

Abstract
In this study, we show that methylselenol, a selenometabolite implicated in cancer prevention, did not directly inactivate protein kinase C (PKC). Nonetheless, its oxidation product, methylseleninic acid (MSA), inactivated PKC at low micromolar concentrations through a redox modification of vicinal cysteine sulfhydryls in the catalytic domain of PKC. This modification of PKC that occurred in both isolated form and in intact cells was reversed by a reductase system involving thioredoxin reductase, a selenoprotein. PKC isoenzymes exhibited variable sensitivity to MSA with Ca2+-dependent PKC isoenzymes (α, β, and γ) being the most susceptible, followed by isoenzymes δ and ε. Other enzymes tested were inactivated only with severalfold higher concentrations of MSA than those required for PKC inactivation. This specificity for PKC was further enhanced when MSA was generated within close proximity to PKC through a reaction of methylselenol with PKC-bound lipid peroxides in the membrane. The MSA-methylselenol redox cycle resulted in the catalytic oxidation of sulfhydryls even with nanomolar concentrations of selenium. MSA inhibited cell growth and induced apoptosis in DU145 prostate cancer cells at a concentration that was higher than that needed to inhibit purified PKCα but in a range comparable with that required for the inhibition of PKCε. This MSA-induced growth inhibition and apoptosis decreased with a conditional overexpression of PKCε and increased with its knock-out by small interfering RNA. Conceivably, when MSA is generated within the vicinity of PKC, it specifically inactivates PKC isoenzymes, particularly the promitogenic and prosurvival ε isoenzyme, and this inactivation causes growth inhibition and apoptosis.
Epidemiological, experimental, and preliminary clinical data suggest that dietary supplementation with selenium can prevent cancer (1–4). Supplemental selenium may reduce the incidence and mortality of prostate, lung, and colorectal cancers, but it has not been shown to be effective against breast and skin cancers in humans (4). Therefore, it is imperative to know why selenium prevents cancers in certain tissues but not in others. Currently, large scale trials are underway to evaluate the efficacy of selenium in prostate cancer prevention (5–7). Nevertheless, the chemopreventive mechanism of selenium and the molecular targets upon which selenium acts are largely unknown.
The cancer-preventive efficacy of selenium depends on its chemical form (8). Because naturally occurring organic forms of selenium such as selenomethionine (–2 Se) and and Se-methylselenocysteine (–2 Se) are safer than inorganic selenite (+4 Se), they are the forms of selenium considered to be ideal for dietary supplementation. These parent selenocompounds are ultimately metabolized to methylselenol,2 a postulated intermediate that carries out the chemopreventive actions of selenium (1, 9). When selenium is given to experimental animals at a nutritionally adequate dose (0.1 ppm), its physiological functions are believed to be mediated primarily by selenoproteins such as thioredoxin reductase (TR)3 and glutathione peroxidase (10–12). Nonetheless, when selenium is given at high doses (>5 ppm), its toxicity is thought to be mediated by selenometabolites (10). When selenium is given at cancer-preventive intermediate doses (1–3 ppm), it is unclear whether selenium carries out its preventive actions through selenometabolites, selenoproteins, or a combination of both.
Selenometabolites are well known to inhibit cell growth and induce apoptosis at micromolar concentrations (13–15). However, at cancer-preventive doses, the majority (9) of selenium in circulation is present as selenoproteins or is bound to proteins, and only a limited amount (<5%) is present as selenometabolites (10). Cancer cell growth inhibition and induction of apoptosis are considered to be important for the chemopreventive actions of selenium, but it is challenging to demonstrate how these actions can occur in vivo when such low concentrations of selenium are available to tissues. In this context, it is noteworthy to mention that the concentration of selenium required for in vitro inhibition of tumor promotion is lower than that required for growth inhibition or apoptosis of established malignant cells (16–18). Furthermore, it is crucial to determine which mechanism(s) selenium, at cancer-preventive concentrations, utilizes to selectively kill precancer or cancer cells without causing global toxicity to the host. Moreover, it is also important to identify which specific molecular targets selenium uses to inhibit tumor promotion and progression.
Protein kinase C (PKC) may be a potential target involved in both tumor promotion and progression (19, 20). Therefore, it is possible that chemopreventive agents such as selenium act on the same target as tumor promoters do but induce an opposing response (21). In this scenario, the cancer-preventive agent efficiently counteracts tumor promoter-induced effects. Moreover, several of the tumor promotion or cancer prevention mechanisms may involve redox-sensitive targets (19).
PKC is a unique target for oxidants (19). Whether PKC is activated or inactivated by oxidation depends on the type of oxidant, the site of oxidation, and the extent of modification (22). The regulatory domain of PKC contains a C1 module with a cysteine-rich region that coordinates four atoms of zinc (23). Selective modification of this autoinhibitory domain is induced by treatment with low concentrations of oxidants, resulting in a cofactor-independent activation of PKC (19, 22). Alternatively, some oxidants, alkylating agents, and certain cancer-preventive agents modify cysteine residues present within the catalytic domain, leading to the inactivation of PKC (24–26). Because PKC is a family of more than 11 phospholipid-dependent serine/threonine protein kinases that vary in structure (27–29), these isoenzymes may have difference(s) in susceptibility to redox-modifying agents.
Generally, PKC isoenzymes are divided into three categories based on the cofactors that are required for optimal catalytic activity (27–29). Conventional PKCs (α, β, and γ) are calcium-dependent and are stimulated by a second messenger diacylglycerol. Novel PKCs (δ, ε, η, and θ) are also activated by diacylglycerol but in a calcium-independent fashion. Atypical PKCs (ζ and λ/ι) require neither calcium nor diacylglycerol for optimal activity. Although most cells express more than one type of PKC, differences among the isoenzymes with respect to activation conditions and subcellular locations suggest that individual PKC isoenzymes mediate distinct cellular processes (27–29). For example, in most cell types, PKCε is a pro-mitogenic and pro-survival kinase, whereas PKCδ is an antiproliferative and proapoptotic kinase (28, 30, 31). Opposing actions of PKCα and PKCδ were also reported (32).
In this study, we show that methylseleninic acid4 (MSA), which is locally generated by the reaction of methylselenol with lipid peroxides, specifically inactivates PKC isoenzymes. MSA does this by inducing a redox modification of the cysteine-rich region in the catalytic domain, and this modification can be reversed by the TR system. Additionally, we demonstrate that the overexpression of PKCε, a prosurvival isoenzyme that is moderately sensitive to MSA, results in a resistance to selenium-induced apoptosis in prostate cancer cells.
EXPERIMENTAL PROCEDURES
Materials—5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB), protein kinase A (500 Sigma units/mg protein), N-acetyl-l-cysteine, aprotinin, leupeptin, dithiothreitol (DTT), pepstatin A, ebselen, phosphatidylcholine, phosphatidylserine, 1,2-diolein, (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (MTT), and hydrogen peroxide were from Sigma. Benzeneselenol and cumene hydroperoxide were from Aldrich. Auranofin and [γ-32P]ATP (specific activity 20 Ci/mmol) were from MP Biochemicals. [20-3H]Phorbol 12,13-dibutyrate (specific activity 20 Ci/mmol) was from PerkinElmer Life Sciences.
The MSA used in these experiments was a kind gift from Dr. Howard Ganther, University of Wisconsin, Madison. The homogeneous phosphorylase kinase (PK) purified from rabbit skeletal muscle was a generous gift from Dr. Balawant Khatra, California State University, Long Beach. Protein phosphatase 2A (PP2A) was partially purified from rabbit skeletal muscle as described previously (33). TR and thioredoxin were purified to homogeneity from calf thymus (34, 35). TR has a specific activity of 22 units/mg protein, where 1 unit oxidizes 1 μmol of NADPH/min.
PKC isoenzymes α, β, γ, δ, and ε were partially purified from rat brains with specific activities of ∼540, 460, 490, 220, and 360 units/mg protein, respectively (22, 36, 37). PKCζ, with a specific activity of 520 units/mg protein, was partially purified from rabbit kidneys (38). In some cases, a mixture of Ca2+-dependent isoenzymes α, β, and γ (specific activity of 890 units/mg protein) was used, as Ca2+-dependent hydrophobic chromatography can purify these isoenzymes to homogeneity in high amounts (22). The catalytic and regulatory domains of PKC isoenzymes (α, β, and γ) were separated by trypsin treatment and then chromatographically isolated (39). Using previously published procedures, we raised rabbit polyclonal antibodies against PKC isoenzymes by injecting hemocyanin coupled with sequence-specific peptides from the variable regions in PKC isoenzymes (40). These peptides and a PKC-specific substrate peptide that corresponds to a neurogranin amino acid sequence (residues 25–43) were synthesized at the core facility of Norris Comprehensive Cancer Center.
Cell Culture and Treatments—Androgen-independent DU145 prostate carcinoma cells were obtained from the American Type Culture Collection and maintained in Eagle's minimum essential medium supplemented with 10% fetal calf serum, 50 units/ml penicillin, and 0.05 mg/ml streptomycin. MSA was diluted in Hanks' balanced salt solution and added to the culture medium. In some experiments, when benzeneselenol was used as a surrogate to methylselenol, its sulfur analog benzenethiol was used as a control. When agents were dissolved in organic solvents, appropriate solvent controls were used.
PKC Assay—The assays of PKC as well as of other protein kinases were carried out in 96-well plates with fitted filtration disks made of Durapore membrane (41). Briefly, PKC reaction samples containing 20 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 0.33 mm CaCl2, 0.1 mm [γ-32P]ATP (3 million cpm), histone H1 (0.1 mg/ml), 40 μm leupeptin, and 25 μl of PKC sample in a total volume of 125 μl were incubated for 5 min at 30 °C. After arresting the reaction with trichloroacetic acid and ultrafiltration, we determined the radioactivity associated with the protein retained on the membrane. In certain cases, histone H1 was replaced with 5 μm neurogranin substrate polypeptide (residues 25–43), and the above reactions were instead carried out in regular 96-well plates without filtration disks. The reaction was arrested with 10 μl of 1 m phosphoric acid; the samples were applied to Whatman P81 paper (2 × 2 cm), and the papers were washed four times with 75 mm phosphoric acid. Radioactivity retained in the washed paper was counted. The basal activity observed in the absence of cofactors was subtracted from the activity observed in the presence of cofactors. The difference was expressed as PKC activity in units, where 1 unit of enzyme transfers 1 nmol of phosphate to either histone H1 or neurogranin polypeptide per min at 30 °C.
Because assaying PKC by using histone H1 as a substrate is relatively simple, it was utilized for determining the activity of conventional PKC isoenzymes (α, β, and γ). Histone H1, however, is not a good substrate for measuring the activity of novel and atypical isoenzymes. On the other hand, the neurogranin substrate peptide is appropriate for determining the phosphotransferase activity of all PKC isoenzymes (42). Therefore, we used this substrate for determining the combined activity of all PKC isoenzymes in cell extracts.
Phorbol Ester Binding—The phorbol ester binding of isolated PKC was carried out by a multiwell filtration approach using [3H]phorbol 12,13-dibutyrate (PDBu) as a ligand (41). Purified PKC was incubated with selenocompounds in multiwell plates for 5 min at 30 °C. Phosphatidylserine (2.5 μg) was added, followed by a PDBu-binding mixture. Samples were incubated for an additional 10 min at 30 °C, and the [3H]PDBu bound to PKC was determined as described previously (41).
Quantitation of Cysteine Sulfhydryls and Disulfides in MSA-modified PKC—A mixture of PKC isoenzymes (α, β, and γ) that can be purified in large amounts was used for this experiment. Sulfhydryl residues in PKC were quantitated by employing previously published methods using DTNB (43), except a 96-well plate was utilized to enhance the sensitivity of the assay in measuring low amounts of protein sulfhydryls. The disulfides were determined after sulfitolysis using sodium sulfite. The sulfhydryls liberated (one per each disulfide) by this reaction were quantitated with NTSB by utilizing a modified method (44).
The incubations were carried out in two sets, each one in a 96-well plate. Highly purified PKC (0.5 nmol) free from thiol agents was incubated with MSA in 20 mm Tris-HCl, pH 7.4, 1 mm EDTA for 25 min at 30 °C, in a total volume of 180 μl. Then 20 μl of 4 mm DTNB solution was added to one set of samples. To another set of samples, 20 μl of 4 mm NTSB, 100 mm sulfite solution was added. After incubating the samples in the dark for 10 min, the absorbance of the thionitrobenzoic acid formed in the reactions was determined at 405 nm by using a Thermomax microplate reader (Molecular Devices, Sunnyvale, CA).
Isolation of PKC from Cells Treated with MSA—DU145 cells were grown in 100-mm Petri dishes in minimum essential medium supplemented with 10% fetal calf serum. Cells were serum-starved at confluency by incubation with serum-free medium (0.1% serum) for 24 h. Cells were treated with MSA and then homogenized in buffer (20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.5 mm phenylmethylsulfonyl fluoride, 150 nm pepstatin A, 1% Igepal CA-630). Unless otherwise indicated, mercapto-compounds were omitted from all of the buffers used for cell homogenization and chromatographic isolation of PKC. The cell extracts were subjected to DEAE-cellulose chromatography as described previously (22).
Western Immunoblotting for PKC Isoenzymes—Cells were serum-starved overnight and treated with MSA. Cell extracts were subjected to SDS-PAGE. Electrophoretically separated proteins were transferred to a polyvinylidene fluoride membrane. The membranes were blocked with 5% dry milk and subsequently incubated with the indicated PKC isoenzyme-specific primary antibodies followed by goat anti-rabbit secondary antibodies conjugated with horseradish peroxidase. The immuno-reactive bands were visualized by the enhanced chemiluminescence Western blot detection kit (Pierce). These bands were analyzed by densitometric scanning using the Omega 12 IC Molecular Imaging System and UltraQuant software.
Stable Transfection of PKCε—The metallothionein expression vector (45) used in these experiments was a kind gift from Dr. Wayne Anderson, NCI, National Institutes of Health, Bethesda. The cells were transfected with either a metallothionein-driven PKCε expression vector (to overexpress PKCε) or an empty vector (as a control) using Lipofectamine 2000 according to the manufacturer's recommended procedure. One day after transfection, DU145 cells were plated at a lower density and grown in a selection medium containing 450 μg/ml G418. After 4 weeks in the selection medium, single colonies were picked, expanded, and screened for the presence of PKCε by using Western blot analysis. Zinc acetate (75 μm) was used for the optimal expression of PKCε in these transfectants.
Transient Transfection of DU145 Cells with PKCε siRNA—DU145 cells (1 × 104/ml) were plated in a 6-well plate. After 24 h, 50 nm PKCε siRNA oligonucleotides (three predesigned Silencer oligonucleotides from Ambion) were transfected into DU145 cells with Lipofectamine 2000 according to the manufacturer's instructions. As a negative control, we used scrambled siRNA that did not exhibit homology to any encoding region but had similar GC content. The efficiency of transfection and knock-out of PKCε was determined by Western immunoblotting. In the conditions used, immunoblot of other PKC isoenzymes such as α, β, γ, and δ were not affected by the transfection with PKCε siRNA oligonucleotides, demonstrating the specificity involved in the knock-out procedure. The experiments were continued with the PKCε siRNA oligonucleotide that produced the greatest knock-out (a decrease of ∼80% from the control).
Cell Growth Assay—Cells grown in 96-well plates were treated with various concentrations of MSA for 48 h. The cells were then fixed in 10% trichloroacetic acid. After washing them with water, the cells were stained with a 0.4% solution of sulforhodamine B for 30 min. The excess dye was removed by washing the cells with 1% acetic acid, and the dye bound to the cells was later dissolved in 10 mm Tris base. The absorbance was read at 550 nm as a growth index (46).
Cell Viability Assay—We assessed the cytotoxicity of MSA using the MTT reduction assay. Initially, DU145 cells were grown in 96-well plates and treated with MSA for 24 h. Then an MTT solution (5 mg/ml) in Hanks' balanced salt solution was added, and the plates were incubated for 4 h. The formazan formed was dissolved in dimethyl sulfoxide, and the absorbance was read at 550 nm (47).
Apoptosis Assay—To assess morphological changes in the chromatin structure of DU145 cells undergoing apoptosis, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were grown on culture slides, treated with MSA for 24 h, and then fixed with 4% paraformaldehyde in phosphate-buffered saline for 20 min at room temperature. After rinsing them with phosphate-buffered saline, cells were stained with DAPI (10 μg/ml) for 5 min. The morphology of the nuclei was observed using a fluorescence microscope (Nikon Eclipse TE300) at an excitation wavelength of 345 nm. Apoptotic nuclei were identified by chromatin condensation and fragmentation. The incidence of apoptosis in each preparation was analyzed by counting 500 cells and determining the percentage of apoptotic nuclei (48).
Caspase-3 Assay—Enzyme activity was determined by using tetrapeptide substrate (N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide) with an assay kit obtained from Biomol (Plymouth Meeting, PA). Cells were seeded in 60-mm Petri dishes and allowed to grow for 24 h. The cells were then changed to serum-free medium and treated with MSA for the indicated time. Then cells were homogenized, and an aliquot of the cell extract (50 μg protein) was subjected to caspase-3 activity determination according to the manufacturer's instructions.
RESULTS
Inactivation of PKC Catalytic Activity and Phorbol Ester Binding by MSA—Initially, the thiol agents present in a PKC (α isoenzyme) preparation isolated from rat brains were removed by PD-10 gel filtration, and then the protein fraction was incubated with increasing concentrations of MSA. As shown in Fig. 1A, MSA inhibited the PKC activity with an IC50 of 0.25 μm. Thiol agents 1 mm DTT or 5 mm 2-mercaptoethanol reversed this inactivation. When this inactivation occurred in the absence of thiol agents, the enzyme activity did not recover, even after removing the MSA by subjecting the treated sample to a centrifuge gel filtration using Bio-Spin 6 (Bio-Rad) columns. Subsequent exposure to 1 mm DTT reversed this modification, suggesting the involvement of a sulfhydryl redox mechanism. Although DTT almost completely regenerated PKC activity when a low concentration (<1 μm) of MSA was used for modification, it only partially regenerated the kinase activity when a high concentration (>2 μm) of MSA was used.
FIGURE 1.

Reversal of redox modification of PKC resulting in loss of kinase activity and phorbol ester binding. A, effect of MSA on the phosphotransferase activity of PKC. Desalted PKC (α isoenzyme) was incubated in two sets in multiwell filtration plates, with various concentrations of MSA for 5 min at 30 °C in the absence of thiol agents. Then, to one set of samples, 1 mm DTT was added. To both sets of samples, the lipids along with a prewarmed reaction mixture were added, and the samples were incubated for 5 min at 30 °C to determine histone phosphotransferase activity. B, effect of MSA on the phorbol ester binding of PKC. PKC was preincubated with MSA as described in A. Phosphatidylserine, followed by [3H]PDBu binding mixture, was added, and then [3H]PDBu bound to PKC was determined as described previously (41). Each value is the mean ± S.E. of triplicate estimations.
MSA inhibited phorbol ester binding at a higher concentration than that required for inhibiting PKC activity (Fig. 1B). This inactivation was also either fully blocked or partially reversed by 1 mm DTT. MSA also inhibited the cofactor-independent activity of a proteolytically derived PKC catalytic fragment. However, the phorbol ester binding of the proteolytically derived regulatory domain was virtually unaffected by the low (<1 μm) concentration of MSA. Nonetheless, a high (>2 μm) concentration of MSA did decrease phorbol ester binding of the regulatory domain.
Regeneration of Oxidatively Inactivated PKC by the TR System—First we treated purified PKCα with a low concentration of MSA, desalted it using a Bio-Spin 6 column, and then incubated it with a TR system comprised of TR, MSA-oxidized thioredoxin,5 and NADPH. We then determined the PKC activity and phorbol ester binding. When a low (<2 μm) concentration of MSA was used for PKC modification, the TR system regenerated PKC activity to its full extent, but when a high (>5 μm) concentration of MSA was used for PKC modification, the TR system only partially regenerated the modified PKC (Fig. 2). Similarly, the redox modification of PKCε was also reversed by the TR system (data not shown).
FIGURE 2.

Reversal of MSA-induced PKC modification by the TR system. Approximately 20 units of purified PKC (α isoenzyme) in 0.2 ml were incubated witha 2 or 10 μm concentration of MSA for 5 min at 30 °C. Then MSA was removed by using the Bio-Spin 6 column. The MSA-treated PKC samples were incubated with the TR system comprised of TR (0.1 μm), MSA-modified thioredoxin (∼0.1 μm), and 0.2 mm NADPH. PKC activity and phorbol ester binding in the incubated samples were then determined. The data are means ± S.E. of triplicate estimations. The values obtained by incubation with the TR system were compared with the respective values obtained by incubation without the TR system (**, p < 0.01, evaluated by paired t test).
MSA-induced Oxidation of Cysteine Residues in PKC—Ca2+-dependent PKC isoenzymes α, β, and γ, containing 16–20 (average) cysteine residues in their sequences, exhibited equal susceptibility to MSA. Therefore, we used a mixture of all three isoenzymes, which can be purified in high amounts, to quantitate the sulfhydryls. Approximately 18 sulfhydryls were titrated with DTNB after the SDS denaturation of the untreated protein (control). As shown in Table 1, when PKC was first incubated with a low amount of MSA (1 nmol/nmol of protein), nearly four cysteine sulfhydryls were not titrated with DTNB, even after the denaturation of PKC with SDS. Two disulfides were measured in modified PKC, correlating with the loss of four DTNB-titratable sulfhydryls. During this period, PKC activity decreased but recovered substantially after treatment with DTT. Phorbol ester binding did not decrease during this time. When MSA was used at high amounts (5 mol/mol of protein) to modify PKC, seven to eight sulfhydryls in the modified PKC were not detectable with DTNB, and approximately three to four disulfides were formed. Concomitantly, this modification resulted in a loss of both PKC activity and phorbol ester binding, which was only partially recovered by treatment with DTT.
TABLE 1.
MSA-induced oxidation of cysteine residues and formation of disulfides in PKC
This experiment was carried out using purified PKC containing a mixture of α, β, and γ isoenzymes. The purity of the preparation was established by SDS-gel electrophoresis that showed a single band corresponding to a molecular weight of approximately 80,000 (all three isoenzymes have similar molecular weights). PKC (0.5 nmol) was incubated with the indicated amounts of MSA in two sets in the wells of 96-well microtiter plates. DTNB was added to one set of samples. To another set of samples, NTSB/sulfite solution was added. After incubating the samples for 10 min, the absorbance of thionitrobenzoic acid formed was determined at 405 nm. The sulfhydryls and disulfides were quantitated using N-acetylcysteine and cystine, respectively, as the standards. The values represent means of triplicate estimations.
| Cysteine sulfhydryls titrated | Disulfides formed | PKC activity | PDBu binding | ||||
|---|---|---|---|---|---|---|---|
| nmol/nmol of enzyme | units × 100 | pmol | |||||
| Modification | DTNB | Sulfite/NTSB | a | No DTT | DTT | No DTT | DTT |
| Control | 12.27 ± 0.26 | 11.97 ± 0.23 | 0 | 1.82 | 2.12 | 0.72 | 0.76 |
| MSA (0.5 nmol) | 8.50 ± 0.15 | 10.27 ± 0.26 | 1.8 | 0.32 | 1.64 | 0.66 | 0.71 |
| MSA (2.5 nmol) | 5.17 ± 0.23 | 8.83 ± 0.15 | 3.7 | 0.06 | 0.28 | 0.11 | 0.23 |
In the 3rd column (Sulfite/NTSB), NTSB quantitated the readily accessible free thiols present in PKC as well as those thiols liberated by sulfitolysis of the disulfide bonds formed in PKC by MSA-induced oxidation. In the 2nd column (DTNB), only the accessible free thiols were determined. Subtraction of this column from the 3rd column gives the number of thiols liberated from disulfides (one thiol per each disulfide). This difference is presented as disulfides formed in the 4th column.
Susceptibility of Various PKC Isoenzymes to MSA—As shown in Fig. 3, MSA inactivated Ca2+-dependent isoenzymes at lower concentrations (IC50 ∼ 0.25 μm) and PKCδ and PKC ε at higher concentrations (IC50 ∼ 1.5 μm). Clearly, PKCζ was the least susceptible isoenzyme tested (IC50 3.5 μm).
FIGURE 3.

Variation in sensitivity of PKC isoenzymes to MSA. Purified PKC isoenzyme preparations were diluted in a buffer containing 1 mg/ml bovine serum albumin and incubated with 0.1–10 μm concentrations of MSA for 5 min at 30 °C. PKC activity was then determined using a neurogranin substrate polypeptide. The values are expressed as mean ± S.E. of triplicate estimations.
PKC as a Specific Target for MSA—As shown in Fig. 4, although PKC (a mixture of α, β, and γ) was inactivated by MSA at low concentrations (IC50 = <1 μm), the other enzymes tested were inactivated only at high concentrations of this agent. In the absence of thiol agents, protein kinase A (PKA) was inactivated only at higher concentrations of MSA with an IC50 of 50 μm, which was ∼100-fold higher than that required to inactivate PKC. Furthermore, this inactivation required the presence of cAMP. PP2A required a much higher concentration of MSA (IC50 = 90 μm) for inactivation. PK was not inactivated by MSA even at these high concentrations. Both PKA and PP2A, which both have only one critical sulfhydryl in the catalytic region, require a higher concentration of MSA for inactivation than PKC requires.
FIGURE 4.

Susceptibility of other enzymes to MSA-induced inactivation. In a manner similar to the treatment of PKC, PKA and PK were preincubated with MSA, and the activities were determined. Instead of Ca2+/lipids and histone H1, 5 μm cAMP and unfractionated histone were used to determine PKA activity; 0.1 mm CaCl2 and phosphorylase b were used in the PK assay. PP2A activity was determined by the multiwell filtration method using PKC-phosphorylated histone H1 as the substrate (86). Determinations of enzyme activities were carried out in triplicate and are expressed as percentages of the control values.
PKC Inactivation by Locally Generated MSA Resulting from the Reaction of Methylselenol with Lipid Peroxides—It is possible that, in addition to PKC, MSA could react with other proteins that have cysteine-rich regions. Nevertheless, if MSA is formed in close proximity to PKC, it may selectively react with the kinase. To test this hypothesis, PKCα was incubated with linoleic acid hydroperoxide and then treated with increasing concentrations of methylselenol. Methylselenol induces the reduction of fatty acid hydroperoxide and in turn oxidizes into MSA. Because linoleic acid binds to PKC, the locally generated MSA could specifically inactivate PKC. Given the fact that methylselenol is volatile and readily oxidizable in air, we used MSA (nonvolatile form), which readily generates methylselenol upon reaction with DTT. First, the rapid conversion of MSA to methylselenol was demonstrated using 1-chloro-2,4-dinitrobenzene, which can efficiently trap selenol (49). Then there was an inactivation of PKC at a concentration as low as 50 nm methylselenol (Fig. 5A). In the absence of hydroperoxide, such low concentrations of exogenous MSA were not effective in inactivating PKC. The combination of hydroperoxide and nascently produced methylselenol did not affect the activity of PKA, again suggesting the specificity involved in PKC inactivation by methylselenol.
FIGURE 5.

Low concentration of methylselenol inactivates PKC bound to lipid peroxides. A, inactivation of PKC by methylselenol in the presence of linoleic acid hydroperoxide. Linoleic acid hydroperoxide was prepared from linoleic acid as described previously (87). Various concentrations of nascent methylselenol, prepared by mixing MSA and DTT (1:2 mol/mol) were incubated with PKCα in the presence of either linoleic acid (LA) or linoleic acid hydroperoxide (25 μm). This reaction was carried out in a total volume of 0.5 ml in microcentrifuge tubes with fitted rubber stoppers. The samples were incubated for 15 min at room temperature while shaking on an end-to-end rotator. Then the residual PKC activity was determined using histone H1 as the substrate. In some experiments, methylselenol was replaced with benzeneselenol. B, methylselenol inactivates PKC bound to peroxidized phospholipid vesicles. Phospholipid vesicles (phosphatidylcholine and phosphatidylserine (4:1)) were prepared for binding PKC. These vesicles were subjected to peroxidation as described previously (88). PKCα was incubated with 50 nm methylselenol in the presence of either unoxidized phospholipids or oxidized phospholipids. CaCl2 (1 mm) was included to facilitate the binding of PKC to lipid vesicles. Incubations and PKC determinations were carried out as in A. The values are expressed as mean ± S.E. of triplicate estimations. The values obtained from methylselenol in the presence of peroxidized lipids were compared with the values of the respective controls obtained from peroxidized lipids alone (*, p < 0.01, evaluated by paired t test).
Because volatile methylselenol was prepared by mixing MSA and DTT, the unreacted components may have affected the results. For this reason, benzeneselenol, a nonvolatile surrogate for methylselenol, was directly used for the inactivation of PKC in the presence of the fatty acid hydroperoxide. As shown in Fig. 5A, benzeneselenol was somewhat more effective than methylselenol in peroxide-dependent inactivation of PKC. However, the sulfhydryl analog of benzeneselenol, benzenethiol, did not induce this PKC inactivation (data not shown).
PKCα is activated by liposomes or peroxidized liposomes made of phosphatidylcholine and phosphatidylserine (4:1) (50). We determined whether selenol can inactivate PKC that is bound to these peroxidized phospholipid vesicles. PKC was incubated with peroxidized phospholipid vesicles and then treated with 50 nm methylselenol. PKC was inactivated by methylselenol only when peroxidized lipid vesicles were used, but not when control unoxidized lipid vesicles were used (Fig. 5B). PKCε was also inactivated by methylselenol (100 nm) in the presence of peroxidized phospholipid vesicles.
Methylselenol Catalyzes the Oxidation of Sulfhydryls in the Presence of Peroxides—The reaction of methylselenol with peroxides generates MSA, which, after oxidizing sulfhydryls, is converted back into methylselenol. This methylselenol-MSA redox cycle results in the catalytic oxidation of sulfhydryls. As the goal of this study is to elucidate how a low concentration of methylselenol can oxidize the sulfhydryls present in PKC, understanding the mechanism for the catalytic oxidation of sulfhydryls is vital. Low molecular weight compounds such as ebselen and selenocysteine are known to catalyze the oxidation of sulfhydryls such as glutathione in conjunction with hydrolyzing peroxides (glutathione peroxidase-mimetic action) (51, 52). Therefore, we tested whether methylselenol could also catalyze this reaction. As shown in Fig. 6, when DTT was used as a reductant, methylselenol catalyzed the oxidation of DTT in the presence of hydrogen peroxide and cumene hydroperoxide. In all of these cases, methylselenol had nearly 90% of the catalytic activity of ebselen, and 2–3-fold higher than that of selenocystine (selenocysteine).
FIGURE 6.

Peroxidatic thiol oxidation catalyzed by methylselenol and its comparison with ebselen and selenocystine. A mixture containing buffer (50 mm phosphate, pH 7.4, 0.5 mm EDTA, 1 mm sodium azide, 0.2% Triton X-100) 1 mm peroxide, and 0.5 mm DTT in a total volume of 1 ml was placed in microcentrifuge tubes with fitted rubber stoppers. Selenocompounds (1 μm) were then injected into the tubes, and the tubes were incubated at 30 °C for 5 min. The amount of the thiol left that remained oxidized was quantitated using DTNB. The data are means ± S.E. of triplicate estimations.
Inactivation of PKC Isoenzymes in MSA-treated DU145 Prostate Cancer Cells—We determined whether the redox modification of PKC isoenzymes that occurred in the test tube could also occur in intact prostate cancer cells. For this experiment, the androgen-independent DU145 prostate carcinoma cell line was preferred as this has previously been shown to be highly sensitive to selenium (53). Serum-starved DU145 cells were treated with a 5 μm concentration of MSA for 120 min. A substantial decrease in activity of PKC (all isoenzymes) occurred within 5–15 min, which then significantly recovered over a 90-min time period to a level still below that of the control (Fig. 7). There was no increase in membrane-associated PKC activity during this time. The modification of PKC, occurring in response to MSA, was completely reversed by directly homogenizing the cells in a buffer containing 1 mm DTT or 5 mm 2-mercaptoethanol. MSA also inactivated PKC in the androgen-dependent LNCaP cell line (data not shown).
FIGURE 7.

Inactivation of PKC in MSA-treated DU145 cells and its enhancement by TR inhibition. Serum-starved DU145 cells at confluency were initially treated with 100 nm auranofin or vehicle (ethanol) for 1 h, and then treated with MSA (5 μm) for the indicated times. Cells were homogenized in buffer containing 1% Igepal CA-630 to extract total PKC from both the cytosol and the membrane. The cell extracts were subjected to DEAE-cellulose chromatography, and PKC isoenzymes were eluted as described under “Experimental Procedures.” PKC activity was determined using neurogranin as a substrate. The values are the mean ± S.E. of triplicate estimations.
Possible Role of the TR System in Reversing MSA-induced PKC Modification in Intact DU145 Cells—The extent of MSA-induced decrease in PKC activity in intact DU145 cells was not as great as that seen with purified enzyme in a test tube. The low degree of modification could be interpreted in two ways. One possibility is that these agents induced only a limited modification of PKC in the cells treated with MSA. The alternative possibility is that the PKC modification was extensive, but it was readily reversed by an endogenous reductive mechanism. We tested the later possibility by using auranofin, a potent inhibitor of TR (IC50 = 20 nm). Auranofin inhibits other selenoenzymes such as glutathione peroxidase, and other sulfhydryl enzymes such as glutathione reductase and PKC only at micromolar concentrations (54, 55). Because the TR system can reverse the MSA-induced redox modification of PKC in purified form (Fig. 2), we determined whether the inhibition of TR by auranofin could result in enhanced inactivation of PKC in MSA-treated DU145 cells. As shown in Fig. 7, the inhibition of TR in intact cells by a pretreatment with auranofin enhanced the rate of MSA-induced inactivation of PKC, suggesting that the TR system is likely to be involved in the reversal of the intracellular PKC redox modification.
Reversal of PKC Redox Modification by Endogenous Reductase and NADPH in the Cell Homogenates—Because purified PKC after oxidation is regenerated by the TR system, and NADPH provides necessary reducing equivalents for TR or other protein-disulfide reductases, the addition of NADPH to cell homogenates may reverse the MSA-mediated modification of PKC. Considering that TR also converts nonvolatile MSA to volatile methylselenol (56), TR may prevent PKC modification by eliminating MSA. To avoid this possibility, the cell homogenates were initially treated with MSA, and the excess MSA was then removed by gel filtration. The desalted cell extracts were incubated with DTT (positive control) or NADPH. These agents regenerated ∼60–70% of the PKC activity (Fig. 8). Nevertheless, NADPH failed to regenerate PKC activity when the desalted cell extracts were preincubated with the TR-specific inhibitor, auranofin. This further supports the argument that the endogenous reductase that regenerated the activity of oxidatively modified PKC is indeed TR.
FIGURE 8.

Reversal of PKC redox modification by an endogenous reductase and NADPH in the cell homogenates. Cells grown to confluency were homogenized in 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.5 mm phenylmethylsulfonyl fluoride (2 ml of buffer per 100-mm Petri dish) and centrifuged at 10,000 × g. The soluble fraction was incubated with MSA (5 μm) for 10 min at 30 °C. Then MSA was removed by gel filtration. Aliquots of this desalted cell extract were treated with DTT (1 mm) or NADPH (1 mm) for 10 min at 30 °C, and PKC isoenzymes (combined) were isolated by using a small DEAE-cellulose column. PKC activity was determined using neurogranin polypeptide as a substrate. The values are expressed as mean ± S.E. of triplicate estimations.
Effect of TR Inhibition on MSA-induced Cell Growth Inhibition and Apoptosis—We have observed that treating DU145 cells with PKC-specific inhibitors calphostin C (200 nm) and bisindolylmaleimide (2 μm) results in a decrease in cell growth and induction of apoptosis (data not shown). Similarly, it is possible that MSA-induced PKC inactivation may play a causal role in MSA-induced cell growth inhibition and cell death. Considering that TR can reverse the MSA-induced PKC inactivation, auranofin, by inhibiting TR, can indirectly enhance PKC inactivation and thus potentiate the action of MSA. To test this hypothesis, we have determined the effects of MSA in the presence and absence of auranofin on DU145 cell proliferation using a sulforhodamine B growth assay. The proliferation of DU145 cells was reduced significantly upon a 48-h exposure to MSA alone in a concentration-dependent manner with an IC50 of ∼1 μm (Fig. 9A). In the presence of auranofin, the concentration of MSA needed was 2–3-fold lower for a 50% inhibition of cell growth. This suggests that TR may oppose the selenocompound inhibitory effect on cell growth, presumably at the site of PKC.
FIGURE 9.

TR inhibition enhances MSA-induced cell growth inhibition and apoptosis. A, MSA-induced cell growth inhibition in the presence and absence of TR-specific inhibitor auranofin. Du145 cells were seeded in 96-well culture plates, and after 24 h cells were treated with the indicated concentrations of MSA along with auranofin (50 nm) or vehicle control (ethanol) for 48 h. Then cells were stained with sulforhodamine B, and the absorbance was measured at 550 nm. B, cytotoxicity of MSA in the presence and absence of auranofin. Du145 cells grown in multiwell plates were treated with MSA with and without auranofin for 24 h and then incubated with MTT for 4 h. The formazan developed was dissolved, and the absorbance was read at 550 nm. C, apoptosis induced by MSA in the presence and absence of auranofin. DU145 cells, grown on culture slides, were treated with MSA with and without auranofin, and then stained with DAPI. The incidence of apoptosis in each sample was analyzed by counting 500 cells and determining the percentage of apoptotic nuclei. D, effect of auranofin on the MSA-induced caspase-3 activation. Cells were grown in 60-mm Petri dishes and treated with MSA in the presence and absence of auranofin, and the caspase-3 activity (expressed as absorbance at 405 nm) was determined as described under “Experimental Procedures.” The values are the mean ± S.E. of three independent experiments. *, values for auranofin control are statistically different from control without auranofin (paired t test, p < 0.05). **, values obtained with MSA (2 μm) in the presence of auranofin were compared with the values obtained with MSA alone (paired t test, p < 0.01).
Cytotoxicity and cell viability of DU145 cells were determined by an MTT reduction assay (47). As shown in Fig. 9B, the viability of DU145 cells was reduced substantially upon a 24-h exposure to MSA in a concentration-dependent manner with an IC50 of about 5 μm. Auranofin potentiated the action of MSA. When auranofin is present, MSA was needed at a 5-fold lower concentration than in its absence. Cell viability results are in clear agreement with the results obtained from the cell proliferation assay.
Next, we assessed whether the loss of cell viability induced by MSA correlates with the induction of apoptosis, a common property of many cancer chemopreventive agents (57). We identified MSA-induced apoptosis through the observation of chromatin condensation and fragmentation by DAPI staining. As shown in Fig. 9C, auranofin potentiated the action of MSA in inducing apoptosis.
Proteolytic activation of caspase-3 is a marker for apoptosis in some cell types. Using a caspase-3 enzymatic assay, we observed activation of this enzyme in DU145 cells treated with a 2 μm concentration of MSA (Fig. 9D). Auranofin further enhanced the caspase-3 activity induced by a low concentration (2 μm) of MSA. However, at high concentrations (5 and 10 μm), the MSA-increased caspase-3 activity was not proportional to increase in apoptosis, and auranofin did not further enhance the caspase-3 activity induced by a high concentration of MSA. This suggests that although caspase-3 plays an important role in apoptosis, its activation was not proportional to the extent of apoptosis.
Although auranofin enhanced MSA-induced cell growth inhibition, cytotoxicity, and apoptosis, it alone also inhibited cell growth and induced apoptosis in a concentration-dependent manner. Auranofin, at the 50 nm concentration used in the above experiments, decreased cell growth and viability by ∼40% and induced small but significant apoptosis (8% of cells) in DU145 cells. This suggests that TR may play a role in cell growth and cell survival, and inhibition of this enzyme may result in suppression of cell growth and induction of cell death.
Expression of PKCε and Its Relation to MSA-induced Cell Growth Inhibition and Apoptosis—Initially, a detailed characterization of PKCα was preferred because of its high sensitivity to MSA and the availability of a sufficient amount of purified enzyme. However, the concentration of MSA required to inhibit DU145 cell growth was certainly higher than that to inhibit purified PKCα, and it was in a range comparable with that required for the inhibition of purified PKCε. Because PKCε is a promitogenic and prosurvival enzyme, we focused on it as a target for MSA-induced inactivation and its relation to MSA-induced cell growth inhibition and cell death.
To further evaluate the role of PKCε in MSA-induced growth inhibition and cell death, two different approaches were used to determine whether altering the expression of PKCε could affect the sensitivity of DU145 cells to MSA. In the first approach, we overexpressed PKCε (nearly 3-fold) by stable transfection, and in the second approach, we suppressed its levels (by ∼80%) by using siRNA (Fig. 10A). In DU145 cells overexpressing PKCε, the concentration of MSA required for 50% growth inhibition was approximately three to four times higher than that required in control cells transfected with an empty vector. Conversely, with a knockdown of PKCε by siRNA, the concentration of MSA required for 50% growth inhibition was nearly 2-fold lower than that of the control DU145 cells transfected with scrambled siRNA.
FIGURE 10.

PKCε levels in DU145 cells stably transfected with PKCε vector or transiently transfected with siRNA and the correlation with MSA-induced cell growth inhibition and apoptosis. A, Western immunoblotting of PKCε in DU145 cells stably transfected with either the control vector or a metallothionein-driven expression vector and transiently transfected with either control scrambled siRNA or PKCε siRNA oligonucleotides as described under “Experimental Procedures.” Where indicated, zinc acetate was added to the growth medium. Immunoblot analysis was used to determine possible changes in PKCε or β-actin as the loading control was carried out. B, DU145 cell sensitivity to MSA-induced cell growth inhibition under the conditions presented in A. Cell growth was analyzed by the sulforhodamine B assay. C, MSA-induced apoptosis and its relation to the expression of PKCε. Apoptosis was measured by identifying chromatin condensation and fragmentation after DAPI staining. The results obtained with the control cells transfected with empty vector and the control cells transiently transfected with scrambled siRNA were not significantly different from that of wild-type control cells. These results were not shown in B and C. The data are means ± S.E. of three independent experiments.
Cell viability was affected differently in cells expressing varying amounts of PKCε. For example, at a 2 μm concentration of MSA, the viability of wild-type cells decreased by 25%. Conversely, in DU145 cells overexpressing PKCε, the viability decreased by only 10%. Furthermore, in DU145 cells underexpressing PKCε, the viability decreased by as much as 50% (data not shown).
At a concentration of 5 μm, MSA induced apoptosis in nearly 20% of wild-type DU145 cells (Fig. 10C). In cell transfectants overexpressing PKCε, MSA induced apoptosis in only 8% of cells at this concentration. In cells where PKCε was knocked out by siRNA transfection, there was an increase in apoptosis to nearly 37%. Caspase-3 was induced by MSA treatment to the same extent in wild-type cells, cells expressing high amounts of PKCε, and cells expressing low amounts of PKCε (data not shown).
DISCUSSION
In proposing a chemopreventive mechanism for selenium, the following five important criteria must be considered. First, the molecular target for selenium must be specific; second, the mechanism must explain how the very low levels of selenometabolites available to tissues can induce inactivation of the specific target; third, the proposed target must account for how its inactivation can induce growth inhibition and apoptosis; fourth, the mechanism must explain how a resistance to supplementary selenium can develop in advanced cancers; finally, the mechanism should clarify whether any relationship exists between the functions of the two forms of selenium, selenometabolites and selenoproteins. Our present studies reveal that PKC isoenzymes are likely the targets for selenium, and the proposed redox mechanism appears to meet all five of the above criteria. The experimental evidence to support this notion is discussed below.
First, to evaluate PKC isoenzymes as appropriate molecular targets for selenium, we determined the molecular basis for the specificity involved in MSA-induced inactivation. A complete reduction of one molecule of MSA to methylselenol requires four thiols. Upon Reaction 1 with thiols or other reducing agents, MSA is expected to undergo a four-electron reduction to methylselenol by way of an intermediate selenylsulfide (58).
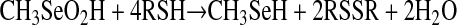 |
REACTION 1 |
Thus, two pairs of juxtaposed sulfhydryl residues may be well suited for reduction of MSA to methylselenol. By possessing a cluster of cysteine residues in their catalytic domains, PKC isoenzymes are well suited for this reduction, and thus are likely specific targets for MSA-induced oxidation of protein thiols.
The presence of two types of cysteine-rich regions in PKC creates the different reactivities of its regulatory and catalytic domains to MSA. Even though the regulatory domain contains 12 cysteine residues (thiolates), they coordinate the binding of four redox-inert zinc ions. This makes the site less susceptible to the addition reaction (formation of selenosulfide) that is expected to occur with MSA. Conversely, although the catalytic domain has only six or seven cysteine residues, they do not coordinate the binding of zinc or other metal ions, making them more susceptible to the initial addition reaction followed by oxidation. Recent studies have elegantly shown that the function of estrogen receptor-β, which contains the zinc-thiolate structure, was not affected in MSA-treated prostate carcinoma cells (59). This further supports the notion that zinc thiolates react less efficiently with selenium.
The catalytic domain of Ca2+-dependent PKC isoenzymes, besides containing the cysteine residue located close to the peptide recognition site as in the other protein kinases such as PKA and PK, has three additional conserved cysteine residues. These cysteines are unique to PKC, with the homologous cysteine residues being absent in other protein kinases (60). A requirement for the cluster of four cysteine residues to readily reduce MSA creates specificity for the interaction of MSA with PKC, particularly the Ca2+-dependent isoenzymes. MSA may oxidize four conserved cysteine residues present in the catalytic domain. Although this interpretation is based on the equal susceptibility of all three Ca2+-dependent isoenzymes to MSA, certainly further studies are needed to identify specific amino acid residues that are oxidized by MSA.
The conserved cysteine residues, however, are not present in all of the Ca2+-independent PKC isoenzymes. In the catalytic domain of isoenzymes δ, ε, and ζ, one conserved cysteine (corresponding to residue 569 in α isoenzyme numbering) is absent, and in the ζ isoenzyme an additional cysteine (corresponding to residue 383 in α isoenzyme numbering) is also absent. The variation in cysteine residues among these isoenzymes correlates well with their observed relative susceptibility to MSA. Ca2+-dependent isoenzymes (α, β, and γ) are the most susceptible among the isoenzymes tested, followed by δ and ε isoenzymes; PKCζ is the least sensitive isoenzyme.
Second, to validate the cancer-preventive mechanism of selenium, we determined how the low levels of selenium available to tissues could induce the inactivation of PKC isoenzymes, the specific targets for selenium. Volatile methylselenol may function more efficiently if it is retained in tumor cells. The formation of nonvolatile MSA after the reaction of methylselenol with peroxide enhances the retention of the active form of selenium in cells. Although cysteine-rich regions make PKC a possible target for MSA, other proteins with cysteine-rich regions are also probable targets for MSA. Therefore, if the MSA that reacts with PKC is formed elsewhere in the cell, its reactivity may not be highly specific for PKC. However, even if a limited amount of MSA is formed in the vicinity of PKC, it may react selectively with PKC. Unsaturated fatty acids, phospholipids, and their peroxides bind to and activate PKC (61, 62). The lipid peroxides do this without reacting directly with the sulfhydryls in PKC. When low amounts of methylselenol or other lipophilic selenols are present in the membrane, they react with these PKC-bound lipid hydroperoxides to produce seleninic acid locally, which can then selectively react with the nearby PKC (Fig. 11). Because the cysteine residues present within the catalytic domain of PKC do not coordinate zinc binding, they readily react with MSA, leading to a loss of kinase activity. However, if MSA is formed at high concentrations, it also reacts with the zinc thiolates present in the regulatory domain of PKC.
FIGURE 11.

Specific inactivation of PKC by locally generated redox-amplified MSA. Methylselenol reduces PKC-bound lipid peroxides and in turn gets converted to MSA. This locally generated MSA specifically oxidizes PKC cysteine sulfhydryls. This oxidative reaction regenerates methylselenol, which repeats its peroxide-reducing action. The redox cycle amplifies the action of MSA and increases its sensitivity to oxidize PKC sulfhydryls. Although the PKC regulatory domain has 12 cysteine thiol residues, these residues coordinate four zinc ions, which renders them to not be readily oxidized by MSA. Conversely, the catalytic domain cysteine sulfhydryls do not coordinate metal ions, and thus, they are readily oxidized by MSA. Sulfhydryl-oxidized PKC loses its activity. The TR system reduces disulfides formed in PKC catalytic domain and regenerates PKC activity.
During these oxidation processes, methylselenol regenerates and repeats the reduction of lipid peroxide. This catalytic redox cycle enables low concentrations of methylselenol to act as high concentrations of selenol.
Previously, we have reported the chemical modification of PKC with selenite (39). Although the MSA-induced modification of PKC resembles to a certain extent the selenite-induced modification of PKC, there is a significant difference between these reactions. Selenite and MSA react directly with PKC but lack high selectivity because they also react with other cysteine-rich proteins. Contrarily, when MSA is generated by the reaction of methylselenol with PKC-bound lipid peroxides, it reacts with PKC very specifically. Selenite is converted into selenide, which generates reactive oxygen species (63, 64) that nonspecifically affect a variety of proteins. Other studies have shown a direct inactivation of AP-1, p53, and c-Jun N-terminal kinase by selenite (65–67). Furthermore, redox-sensitive NF-κB-regulated gene expression was decreased in MSA-treated prostate carcinoma cells (68). It is interesting to know if these important targets are also directly modified by MSA or methylselenol and lipid peroxides.
Third, as an additional criterion for the mechanism of selenium action, we assessed how PKC inactivation leads to growth inhibition and cell death. Deregulation in cell growth and escape from cell death are hallmarks of tumor promotion. Because PKC is a receptor for a variety of tumor promoters, its inactivation by selenium may prevent the tumor promotion process (19). In addition, selenium may suppress cell growth and induce apoptosis of established tumor cells (13–15). Because greater amounts of peroxides are present in cells undergoing tumor promotion and in cancer cells (69, 70), selenium may be retained well and inactivate PKC to a greater degree in these cells than in normal cells. This may explain previous observations that tumor cells are more susceptible to selenium toxicity than normal cells are (15, 71).
Various PKC isoenzymes respond differently to stimuli that influence cell growth and death. The role of PKCα in cell growth and cell death varies depending upon cell type and the stimuli (30). Its activation is involved in phorbol ester-induced apoptosis in androgen-dependent LNCaP prostate cancer cells (30). Its inhibition in androgen-independent PC3 cells appears to be associated with growth suppression (72). Specific inhibition of PKCα in androgen-independent DU145 cells resulted in increased sensitivity to chemotherapeutic drugs (73), suggesting that, in addition to PKCα inactivation, induction of an additional apoptotic signal is required. This study suggests that PKCα inhibition alone is not sufficient for MSA-induced apoptosis and that inactivation of PKCε is required as well.
PKCε has oncogenic potential and is a promitogenic and prosurvival enzyme (28, 74). Previous studies have shown that growth suppression of lung cancer cells by kinase-dead PKCε was associated with the specific induction of p21Cip1 and the inactivation of cyclin Cdk2 complexes (75). The fact that PKCε expression significantly increases prostate cancer in a manner correlating with the aggressiveness of the disease suggests that PKCε is probably linked to the maintenance of androgen-independent prostate cancer (76). Therefore, inactivation of this PKC isoenzyme decreases cell proliferation and induces apoptosis.
Fourth, we determined whether this mechanism could explain how a resistance to selenium develops in some advanced tumor cells. These cells may have a greater adaptive potential to the toxicity of selenium by virtue of having high levels of PKCε, a prosurvival isoenzyme that is relatively resistant to selenium. In this study, this hypothesis has been supported by the fact that an overexpression of PKCε by transfection converted MSA-sensitive DU145 cells to MSA-resistant ones. Because of excess expression, a sufficient amount of PKCε may be spared from selenium-induced inactivation and in turn make advanced tumor cells refractory to selenium-induced cell death. Conversely, knocking out the level of PKCε using siRNA enhanced the cellular sensitivity to MSA. It is therefore likely that the expression of molecular targets such as PKCε determines at least in part whether or not cells respond to selenium at dietary supplementary concentrations.
Finally, to understand the relationship between the actions of selenometabolites and selenoproteins, we assessed the ability of the TR system to reverse the redox modification of PKC induced by selenometabolites. Studies carried out with PKC in purified form or PKC in intact cells have shown a reversal of the redox modification of PKC by the TR system. It is intriguing that interaction of selenium with the cysteine-rich regions of proteins is nullified by the selenoprotein TR along with thioredoxin. In this context, it is also interesting to note that thioredoxin-related proteins bind to the PKC catalytic domain with a high affinity and reverse the redox modification of PKC (77–79). Although reduced thioredoxin alone can regenerate PKC sulfhydryls, this protein is oxidized by MSA.6 Therefore, under oxidizing conditions, TR is a limiting factor for providing reducing equivalents necessary for the regeneration of PKC activity. There have been previous reports that show severalfold higher amounts of TR in tumor cells compared with the amounts in their normal counterparts (80, 81). An overexpression of TR could reverse selenium-induced inactivation of PKC; thus, a resistance to selenium may be developed during tumor progression.
It is likely that a relationship exists between the actions of selenoproteins and selenometabolites. It is important to understand this relationship to clarify the complexity of selenium action as a prooxidant and an antioxidant (26). Selenometabolites, as prooxidants, produce free radicals and cause lipid peroxidation and DNA damage (63, 82, 83). Selenoproteins, as antioxidants, negate the action of selenometabolites. For example, selenocompounds such as selenite produce hydrogen peroxide and lipid peroxides (63, 83), which are removed by the selenoprotein glutathione peroxidase. Similarly, selenometabolite-induced protein thiol oxidation is reversed by the TR system as shown in this study. Furthermore, TR converts protein sulfhydryl-oxidizing MSA into volatile methylselenol, which cannot oxidize protein thiols. Conversely, selenoproteins may also support the actions of selenometabolites. For instance, a high level of TR may increase the conversion of selenite to selenide, which can then undergo the redox cycle, induce oxidative stress, inhibit lipoxygenases, and induce apoptosis (63, 64, 84).
Other studies have shown that various skin tumor promoters, which are known to activate PKC isoenzymes, increase the expression of TR and thioredoxin (85). If selenium causes a partial inactivation of PKC, the induced TR/thioredoxin system may reverse the selenium-mediated inactivation of PKC. One can speculate that these types of reversal actions might contribute to the failure of selenium in preventing skin cancers (4). Conceivably, knowledge of the relationship between selenometabolites and selenoproteins may aid in understanding why certain cancers are prevented by selenium and others are not.
In summary, this study suggests that the locally generated redox-amplified active species of selenium may affect the targets involved in tumor promotion and that PKC isoenzymes are qualified in many respects to be important molecular targets for the chemopreventive actions of selenometabolites.
Acknowledgments
We thank Dr. Howard Ganther for kindly providing MSA, Dr. Wayne Anderson for providing metallothionein-driven PKCε expression vector, and Dr. Balawant Khatra for providing PK.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Methylselenol exists predominantly as its ionized form methylselenolate at pH 7.4.
The abbreviations used are: TR, thioredoxin reductase; PKC, protein kinase C; MSA, methylseleninic acid; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); DTT, dithiothreitol; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; PDBu, phorbol 12,13-dibutyrate; siRNA, small interfering RNA; DAPI, 4′,6-diamidino-2-phenylindole; PKA, protein kinase A; PP2A, protein phosphatase 2A; PK, phosphorylase kinase; NTSB, 2-nitro-5-thiosulfobenzoate.
Methylseleninic acid exists predominantly as its ionized form methylseleninate at pH 7.4.
Reduced thioredoxin alone regenerated the MSA-modified PKC and restored its kinase activity without the need for TR/NADPH (data not shown). Therefore, we used MSA-oxidized thioredoxin, which is expected to be generated in the cells treated with MSA. Under these conditions, the system depends on TR for reducing equivalents.
R. Gopalakrishna and U. Gundimeda, unpublished results.
References
- 1.Ip, C., and Ganther, H. E. (1990) Cancer Res. 50 1206–1211 [PubMed] [Google Scholar]
- 2.Clark, L. C. (1985) Fed. Proc. 44 2584–2589 [PubMed] [Google Scholar]
- 3.Greeder, G. A., and Milner, J. A. (1980) Science 209 825–827 [DOI] [PubMed] [Google Scholar]
- 4.Clark, L. C., Combs, G. F., Jr., Turnbull, B. W., Slate, E. H., Chalker, D. K., Chow, J., Davis, L. S., Glover, R. A., Graham, G. F., Gross, E. G., Krongrad, A., Lesher, J. L., Jr., Park, H. K., Sanders, B. B., Jr., Smith, C. L., and Taylor, J. R. (1996) J. Am. Med. Assoc. 276 1957–1963 [PubMed] [Google Scholar]
- 5.Klein, E. A., Thompson, I. M., Lippman, S. M., Goodman, P. J., Albanes, D., Taylor, P. R., and Coltman, C. (2001) J. Urol. 166 1311–1315 [DOI] [PubMed] [Google Scholar]
- 6.Thompson, I. M., Coltman, C. A., Jr., and Crowley, J. (1997) Prostate 33 217–221 [DOI] [PubMed] [Google Scholar]
- 7.Karp, J. E., Chiarodo, A., Brawley, O., and Kelloff, G. J. (1996) Cancer Res. 56 5547–5556 [PubMed] [Google Scholar]
- 8.El-Bayoumy, K., Rao, C. V., and Reddy, B. S. (2001) Nutr. Cancer 40 18–27 [DOI] [PubMed] [Google Scholar]
- 9.Ip, C., Thompson, H. J., Zhu, Z., and Ganther, H. E. (2000) Cancer Res. 60 2882–2886 [PubMed] [Google Scholar]
- 10.Combs, G. F., Jr., and Gray, W. P. (1998) Pharmacol. Ther. 79 179–192 [DOI] [PubMed] [Google Scholar]
- 11.Gladyshev, V. N., Jeang, K. T., and Stadtman, T. C. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 6146–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burk, R. F., Hill, K. E., and Motley, A. K. (2003) J. Nutr. 133 S1517–S1520 [DOI] [PubMed] [Google Scholar]
- 13.Sinha, R., Unni, E., Ganther, H. E., and Medina, D. (2001) Biochem. Pharmacol. 61 311–317 [DOI] [PubMed] [Google Scholar]
- 14.Jiang, C., Wang, Z., Ganther, H., and Lu, J. (2001) Cancer Res. 61 3062–3070 [PubMed] [Google Scholar]
- 15.Nilsonne, G., Sun, X., Nystrom, C., Rundlof, A.-K., Potamitou Fernandes, A., Bjornstedt, M., and Dobra, K. (2006) Free Radic. Biol. Med. 41 874–885 [DOI] [PubMed] [Google Scholar]
- 16.Gopalakrishna, R., Chen, Z. H., and Gundimeda, U. (1997) Arch. Biochem. Biophys. 348 37–48 [DOI] [PubMed] [Google Scholar]
- 17.Borek, C., Ong, A., Mason, H., Donahue, L., and Biaglow, J. E. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 1490–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma, S., Stutzman, J. D., Kelloff, G. J., and Steele, V. E. (1994) Cancer Res. 54 5848–5855 [PubMed] [Google Scholar]
- 19.Gopalakrishna, R., and Jaken, S. (2000) Free Radic. Biol. Med. 28 1349–1361 [DOI] [PubMed] [Google Scholar]
- 20.Blumberg, P. M. (1991) Mol. Carcinog. 4 339–344 [DOI] [PubMed] [Google Scholar]
- 21.Gopalakrishna, R., and Gundimeda, U. (2001) Nutr. Cancer 40 55–63 [DOI] [PubMed] [Google Scholar]
- 22.Gopalakrishna, R., and Anderson, W. B. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 6758–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kazanietz, M. G., Wang, S., Milne, G. W., Lewin, N. E., Liu, H. L., and Blumberg, P. M. (1995) J. Biol. Chem. 270 21852–21859 [DOI] [PubMed] [Google Scholar]
- 24.Gopalakrishna, R., and Gundimeda, U. (2002) J. Nutr. 132 S3819–S3823 [DOI] [PubMed] [Google Scholar]
- 25.Chu, F., Chen, L. H., and O'Brian, C. A. (2004) Carcinogenesis 25 585–596 [DOI] [PubMed] [Google Scholar]
- 26.Drake, E. N. (2006) Med. Hypotheses 67 318–322 [DOI] [PubMed] [Google Scholar]
- 27.Nishizuka, Y. (1992) Science 258 607–614 [DOI] [PubMed] [Google Scholar]
- 28.Griner, E. M., and Kazanietz, M. G. (2007) Nat. Rev. Cancer 7 281–294 [DOI] [PubMed] [Google Scholar]
- 29.Newton, A. C. (1997) Curr. Opin. Cell Biol. 9 161–167 [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Guerrico, A. M., Meshki, J., Xiao, L., Benavides, F., Conti, C. J., and Kanzanietz, M. G. (2005) J. Biochem. Mol. Biol. 38 639–645 [DOI] [PubMed] [Google Scholar]
- 31.Murriel, C. L., and Mochly-Rosen, D. (2003) Arch. Biochem. Biophys. 420 246–254 [DOI] [PubMed] [Google Scholar]
- 32.Deucher, A., Efimova, T., and Eckert, R. L. (2002) J. Biol. Chem. 277 17032–17040 [DOI] [PubMed] [Google Scholar]
- 33.Killilea, S. D., Mellgren, R. L., Aylward, J. H., Metieh, M. E., and Lee, E. Y. (1979) Arch. Biochem. Biophys. 193 130–139 [DOI] [PubMed] [Google Scholar]
- 34.Holmgren, A. (1977) J. Biol. Chem. 252 4600–4606 [PubMed] [Google Scholar]
- 35.Luthman, M., and Holmgren, A. (1982) Biochemistry 21 6628–6633 [DOI] [PubMed] [Google Scholar]
- 36.Ogita, K., Miyamoto, S., Yamaguchi, K., Koide, H., Fujisawa, N., Kikkawa, U., Sahara, S., Fukami, Y., and Nishizuka, Y. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 1592–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saido, T. C., Mizuno, K., Konno, Y., Osada, S., Ohno, S., and Suzuki, K. (1992) Biochemistry 31 482–490 [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi, H., and Exton, J. H. (1992) J. Biol. Chem. 267 16347–16354 [PubMed] [Google Scholar]
- 39.Gopalakrishna, R., Gundimeda, U., and Chen, Z. H. (1997) Arch. Biochem. Biophys. 348 25–36 [DOI] [PubMed] [Google Scholar]
- 40.Kosaka, Y., Ogita, K., Ase, K., Nomura, H., Kikkawa, U., and Nishizuka, Y. (1988) Biochem. Biophys. Res. Commun. 151 973–981 [DOI] [PubMed] [Google Scholar]
- 41.Gopalakrishna, R., Chen, Z. H., Gundimeda, U., Wilson, J. C., and Anderson, W. B. (1992) Anal. Biochem. 206 24–35 [DOI] [PubMed] [Google Scholar]
- 42.Chen, S. J., Klann, E., Gower, M. C., Powell, C. M., Sessoms, J. S., and Sweatt, J. D. (1993) Biochemistry 32 1032–1039 [DOI] [PubMed] [Google Scholar]
- 43.Ellman, G. L. (1959) Arch. Biochem. Biophys. 82 70–77 [DOI] [PubMed] [Google Scholar]
- 44.Thannhauser, T. W., Konishi, Y., and Scheraga, H. A. (1987) Methods Enzymol. 143 115–119 [DOI] [PubMed] [Google Scholar]
- 45.Olah, Z., Lehel, C., Jakab, G., and Anderson, W. B. (1994) Anal. Biochem. 221 94–102 [DOI] [PubMed] [Google Scholar]
- 46.Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., Warren, J. T., Bokesch, H., Kenney, S., and Boyd, M. R. (1990) J. Natl. Cancer Inst. 82 1107–1112 [DOI] [PubMed] [Google Scholar]
- 47.Mosmann, T. (1983) J. Immunol. Methods 65 55–63 [DOI] [PubMed] [Google Scholar]
- 48.Fujii, T., Garcia-Bermejo, M. L., Bernabo, J. L., Caamano, J., Ohba, M., Kuroki, T., Li, L., Yuspa, S. H., and Kazanietz, M. G. (2000) J. Biol. Chem. 275 7574–7582 [DOI] [PubMed] [Google Scholar]
- 49.Cotgreave, I. A., Morgenstern, R., Engman, L., and Ahokas, J. (1992) Chem. Biol. Interact. 84 69–76 [DOI] [PubMed] [Google Scholar]
- 50.Boni, L. T., and Rando, R. R. (1985) J. Biol. Chem. 260 10819–10825 [PubMed] [Google Scholar]
- 51.Sies, H., and Masumoto, H. (1997) Adv. Pharmacol. 38 229–246 [DOI] [PubMed] [Google Scholar]
- 52.Wendel, A., Fausel, M., Safayhi, H., Tiegs, G., and Otter, R. (1984) Biochem. Pharmacol. 33 3241–3245 [DOI] [PubMed] [Google Scholar]
- 53.Webber, M. M., Perez-Ripoll, E. A., and James, G. T. (1985) Biochem. Biophys. Res. Commun. 130 603–609 [DOI] [PubMed] [Google Scholar]
- 54.Gromer, S., Arscott, L. D., Williams, C. H., Jr., Schirmer, R. H., and Becker, K. (1998) J. Biol. Chem. 273 20096–20101 [DOI] [PubMed] [Google Scholar]
- 55.Froscio, M., Murray, A. W., and Hurst, N. P. (1989) Biochem. Pharmacol. 38 2087–2089 [DOI] [PubMed] [Google Scholar]
- 56.Gromer, S., and Gross, J. H. (2002) J. Biol. Chem. 277 9701–9706 [DOI] [PubMed] [Google Scholar]
- 57.Sun, S.-Y., Hail, N., Jr., and Lotan, R. (2004) J. Natl. Cancer Inst. 96 662–672 [DOI] [PubMed] [Google Scholar]
- 58.Ganther, H. E. (1999) Carcinogenesis 20 1657–1666 [DOI] [PubMed] [Google Scholar]
- 59.Parker, T. L., Eggett, D. L., and Christensen, M. J. (2007) J. Nutr. Biochem. 18 746–752 [DOI] [PubMed] [Google Scholar]
- 60.Hanks, S. K., and Quinn, A. M. (1991) Methods Enzymol. 200 38–62 [DOI] [PubMed] [Google Scholar]
- 61.Sweetman, L. L., Zhang, N. Y., Peterson, H., Gopalakrishna, R., and Sevanian, A. (1995) Arch. Biochem. Biophys. 323 97–107 [DOI] [PubMed] [Google Scholar]
- 62.O'Brian, C. A., Ward, N. E., Weinstein, I. B., Bull, A. W., and Marnett, L. J. (1988) Biochem. Biophys. Res. Commun. 155 1374–1380 [DOI] [PubMed] [Google Scholar]
- 63.Spallholz, J. E. (1997) Biomed. Environ. Sci. 10 260–270 [PubMed] [Google Scholar]
- 64.Holmgren, A. (2006) Free Radic. Biol. Med. 41 862–865 [DOI] [PubMed] [Google Scholar]
- 65.Spyrou, G., Bjornstedt, M., Kumar, S., and Holmgren, A. (1995) FEBS Lett. 368 59–63 [DOI] [PubMed] [Google Scholar]
- 66.Seo, Y. R., Kelley, M. R., and Smith, M. L. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 14548–14553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park, H. S., Park, E., Kim, M. S., Ahn, K., Kim, I. Y., and Choi, E. J. (2000) J. Biol. Chem. 275 2527–2531 [DOI] [PubMed] [Google Scholar]
- 68.Christensen, M. J., Nartey, E. T., Hada, A. L., Legg, R. L., and Barzee, B. R. (2007) Nutr. Cancer 58 197–204 [DOI] [PubMed] [Google Scholar]
- 69.Fleshner, N., Fair, W. R., Huryk, R., and Heston, W. D. (1999) J. Urol. 161 1651–1654 [PubMed] [Google Scholar]
- 70.Ghosh, J., and Myers, C. E. (1997) Biochem. Biophys. Res. Commun. 235 418–423 [DOI] [PubMed] [Google Scholar]
- 71.Ghosh, J. (2004) Biochem. Biophys. Res. Commun. 315 624–635 [DOI] [PubMed] [Google Scholar]
- 72.Lamm, M. L., Long, D. D., Goodwin, S. M., and Lee, C. (1997) Endocrinology 138 4657–4664 [DOI] [PubMed] [Google Scholar]
- 73.Orlandi, L., Binda, M., Folini, M., Bearzatto, A., Villa, R., Daidone, M. G., and Zaffaroni, N. (2003) Prostate 54 133–143 [DOI] [PubMed] [Google Scholar]
- 74.Cacace, A. M., Guadagno, S. N., Krauss, R. S., Fabbro, D., and Weinstein, I. B. (1993) Oncogene 8 2095–2104 [PubMed] [Google Scholar]
- 75.Bae, K.-M., Wang, H., Jiang, G., Chen, M. G., Lu, L., and Xiao, L. (2007) Cancer Res. 67 6053–6063 [DOI] [PubMed] [Google Scholar]
- 76.Wu, D., Foreman, T. L., Gregory, C. W., McJilton, M. A., Wescott, G. G., Ford, O. H., Alvey, R. F., Mohler, J. L., and Terrian, D. M. (2002) Cancer Res. 62 2423–2429 [PubMed] [Google Scholar]
- 77.Watson, J. A., Rumsby, M. G., and Wolowacz, R. G. (1999) Biochem. J. 343 301–305 [PMC free article] [PubMed] [Google Scholar]
- 78.Witte, S., Villalba, M., Bi, K., Liu, Y., Isakov, N., and Altman, A. (2000) J. Biol. Chem. 275 1902–1909 [DOI] [PubMed] [Google Scholar]
- 79.Kahlos, K., Zhang, J., Block, E. R., and Patel, J. M. (2003) Mol. Cell. Biochem. 254 47–54 [DOI] [PubMed] [Google Scholar]
- 80.Tamura, T., and Stadtman, T. C. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 1006–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Berggren, M., Gallegos, A., Gasdaska, J. R., Gasdaska, P. Y., Warneke, J., and Powis, G. (1996) Anticancer Res. 16 3459–3466 [PubMed] [Google Scholar]
- 82.Wycherly, B. J., Moak, M. A., and Christensen, M. J. (2004) Nutr. Cancer 48 78–83 [DOI] [PubMed] [Google Scholar]
- 83.Moak, M. A., and Christensen, M. J. (2001) Biol. Trace Elem. Res. 79 257–269 [DOI] [PubMed] [Google Scholar]
- 84.Bjornstedt, M., Odlander, B., Kuprin, S., Claesson, H. E., and Holmgren, A. (1996) Biochemistry 35 8511–8516 [DOI] [PubMed] [Google Scholar]
- 85.Kumar, S., and Holmgren, A. (1999) Carcinogenesis 20 1761–1767 [DOI] [PubMed] [Google Scholar]
- 86.Gopalakrishna, R., Gundimeda, U., Wilson, J. C., and Chen, Z. H. (1993) Anal. Biochem. 212 296–299 [DOI] [PubMed] [Google Scholar]
- 87.Forman, H. J., and Kim, E. (1989) Arch. Biochem. Biophys. 274 443–452 [DOI] [PubMed] [Google Scholar]
- 88.Salgo, M. G., Corongiu, F. P., and Sevanian, A. (1993) Arch. Biochem. Biophys. 304 123–132 [DOI] [PubMed] [Google Scholar]