using indirect measurements and enzymatic activity assays suggest that a number of cellular PTPs, including PTP1B (20, 25, 26), PTEN (21), SHP1 (22), and SHP2 (22), may be inactivated through S-nitrosylation in response to stress stimulation. The exact chemical process of S-nitrosylation and its reversibility in normal physiological or pathophysiological conditions are unknown.
In this study, we investigated the mechanism of RNS-mediated S-nitrosylation of PTPs, including the specificity and reversibility of this process in the regulation of enzyme activity. Instead of relying on indirect measures, as described in previous studies (20-22, 26), we used advanced mass spectrometry (MS) techniques along with x-ray crystallography to study whether the same ROS-sensitive Cys residue (Cys-215) at the active site might be the primary S-nitrosylation site in human PTP1B, a prototype in the PTP superfamily. We also investigated whether an increased level of cellular NO prior to exposure of cells with ROS could inhibit permanent oxidation and inactivation of endogenous PTP1B.
MATERIALS AND METHODS
Preparation of S-Nitrosylated PTP1B for Solution-based Trypsin Digestion—The C-terminally truncated, 37-kDa human PTP1B was purified as described previously (27). PTP1B was treated with H2O2 at 37 °C for 10 min or treated with S-nitroso-N-penicillamine (SNAP) (Calbiochem), S-nitrosoglutathione (GSNO) (Alexis Biochemicals), DETA-NONOate (Sigma), or N-acetylpencillamine (NAP) (Fluka) at 37 °C for 20 min. For analyzing Cys modifications by mass spectrometry, PTP1B was subsequently digested with trypsin (Promega) according to the manufacturer's instructions.
MS Analysis for Identifying S-Nitrosylated Sites of PTP1B—Direct nano-ESI-MS analysis was performed on a Micromass Q-TOF Ultima™ API mass spectrometer (Micromass; Waters) fitted with a nano-LC sprayer. The tryptic peptides were separated on a C18 capillary column connected online to a QSTAR-XL hybrid quadrupole time-of-flight mass spectrometer (Applied Biosystems/MDS Sciex). All tandem mass spectra were interpreted manually.
Quantitative MS for Determining Cys Residue Susceptibility to S-Nitrosylation—PTP1B was treated with 1 mm or 0.01 mm SNAP at 37 °C for 20 min, followed by incubation with 10 mm iodoacetic acid for 1 h. The PTP1B was then treated with ascorbate (5 mm; Sigma) for 1 h. Subsequently, PTP1B that had been treated with 1 mm SNAP or 0.01 mm SNAP was labeled with the light or heavy acid-cleavable isotope-code affinity tag (cICAT) reagent (0.1 unit; Applied Biosystems) for 1 h. Two pools of samples were mixed together at a 1:1 ratio and then digested with trypsin. The peptide samples were subjected to MALDI-MS and MS/MS analyses using an Applied Biosystems 4700 proteomics analyzer mass spectrometer (Applied Biosystems), as described previously (17, 25).
Crystallization and Structural Analysis of PTP1B S-Nitrosylation—Crystallization of the 37-kDa PTP1B was carried out as described previously (27). The crystal was incubated with SNAP (1 mm) at room temperature for 20 min and then mixed with the mother liquid containing 29% glycerol as an antifreezing reagent. Samples were then subjected to structural analysis using an x-ray generator. Data were recorded, and all data sets of crystal form P3121 were collected for processing using software packages DENZO and SCALEPACK (28). Data collection and refinement statistics are shown in Table S1.
Cell Culture, Transfection, Immunoprecipitation, and Immunoblotting—COS-7 cells (ATCC) were routinely maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. For transient transfection, cells were incubated with a mixture of plasmid DNA and Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. For immunoprecipitation of endogenous PTP1B, an aliquot of total lysate (750 μg) was incubated with rabbit anti-PTP1B antibody (AF1366; R&D Systems) conjugated to protein G-Sepharose (GE Healthcare) at 4 °C for 3 h. An aliquot of immunocomplex or total lysate (25 μg) was subjected to immunoblotting with an antibody against oxidized PTP active site (anti-oxi-PTP antibody, clone 335636; R&D Systems), PTP1B (FG6, Calbiochem), p-eNOS (phospho-Ser-1177; Cell Signaling), or eNOS (BD Transduction Laboratories).
Measurement of Intracellular NO Level—Intracellular NO was measured using 4-amino-5-methylamino-2′,7′-difluoroflurescencein diacetate (Invitrogen) by an enzyme-linked immunosorbent assay reader equipped with a fluorescence attachment (Infinite 200; Tecan) according to the manufacturer's recommendations.
RESULTS
Identification of S-Nitrosylated Cys Residues in PTP1B—PTP1B was used in this study to investigate the molecular basis for S-nitrosylation of Cys residues in PTPs. A direct nanospray ESI-MS (nESI-MS) mapping of the tryptic digests of purified PTP1B identified peptides that carry five of the six Cys sites (Fig. S1A). Following their sequential order from the N terminus, three tryptic peptides, T4 (and T3-4 with a tryptic miscleavage site), T15, and T28, afforded molecular ions that had become 29 mass units higher after treatment with SNAP, an S-nitrosothiol agent. This shift corresponded to a conversion from Cys-SH to Cys-SNO. One important way of helping identify an S-nitrosylated peptide is the characteristic loss of the NO moiety (30 mass units) to generate a cysteinyl radical under ESI-MS conditions (29). A careful examination of the transformed nESI-MS data revealed that the implicated signal paired with a 30-mass unit difference was indeed present among the spectral peaks for the SNAP-treated PTP1B (Fig. S1A). This characteristic loss of NO was used to develop an automated, data-dependent MS/MS acquisition of the S-nitrosylated peptides in a nano-LC-nESI-MS/MS run (25). Specifically, a difference of 30 mass units registered between any pair of signals during an LC-MS survey scan would trigger a targeted MS/MS analysis of the respective peptide ions (25). Using this method, we identified six pairs of relevant ion signals in the tryptic peptide pool of SNAP-treated PTP1B (Fig. S1B). The corresponding MS/MS data clearly assigned three of these signal pairs to T28 (amino acids 200-221), T4 (amino acids 25-33), and T15 (amino acids 80-103), which confirmed that Cys-32, Cys-92, and Cys-215 were indeed preferred S-nitrosylation sites (Fig. S1, D-F, and Table 1). No other S-nitrosylated Cys-containing tryptic peptides were identified. The remaining two signal pairs were mapped to peptides in PTP1B (T3 and T39; Fig. S1B) that do not contain a Cys residue and thus were not S-nitrosylated. It is possible that an NO-induced modification occurred elsewhere. For example, tryptophan residues are also susceptible to nitrosation (30).
TABLE 1.
Summary of results from mass difference-based MS analyses
|
NO donors (1 mm, 20 min) |
Chemical characteristics |
Peptides selected by mass difference- based program |
S-Nitrosylated Cys confirmed by MS/MS |
|
|---|---|---|---|---|
| Release of NO radicals | Transnitrosylation | |||
| SNAP | Yes | Yes | T4, T3-4, T15, and T28 | Cys-32, Cys-92, and Cys-215 |
| GSNO | Yes | Yes | T4, T3-4, T15, and T28 | Cys-32, Cys-92, and Cys-215 |
| DETA-NONOate | Yes | No | T4, T3-4, and T28 | Cys-32 and Cys-215 |
The same S-nitrosylation sites were similarly identified when GSNO, an endogenous reservoir of NO groups (31), was used as the NO donor instead of SNAP (Fig. S2A and Table 1). A third reagent, DETA-NONOate, which produces only NO radicals (32) and is not capable of transnitrosylation (33), also effected similar site-specific Cys modifications (Fig. S2B and Table 1). Thus, we concluded that, mechanistically, protein S-nitrosylation (correctly termed S-nitrosation from the chemical point of view (34) can occur through direct reaction with derivatives of NO radicals, such as the higher nitrogen oxide N2O3, which is likely to promote the incorporation of a NO moiety to a thiol (35), in addition to transnitrosylation mediated by GSNO or SNAP.
Active Site Cys-215 Most Susceptible to S-Nitrosylation—We next examined the order of S-nitrosylation among the Cys residues using quantitative MS analysis. The facile loss of the NO functional group under MS conditions necessitated the biotin switch method being used so that we could obtain more stable derivatives and incorporate differentiating stable isotopes (36). We modified the method by replacing the alkylating labels in the two steps outlined in Scheme 1. After treating PTP1B with various concentrations of SNAP, which led to different degrees of S-nitrosylation among the susceptible Cys residues, all remaining free thiols were carboxymethylated. The S-nitrosothiols were then selectively reduced back to free thiols by ascorbate and tagged with either the light or heavy cICAT reagent, for samples originally treated with a high or low dose of SNAP, respectively. The two differentially S-nitrosylated samples were mixed together, digested with trypsin, and directly monitored by MALDI-MS analysis.
SCHEME 1.

Schematic illustration of the quantitative MS workflow employed to assess the differential susceptibility of Cys residues to S-nitrosylation. The isotope-coded tag was commercially available cleavable light (12C9) and heavy (13C9) cICAT reagents, which generate labeled signal pairs with 9 Da apart. The S highlighted in red indicates the active site Cys residue of PTPs. See “Materials and Methods” for details.
As expected, only signals corresponding to tryptic peptides containing the implicated S-nitrosylated Cys sites occurred in pairs, because cICAT labels had been incorporated. As shown in Fig. 1A, the three signal pairs we detected were assigned to the labeled peptides, T4, T28, and T15, which carried light/heavy cICAT-labeled Cys-32, Cys-215, and Cys-92, respectively. The monoisotopic peak area ratios of the signal pairs were 1.49, 0.92, and 2.43 for Cys-32, Cys-215, and Cys-92, respectively (Fig. 1, B-D), which indicated that Cys-92 had less S-nitrosylation at a low dose of SNAP (0.01 mm) than at a high dose (1 mm), whereas Cys-215 had a similar degree of S-nitrosylation at both SNAP concentrations. We concluded that PTP S-nitrosylation occurs most readily at the catalytic Cys residue, but the two other Cys sites can likewise be modified at higher doses of SNAP.
FIGURE 1.

Application of the quantitative MS method and structural analysis for identification of the most susceptible Cys residue of PTP1B to S-nitrosylation. A-D, recombinant PTP1B treated with 1 mm SNAP or 0.01 mm SNAP was subjected to differential isotope labeling for quantitative MALDI-MS analysis as described in Scheme 1. The full scan MALDI-MS profile (A) revealed three pairs of cICAT-labeled tryptic peptides with a 9-Da difference, which could be assigned to T4, T28, and T15, as shown in expanded views (B-D), corresponding to the cICAT-labeled peptide pairs containing Cys-32, Cys-215, or Cys-92, respectively. The ratio of light/heavyc ICAT-labeled peak is shown below the spectrum. E, the crystal of PTP1B was soaked with 1 mm SNAP at room temperature for 20 min and subjected tox-ray crystallography. The 2Fo - 2Fc electron density map showed a mixture of reduced and S-nitrosylated states of Cys-215. Other Cys residues (Cys-32, -92, -121, -226, and -231) remained in the completely reduced form. Inset, the expended view of electron density map illustrates the presence of an S-nitrosothiol form of Cys-215.
To provide a structural basis for the site specificity of S-nitrosylation, the native crystal of PTP1B was soaked with 1 mm SNAP for 20 min and analyzed using x-ray crystallography. The electron density map indicated that the active site Cys-215 displayed a mixed conformation that could be assigned to a free thiolate anion form (Cys-S-) and an S-nitrosothiol form (Cys-SNO) (Fig. 1E). Closer inspection revealed that, upon the S-nitrosothiol formation of Cys-215, water-mediated hydrogen bonds and a subtle conformational change on the side chain of residues around the catalytic site collectively orchestrated a network capable of stabilizing the S-nitrosylated structure (Fig. S3). Interestingly, under this condition, Cys-32 and Cys-92, which are exposed on the surface of PTP1B, remained in the reduced form (Fig. 1E). This finding is consistent with quantitative MS analysis, which showed these two Cys residues to be less reactive than the active site Cys-215 (Fig. 1, A-D). Therefore, the NO-mediated modification appears to target primarily the active site Cys residue in PTP1B. In addition, the MS and structural data suggested that S-nitrosylation may regulate PTP1B through a reversible modification of the active site Cys in an S-nitrosothiol form that is unlike the irreversibly oxidized forms, such as the sulfinic acid (Cys-SO2H) or sulfonic acid (Cys-SO3H).
S-Nitrosylation Protects the Active Site against Irreversible Oxidation—Since the observation that NO prevents cells from ROS-induced damage was first reported (37), the cytoprotective effect of NO and RNS has been regarded as one of their most important biological functions (38, 39). We found the NO donor-induced modification of the active site Cys-215 to be fully reversible (Fig. 1). In contrast, it has been recently reported (15, 17, 25) and further confirmed by our current study (Fig. S4A) that when PTP1B is exposed to H2O2, the same Cys-215 site is irreversibly oxidized. Therefore, S-nitrosylation may reversibly trap the Cys residue at the active site and protect it against permanent ROS-induced oxidative damage. We used direct MS analysis to further examine the protective effect of S-nitrosylation. In nano-LC-nESI-MS, the tryptic peptide (T28) carrying the active site Cys-215 in SO2H and SO3H forms can be readily resolved from later-eluting T28 with Cys-215 in the free SH form or SNO form (Fig. 2A). We found that, once formed, S-nitrosothiol could not be further converted to sulfinic or sulfonic acids either by prolonged SNAP treatment (data not shown) or by exposure to subsequent oxidative stress (1 mm H2O2) (Fig. 2A). In a control experiment, the sample was similarly treated with 1 mm H2O2 following mock S-nitrosylation treatment with N-acetylpencillamine, a non-NO donor analogue of SNAP. More T28 was found to carry the sulfinic and sulfonic acid forms than the free thiol form (Fig. 2A). Additional examination of the MS results revealed that, although direct treatment of PTP1B with a low concentration (0.01 mm) of H2O2 led to significant formation of sulfinic acid derivative of Cys-215, when PTP1B was pre-exposed with SNAP and then followed by treatment with a high concentration (1 mm) of H2O2, only a minimal degree of irreversibly oxidized form of Cys-215 was induced (Fig. 2B).
FIGURE 2.

Protective effect of Cys S-nitrosylation on preventing PTP1B from further irreversible oxidation. A, recombinant PTP1B was pretreated with 1 mm N-acetylpencillamine or SNAP for 20 min, followed by 1 mm H2O2 for 10 min, and then digested by trypsin in solution. The tryptic peptides were subjected to LC-nESI-MS analysis. T28 carrying the Cys-215 in Cys-SH, Cys-SO2H, Cys-SO3H, and Cys-SNO forms were first identified by manually examining the nESI-MS profile at the expected retention time. For a semiquantitative assessment, the extracted ion chromatograms for the respective signals are plotted in A without normalization or correcting for nESI-MS response factor. The amount of the irreversibly oxidized SO2H/SO3H form (eluting at 17.7-18.5 min) elicited by 1 mm H2O2 is clearly reduced to basal level in SNAP-pretreated sample, compared with N-acetylpencillamine-pretreated sample. The corresponding nESI-MS profiles for this time point are shown in B, where the signals identified as the irreversibly oxidized T28 (m/z 736.4 and 741.7) are detectable at increasing intensity when PTP1B was treated with increasing concentration of H2O2 but not when it was first S-nitrosylated by SNAP. C, recombinant PTP1B, either directly exposed to H2O2, SNAP, or GSNO or pretreated with SNAP or GSNO followed by H2O2 treatment, was subjected to immunoblotting with an anti-oxidized PTP active site (anti-oxi-PTP) antibody (top) or an anti-PTP1B antibody (FG6) (bottom). The effect of SNAP or GSNO on inhibiting the level of H2O2-induced irreversible oxidation of PTP1B was observed in three independent experiments.
The protective effect of NO was further tested by immunoblotting. The monoclonal anti-oxi-PTP antibody we used recognizes a peptide derived from the conserved signature motif of PTPs with its active site Cys residue in the sulfonic acid form (40). Prior to the application of immunoblotting in subsequent in vitro and in vivo experiments, we examined the performance of this antibody. Our data showed that only the isoform of PTP1B with its Cys-215 in the sulfonic acid state but not in the reduced state, as confirmed by the MS-based analysis, was detected by the anti-oxi-PTP antibody in immunoblotting (Fig. S4B). Using this antibody, we observed that treatment of PTP1B with H2O2 resulted in irreversible oxidation of active site Cys-215 (Fig. 2C). Preincubation of PTP1B with either SNAP or GSNO, both of which have been shown to induce S-nitrosylation of Cys-215 (Table 1), significantly suppressed the level of irreversible Cys oxidation (Fig. 2C). Thus, data collectively indicated that prior exposure of PTP1B to NO/RNS effectively protected the active site Cys-215 against ROS-induced permanent oxidation.
Increased Cellular NO Levels Protect Endogenous PTP1B against Permanent Inactivation by Oxidation—We investigated whether NO-mediated post-translational modification would also protect endogenous PTPs against oxidative stress-induced damage in cells. COS-7 cells that do not express a detectable level of endogenous eNOS (41) were exposed to H2O2 with or without being preincubated with an equal molar mixture of SNAP and l-Cys (to form a plasma membrane-permeable S-nitrosocysteine) (42). The endogenous PTP1B was then immunoprecipitated from COS-7 lysates for immunoblotting analysis using anti-oxi-PTP antibody, whose performance has already been verified (Fig. S4). In the absence of NO, PTP1B was effectively oxidized and therefore permanently inactivated by treatment with H2O2 (Fig. 3A). Pretreatment of S-nitrosocysteine in the culture medium significantly suppressed the degree of irreversible oxidation of PTP1B induced by H2O2 (Fig. 3A).
FIGURE 3.

Increased cellular NO levels suppressed subsequent permanent inactivation of endogenous PTP1B induced by oxidative stress in COS-7 cells. A, COS-7 cells were either treated with 1 mm H2O2 for 10 min or prestimulated with equal molar mixture of SNAP and l-Cys (both 1 mm) for 20 min and followed by H2O2 treatment for an additional 10 min. Endogenous PTP1B was immunoprecipitated from total lysate using anti-PTP1B antibody (AF1366) and then subjected to immunoblotting with anti-oxi-PTP or anti-PTP1B antibody (FG6). B, COS-7 transfectants ectopically expressing human eNOS were stimulated with ATP for the indicated times. Aliquots of total lysate (25 μg) were subjected to immunoblotting with antibodies to phospho-eNOS (p-Ser1177) and eNOS. C, COS-7 transfectants were preloaded with 4-amino-5-methylamino-2′,7′-difluoroflurescencein diacetate and then subjected to ATP stimulation for 15 min prior to detection of intracellular NO levels. Results obtained are presented as relative fluorescence intensity and expressed as means ± S.E. (n = 3). *, p < 0.05 when compared with untreated eNOS transfectants. **, p < 0.005 when compared with ATP-treated mock transfectants. D, COS-7 transfectants were stimulated with ATP for 10 min, followed by exposure to H2O2 for an additional 10 min. Endogenous PTP1B was immunoprecipitated (IP) and then subjected to immunoblotting with anti-oxi-PTP antibody or anti-PTP1B antibody. Aliquots of total lysate (25 μg) were subjected to immunoblotting with antibodies to eNOS and PTP1B. Similar results were obtained from three independent experiments.
We further examined the effect of the intracellularly produced NO on the regulation of the redox status of endogenous PTP1B. COS-7 cells were transiently transfected with a plasmid that encoded the wild type of human eNOS and then stimulated with ATP, which has been shown to transactivate eNOS in COS-7 transfectants (41). As expected, the phosphorylation level of Ser-1177 in eNOS was rapidly increased in response to ATP stimulation (Fig. 3B), suggesting that ectopically expressed eNOS was activated. Indeed, when 4-amino-5-methylamino-2′,7′-difluoroflurescencein diacetate was used as an intracellular fluorescence indicator to detect NO, we found that the addition of ATP yielded a nitrosated, highly fluorescent DAF-2 triazole (Fig. 3C). With ATP stimulation, control cells and eNOS transfectants were exposed to H2O2, and the endogenous PTP1B was immunoprecipitated from COS-7 lysates for immunoblotting analysis. A brief period of intracellular NO production in eNOS transfectants led to less H2O2 stress-induced oxidation of PTP1B than when there was no NO production (Fig. 3D). Therefore, the active site Cys-215 of endogenous PTP1B can be S-nitrosylated through external or intracellular NO to form a stable modification, which prevents this Cys residue from subsequent oxidation from a ROS burst. To the best of our knowledge, these data demonstrate for the first time that NO and RNS may exert their cytoprotective effect through the S-nitrosylation of critical signaling conductors, in our case in the protection of PTPs against oxidative damage.
DISCUSSION
Like all second messengers, ROS and RNS are generated immediately in response to extracellular stimuli, diffused within a short distance, and then decomposed rapidly by the action of intracellular enzymes. Although it has been proposed that various signaling modulators are targeted by ROS and RNS (9, 10, 43), there has been limited direct in vivo evidence to delineate the underlying mechanism of free radicals-mediated signal transduction. In particular, the role of RNS in regulating signal transduction is emerging. One important area is the regulation of tyrosine phosphorylation signaling through redoxdependent control of PTP activity (16). Using a variety of methods, we have provided insight into the mechanistic details of PTP S-nitrosylation as well as of the protective role of S-nitrosylation against permanent oxidation and inactivation.
Quantitative MS analysis indicated that the active site Cys-215 of PTP1B is the primary site most susceptible to S-nitrosylation. Structural analysis confirmed the selective S-nitrosylation of Cys-215, whereas Cys-32 and Cys-92, both of which are surface-exposed residues, and other Cys residues situated in the interior space (Cys-121, -226, and -231) remained in their reduced form under the condition applied. Our observation thus suggests that solvent accessibility based on the position of Cys residues may not play a critical role in determining the selectivity of S-nitrosylation. Furthermore, after examining the sequence of amino acids adjacent to Cys-215, we did not find a conserved “nitrosylation motif” (44) or “acid/base motif” (45), both of which have been proposed as a potential means of determining the susceptibility of Cys S-nitrosylation in proteins (46). Our findings suggest that a high selectivity of S-nitrosylation, which could be achieved by either SNAP and GSNO-catalyzed transnitrosylation, or N2O3-mediated donation of an NO moiety, such as the reaction directed by DETA-NONOate, may preferentially affect the Cys residue located in a hydrophobic pocket with a unique structure-determined low pKa characteristic. In addition, ESI-MS and x-ray crystallography (Fig. 1) showed that, upon forming S-NO, the active site Cys-215 of PTP1B remained in this modified state. This mode of Cys modification is reported to be reversible in response to reductants (10, 47). Irreversibly oxidized derivatives, such as sulfinic acid or sulfonic acid, were not generated under all types of nitrosative stress used in our experiments. In contrast, even mild oxidative stress can rapidly convert a reduced Cys into irreversibly oxidized states in vitro (Fig. 2) and in cells (Fig. 3). Thus, RNS-mediated S-nitrosylation may regulate the enzymatic activity of PTPs in a reversible manner.
The most significant finding in our current study may be that the preformed S-NO would prevent the active site Cys from subsequent oxidation when subjected to oxidative stress. This finding is consistent with previous descriptions of the cytoprotective effect of NO against irreversible oxidative damage, which have suggested that it is one of the most important biological functions of NO and RNS (37-39). The best example of the NO protective effect was its attenuation of I/R injury to the heart (12, 48). It has been shown that I/R injury occurs concomitantly with ROS bursts, leading to widespread protein oxidation and tissue apoptosis or necrosis (49). Notably, the bioavailability of NO is correlated with prevention of I/R-induced heart injury and myocardial protection (50). Our study has provided new molecular details for the protective role of NO, which presumably acts against ROS-mediated damage on cellular proteins. In this study, we showed that the preexistence of Cys S-nitrosothiol prevented ROS-induced irreversible oxidation of PTP1B, which may be a mechanism for NO-mediated cytoprotective effect under pathological conditions, such as I/R. It has been shown that when the deoxygenation was applied in human red blood cells (RBCs), the anion exchange band 3 protein, which was recently identified as a potential substrate of PTP1B (51, 52), was tyrosine-phosphorylated (53). Interestingly, under the deoxygenation, such as ischemic condition, the bioavailable level of NO is increased in RBCs (54), suggesting that PTP1B may be S-nitrosylated and therefore inactivated, concomitantly with an elevated tyrosine phosphorylation level of band 3. Based on our current finding, we propose that ischemia-induced S-nitrosylation may protect PTP1B against irreversible oxidation in RBCs when reperfusion occurs. The reversible modification of Cys-215 in the S-NO form thus allows the rapid rebound of PTP1B activity for down-regulating tyrosine phosphorylation of band 3 after the oxidative stress in RBCs is gone. Further investigation is required to examine the protective role of NO in RBCs under the condition of I/R.
Recent studies suggest that the nitrite anion
( ), which is present in large
quantities in blood and tissues (0.15-1.0 μm in plasma and
>10 μm in tissues), is a vascular storage pool of NO
(55). Under pathological
hypoxic conditions nitrite is converted to NO through enzymatic or
nonenzymatic actions (55,
56). We propose that, in
response to increased levels of NO through nitrite reduction, endogenous PTPs
and other Cys-mediated enzymes may be shielded by S-nitrosylation
against oxidative damage associated with subsequent reperfusion-induced
formation of ROS. Such enzymes include thioredoxin
(57), peroxiredoxin
(58), and caspases
(10), which are important
regulators of cellular redox status as well as cell survival. Advanced
MS-based techniques, such as those used in this study, can help characterize
the protective role of nitrite in facilitating S-nitrosylation of
these critical signaling regulators under ischemic conditions. Future
investigations may provide additional insight into the intriguing function of
nitrite-dependent formation of protein S-nitrosothiols in the
maintenance of signaling homeostasis after reperfusion and reintroduction of
molecular oxygen to ischemic tissues.
), which is present in large
quantities in blood and tissues (0.15-1.0 μm in plasma and
>10 μm in tissues), is a vascular storage pool of NO
(55). Under pathological
hypoxic conditions nitrite is converted to NO through enzymatic or
nonenzymatic actions (55,
56). We propose that, in
response to increased levels of NO through nitrite reduction, endogenous PTPs
and other Cys-mediated enzymes may be shielded by S-nitrosylation
against oxidative damage associated with subsequent reperfusion-induced
formation of ROS. Such enzymes include thioredoxin
(57), peroxiredoxin
(58), and caspases
(10), which are important
regulators of cellular redox status as well as cell survival. Advanced
MS-based techniques, such as those used in this study, can help characterize
the protective role of nitrite in facilitating S-nitrosylation of
these critical signaling regulators under ischemic conditions. Future
investigations may provide additional insight into the intriguing function of
nitrite-dependent formation of protein S-nitrosothiols in the
maintenance of signaling homeostasis after reperfusion and reintroduction of
molecular oxygen to ischemic tissues.
Supplementary Material
The atomic coordinates and structure factors (code 3EU0) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by Taiwan National Science Council Grants NSC-95-3112-B-002-028, NSC-96-3112-B-002-018 (to T. C. M.), NSC-94-3112-B-009-Y, and NSC-95-3112-B-001-014 (to the National Proteomic Core Facility). This work was also supported by Academia Sinica (to T. C. M., K. H. K., D. L. W., and A. H. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S4 and Table S1.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; NO, nitric oxide; RNS, reactive nitrogen species; NOS, nitric-oxide synthase; I/R, ischemia/reperfusion; PTP, protein-tyrosine phosphatase; PTP1B, protein-tyrosine phosphatase 1B; MS, mass spectrometry; ESI, electrospray ionization; LC, liquid chromatography; MALDI, matrix-assisted laser desorption ionization; SNAP, S-nitroso-N-penicillamine; nESI-MS, nanospray ESI-MS; GSNO, S-nitrosoglutathione; RBC, red blood cell; DETA-NONOate, dipropylenetriamine NONOate; cICAT, cleavable isotope-code affinity tag; NAP, N-acetylpencillamine.
References
- 1.Lambeth, J. D. (2004) Nat. Rev. 4181 -189 [DOI] [PubMed] [Google Scholar]
- 2.Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005) Nat. Rev. Mol. Cell Biol. 6150 -166 [DOI] [PubMed] [Google Scholar]
- 3.Mahadev, K., Motoshima, H., Wu, X., Ruddy, J. M., Arnold, R. S., Cheng, G., Lambeth, J. D., and Goldstein, B. J. (2004) Mol. Cell. Biol. 241844 -1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park, H. S., Jung, H. Y., Park, E. Y., Kim, J., Lee, W. J., and Bae, Y. S. (2004) J. Immunol. 1733589 -3593 [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi, N., Mita, S., Yoshida, K., Honda, T., Kobayashi, T., Hara, K., Nakano, S., Tsubokou, Y., and Matsuoka, H. (2003) Hypertension 421004 -1013 [DOI] [PubMed] [Google Scholar]
- 6.Xu, J. W., Ikeda, K., and Yamori, Y. (2004) Hypertension 44217 -222 [DOI] [PubMed] [Google Scholar]
- 7.Lee, S. R., Kwon, K. S., Kim, S. R., and Rhee, S. G. (1998) J. Biol. Chem. 27315366 -15372 [DOI] [PubMed] [Google Scholar]
- 8.Monteiro, H. P., Gruia-Gray, J., Peranovich, T. M., de Oliveira, L. C., and Stern, A. (2000) Free Radic. Biol. Med. 28174 -182 [DOI] [PubMed] [Google Scholar]
- 9.Meng, T. C., Fukada, T., and Tonks, N. K. (2002) Mol. Cell 9387 -399 [DOI] [PubMed] [Google Scholar]
- 10.Mannick, J. B., Hausladen, A., Liu, L., Hess, D. T., Zeng, M., Miao, Q. X., Kane, L. S., Gow, A. J., and Stamler, J. S. (1999) Science 284651 -654 [DOI] [PubMed] [Google Scholar]
- 11.Bell, R. M., Maddock, H. L., and Yellon, D. M. (2003) Cardiovasc. Res. 57405 -415 [DOI] [PubMed] [Google Scholar]
- 12.Jones, S. P., and Bolli, R. (2006) J. Mol. Cell. Cardiol. 4016 -23 [DOI] [PubMed] [Google Scholar]
- 13.Salmeen, A., Andersen, J. N., Myers, M. P., Meng, T. C., Hinks, J. A., Tonks, N. K., and Barford, D. (2003) Nature 423769 -773 [DOI] [PubMed] [Google Scholar]
- 14.van Montfort, R. L., Congreve, M., Tisi, D., Carr, R., and Jhoti, H. (2003) Nature 423773 -777 [DOI] [PubMed] [Google Scholar]
- 15.Denu, J. M., and Tanner, K. G. (1998) Biochemistry 375633 -5642 [DOI] [PubMed] [Google Scholar]
- 16.Tonks, N. K. (2005) Cell 121667 -670 [DOI] [PubMed] [Google Scholar]
- 17.Lou, Y. W., Chen, Y. Y., Hsu, S. F., Chen, R. K., Lee, C. L., Khoo, K. H., Tonks, N. K., and Meng, T. C. (2008) FEBS J. 27569 -88 [DOI] [PubMed] [Google Scholar]
- 18.Wang, Q., Dube, D., Friesen, R. W., LeRiche, T. G., Bateman, K. P., Trimble, L., Sanghara, J., Pollex, R., Ramachandran, C., Gresser, M. J., and Huang, Z. (2004) Biochemistry 434294 -4303 [DOI] [PubMed] [Google Scholar]
- 19.Zhang, Z. Y., and Dixon, J. E. (1993) Biochemistry 329340 -9345 [DOI] [PubMed] [Google Scholar]
- 20.Li, S., and Whorton, A. R. (2003) Arch. Biochem. Biophys. 410269 -279 [DOI] [PubMed] [Google Scholar]
- 21.Yu, C. X., Li, S., and Whorton, A. R. (2005) Mol. Pharmacol. 68847 -854 [DOI] [PubMed] [Google Scholar]
- 22.Barrett, D. M., Black, S. M., Todor, H., Schmidt-Ullrich, R. K., Dawson, K. S., and Mikkelsen, R. B. (2005) J. Biol. Chem. 28014453 -14461 [DOI] [PubMed] [Google Scholar]
- 23.Mannick, J. B., Schonhoff, C., Papeta, N., Ghafourifar, P., Szibor, M., Fang, K., and Gaston, B. (2001) J. Cell Biol. 1541111 -1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin, Y. F., Raab-Graham, K., Jan, Y. N., and Jan, L. Y. (2004) Proc. Natl. Acad. Sci. U. S. A. 1017799 -7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen, Y. Y., Huang, Y. F., Khoo, K. H., and Meng, T. C. (2007) Methods (San Diego) 42 243-249 [DOI] [PubMed] [Google Scholar]
- 26.Forrester, M. T., Foster, M. W., and Stamler, J. S. (2007) J. Biol. Chem. 28213977 -13983 [DOI] [PubMed] [Google Scholar]
- 27.Barford, D., Keller, J. C., Flint, A. J., and Tonks, N. K. (1994) J. Mol. Biol. 239726 -730 [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276307 -326 [DOI] [PubMed] [Google Scholar]
- 29.Kaneko, R., and Wada, Y. (2003) J. Mass Spectrom. 38526 -530 [DOI] [PubMed] [Google Scholar]
- 30.Zhang, Y. Y., Xu, A. M., Nomen, M., Walsh, M., Keaney, J. F., Jr., and Loscalzo, J. (1996) J. Biol. Chem. 27114271 -14279 [PubMed] [Google Scholar]
- 31.Singh, S. P., Wishnok, J. S., Keshive, M., Deen, W. M., and Tannenbaum, S. R. (1996) Proc. Natl. Acad. Sci. U. S. A. 9314428 -14433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keefer, L. K., Nims, R. W., Davies, K. M., and Wink, D. A. (1996) Methods Enzymol. 268281 -293 [DOI] [PubMed] [Google Scholar]
- 33.Dahm, C. C., Moore, K., and Murphy, M. P. (2006) J. Biol. Chem. 28110056 -10065 [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Ruiz, A., and Lamas, S. (2004) Cardiovasc. Res. 6243 -52 [DOI] [PubMed] [Google Scholar]
- 35.Hogg, N. (2002) Annu. Rev. Pharmacol. Toxicol. 42585 -600 [DOI] [PubMed] [Google Scholar]
- 36.Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001) Nat. Cell Biol. 3 193-197 [DOI] [PubMed] [Google Scholar]
- 37.Wink, D. A., Hanbauer, I., Krishna, M. C., DeGraff, W., Gamson, J., and Mitchell, J. B. (1993) Proc. Natl. Acad. Sci. U. S. A. 909813 -9817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duranski, M. R., Greer, J. J., Dejam, A., Jaganmohan, S., Hogg, N., Langston, W., Patel, R. P., Yet, S. F., Wang, X., Kevil, C. G., Gladwin, M. T., and Lefer, D. J. (2005) J. Clin. Invest. 1151232 -1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb, A., Bond, R., McLean, P., Uppal, R., Benjamin, N., and Ahluwalia, A. (2004) Proc. Natl. Acad. Sci. U. S. A. 10113683 -13688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Persson, C., Kappert, K., Engstrom, U., Ostman, A., and Sjoblom, T. (2005) Methods (San Diego) 35 37-43 [DOI] [PubMed] [Google Scholar]
- 41.Iwakiri, Y., Satoh, A., Chatterjee, S., Toomre, D. K., Chalouni, C. M., Fulton, D., Groszmann, R. J., Shah, V. H., and Sessa, W. C. (2006) Proc. Natl. Acad. Sci. U. S. A. 10319777 -19782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, S., and Whorton, A. R. (2005) J. Biol. Chem. 28020102 -20110 [DOI] [PubMed] [Google Scholar]
- 43.Woo, H. A., Chae, H. Z., Hwang, S. C., Yang, K. S., Kang, S. W., Kim, K., and Rhee, S. G. (2003) Science (New York) 300653 -656 [DOI] [PubMed] [Google Scholar]
- 44.Greco, T. M., Hodara, R., Parastatidis, I., Heijnen, H. F., Dennehy, M. K., Liebler, D. C., and Ischiropoulos, H. (2006) Proc. Natl. Acad. Sci. U. S. A. 1037420 -7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stamler, J. S., Toone, E. J., Lipton, S. A., and Sucher, N. J. (1997) Neuron 18 691-696 [DOI] [PubMed] [Google Scholar]
- 46.Derakhshan, B., Hao, G., and Gross, S. S. (2007) Cardiovasc. Res. 75210 -219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hao, G., Xie, L., and Gross, S. S. (2004) J. Biol. Chem. 27936192 -36200 [DOI] [PubMed] [Google Scholar]
- 48.Cohen, M. V., Yang, X. M., and Downey, J. M. (2006) Cardiovasc. Res. 70231 -239 [DOI] [PubMed] [Google Scholar]
- 49.McCord, J. M., Roy, R. S., and Schaffer, S. W. (1985) Adv. Myocardiol. 5183 -189 [PubMed] [Google Scholar]
- 50.Kanno, S., Lee, P. C., Zhang, Y., Ho, C., Griffith, B. P., Shears, L. L., II, and Billiar, T. R. (2000) Circulation 1012742 -2748 [DOI] [PubMed] [Google Scholar]
- 51.Zipser, Y., Piade, A., Barbul, A., Korenstein, R., and Kosower, N. S. (2002) Biochem. J. 368137 -144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciana, A., Minetti, G., and Balduini, C. (2004) Bioelectrochemistry (Amst.) 62 169-173 [DOI] [PubMed] [Google Scholar]
- 53.Barbul, A., Zipser, Y., Nachles, A., and Korenstein, R. (1999) FEBS Lett. 455 87-91 [DOI] [PubMed] [Google Scholar]
- 54.Datta, B., Tufnell-Barrett, T., Bleasdale, R. A., Jones, C. J., Beeton, I., Paul, V., Frenneaux, M., and James, P. (2004) Circulation 1091339 -1342 [DOI] [PubMed] [Google Scholar]
- 55.Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., Yang, B. K., Waclawiw, M. A., Zalos, G., Xu, X., Huang, K. T., Shields, H., Kim-Shapiro, D. B., Schechter, A. N., Cannon, R. O., III, and Gladwin, M. T. (2003) Nat. Med. 91498 -1505 [DOI] [PubMed] [Google Scholar]
- 56.Zweier, J. L., Samouilov, A., and Kuppusamy, P. (1999) Biochim. Biophys. Acta 1411250 -262 [DOI] [PubMed] [Google Scholar]
- 57.Haendeler, J., Hoffmann, J., Tischler, V., Berk, B. C., Zeiher, A. M., and Dimmeler, S. (2002) Nat. Cell Biol. 4743 -749 [DOI] [PubMed] [Google Scholar]
- 58.Diet, A., Abbas, K., Bouton, C., Guillon, B., Tomasello, F., Fourquet, S., Toledano, M. B., and Drapier, J. C. (2007) J. Biol. Chem. 28236199 -36205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.