

Abstract
Background
Chronic alcohol consumption causes alcoholic liver disease, which is associated, or initiated, with dysregulated lipid metabolism. Very recent evidence suggested that dysregulated cholesterol metabolism plays an important role in the pathogenesis of alcoholic fatty liver diseases, however, the effects of chronic alcohol exposure on cholesterol homeostasis have not been well studied and underlying mechanisms behind are still elusive.
Methods
Male Sprague-Dawley rats weighing 250 ± 5.5 g (mean ± SEM) divided into two groups (8 rats per group) and pair-fed with liquid diets containing (in percent of energy intake) 18% protein, 35% fat, 12% carbohydrate, and 35% either ethanol (ethanol diet) or an isocaloric maltose-dextrin mixture (control diet), according to Lieber and De Carli, for four weeks.
Results
Long-term excessive alcohol feeding to rats caused fatty liver and liver injury, which was associated with disrupted cholesterol homeostasis, characterized by increased hepatic cholesterol levels and hypercholesterolemia. Hepatic cholesterol increases were concomitant with constantly activated SREBP-2 in the liver and increased expression of HMG-CoA reductase, a rate-limiting enzyme for cholesterol de novo synthesis, indicating enhanced cholesterol biosynthesis. Alcohol-induced hypercholesterolemia was accompanied by decreased LDL receptor levels in the liver. Further investigations revealed that chronic alcohol exposure increased hepatic PCSK9 contents, a proprotein convertase to downregulate LDLr via a post-translational mechanism. Moreover, alcohol feeding suppressed ERK activation in the liver. In vitro studies showed that inhibition of ERK activation was associated with decreased LDLr expression in HepG2 cells.
Conclusions
Our study provides the first evidence that both increased PCSK9 expression and suppressed ERK activation in the liver contributes to alcohol-induced hypercholesterolemia in rats.
Keywords: Alcohol, Liver, Cholesterol, LDL receptor, PCSK9, SERBP-2, ERK
Introduction
Chronic ethanol intake is well known to cause dysregulated lipid metabolism. Excessive triglyceride accumulation in hepatocytes due to alcohol abuse not only is the initial stage of disease spectrum of alcoholic liver disease, but also plays a critical role in the disease progression to late stage hepatitis and/or cirrhosis (Maxwell et al., 2003; Song et al., 2004; You et al., 2004). Fat accumulation in the liver originates from an imbalance among the uptake/synthesis and export/oxidation of fatty acids. Correspondingly, deleterious effects of excessive alcohol intake on all these processes have been extensively investigated, and reported to contribute to the development of hepatic steatosis (Donohue, 2007; Fischer et al., 2003; Kharbanda et al., 2009; You et al., 2004). Moreover, chronic alcohol consumption also causes hypertriglyceridemia (increased blood triglyceride level), mainly due to elevated VLDL and chylomicrons levels in the blood (Chait et al., 1972).
In addition to disturbing triglyceride metabolism, chronic consumption of excessive alcohol can also exert adverse effects on cholesterol metabolism. It has been reported previously that ethanol feeding increased hepatic esterified cholesterol levels in rats, and this was associated with both enhanced cholesterol biosynthesis and decreased bile acid excretion (Lefevre et al., 1972). Cholesterol is a lipid molecule required for the growth and viability of mammalian cells and plays important structural and functional roles in membranes. Disturbed cholesterol homeostasis has been well-established to play an etiologic role in the pathogenesis of atherosclerotic cardiovascular disease. The liver plays a central role in the regulation of cholesterol homeostasis. It is the major organ for the clearance of circulating LDL, the major cholesterol-carrying lipoprotein, via LDL receptor (LDLr) regulated mechanism. Importantly, the liver is the only organ in the body for cholesterol disposition, via bile acids synthesis and excretion (Chiang, 2003; Chang et al., 2006; Kang and Davis, 2000). Very recent studies provide strong evidence demonstrating that dysregulated cholesterol metabolism was critically involved in the development of both alcoholic and non-alcoholic fatty liver diseases (Fernandez et al., 2008; Wouters et al., 2008). For instance, it has been reported that chronic alcohol exposure increased hepatic free cholesterol pool, which was associated with mitochondrial dysfunction and consequent sensitization to TNF-α induced hepatotoxicity (Fernandez et al., 2008). Nevertheless, studies with regard to the effects of excessive alcohol intake on cholesterol metabolism are still lacking and underlying mechanisms behind are still elusive. Therefore, defining mechanisms behind the deleterious effects of chronic alcohol consumption on cholesterol metabolism will provide critical information to help us better understand the pathogenesis of alcoholic liver disease. In this study, using a heavy drinking rat model, our aims were to investigate the effects of chronic alcohol exposure on cholesterol homeostasis and to identify mechanisms underlying alcohol-induced disruption of cholesterol homeostasis.
Methods
Animals and treatments
Male Sprague-Dawley rats weighing 250 ± 5.5 g (mean ± SEM) were obtained from the Jackson Laboratory (Bar Harbor, ME). All animals were treated according to the experimental procedures approved by the Institutional Animal Care and Use Committee. Sixteen rats were randomly assigned to 2 groups and pair-fed with liquid diets containing (in percent of energy intake) 18% protein, 35% fat, 12% carbohydrate, and 35% either ethanol (ethanol diet) or an isocaloric maltose-dextrin mixture (control diet), according to Lieber and De Carli (Lieber and De Carli, 1973), for four weeks. Food intake and body weight were recorded daily and weekly, respectively. At the end of the experiment, the rats were killed and plasma and liver tissue samples were harvested for assays.
Cells and culture conditions
HepG2 cells, a human hepatoma cell line, were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured in DMEM containing 10% (v/v) fetal bovine serum, 2 mM glutamine, 5 U/ml penicillin, and 50 μg/ml streptomycin at 37 °C in a humidified O2/CO2 (19:1) atmosphere.
Plasma biochemical assays
Plasma alanine aminotransferases (ALT) and aspartate transaminase (AST) assays were performed with commercially available kits (Infinity, Thermo Electron, Melbourne, Australia). Plasma total cholesterol, high-density lipoprotein (HDL)-cholesterol, and low-density lipoprotein (LDL)-cholesterol levels were measured using a clinical CardioChek analyzer (Polymer Technology Systems, Indianapolis, IN).
Liver triglyceride and cholesterol measurements
Liver tissues were homogenized and hepatic total lipids were extracted according to Bligh and Dyer and redissolved in 2% Triton X-100 in water. Hepatic triglyceride content was determined by enzymatic colorimetric methods using commercially available kits (Sigma).
Preparation of nuclear extracts
To isolate nuclear proteins we used a Nuclear Extraction Kit (Imgenex, San Diego, CA) according to manufacturer’s instructions. Briefly, small pieces of fresh liver tissue were washed twice with 5 ml of ice-cold PBS/PMSF buffer, homogenized in 5 ml 1 × hypotonic buffer supplemented with 1 mM DTT and 1% detergent, and put on ice for 30 min. After centrifugation at 10,000 rpm at 4°Cfor 10 min, supernatants (cytoplasmic fraction) were removed and 500 μl of complete lysis buffer was added to the nuclei and incubated for 30 min at 4°C with rocking. The samples were vortexed and centrifuged at 14,000 rpm for 10 min at 4°C. Supernatants containing nuclear proteins were harvested and transferred to prechilled tubes.
Total RNA isolation
Frozen liver samples (~70 mg) were transferred to 1.0 ml of Trizol (Invitrogen, Carlsbad, CA) and homogenized in a Mixer Mill 300 tissue homogenizer (Retsch, Germany). Total RNA was isolated according to the manufacturer’s protocol with an additional phenol-chloroform extraction. Isolated RNA was re-suspended in RNA storage solution (Ambion, Austin, TX), quantified (A260), and assessed for purity by determining the A260/A280 ratio and by visual inspection of 1.0 μg on a denaturing gel.
Quantitative real-time RT-PCR
Total RNA was extracted from liver tissue as described in the previous paragraph. For each sample, 1.0 μg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit as described by the manufacturer (Applied Biosystems, Foster City, CA). The cDNA (1.0 μl) was used as a template in a 25-μl PCR reaction solution containing 10.5 μl,1 μl 10 μM gene-specific primers, and 12.5 μl 1 × SYBR Green PCR master mix (SuperArray Bioscience, Frederick, MD). PCR amplification was conducted in MicroAmp Optical 96-wellreaction plates (Applied Biosystems) on an Applied Biosystems PRISM 7000 sequence detection system under the following conditions: initial denaturation and enzyme activation for 10 min at 95°C, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each plate contained duplicate standards of purified PCR products of known template concentrations covering seven orders of magnitude to interpolate relative template concentrations of the samples from the standard curves of log copy number vs. threshold cycle (CT). No-template controls (NTC) were also included on each plate. Samples with a CT value within 2 SD of the mean CT values for the NTCs were considered below the limits of detection. The copy number of each unknown sample for each gene was standardized to a house-keeping gene (rat 18s rRNA) to control for differences in RNA loading, quality, and cDNA synthesis. For graphing purposes, the relative expression levels were scaled such that the expression level of the time-matched control group was equal to 1.
Western blotting detections
Liver tissues were lysed in Western lysis buffer consisting of the following: 20 mM Tris·HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 2% Nonidet P-40, 1 mM EDTA, pH 8.0, 20 mM sodium fluoride, 30 mM sodium pyrophosphate, 0.2% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1 mM sodium vanadate, 50 μM leupeptin, and 5 μM aprotinin. Samples were incubated on ice with frequent vortexing for 15 min and centrifuged for 20 min at 18,000 g. The protein content of each supernatant was quantified via a protein assay reagent from Bio-Rad Laboratories (Hercules, CA) in accordance with the manufacturer’s instructions. Proteins were separated by SDS-PAGE and transferred to 0.45μm Immobilin-P polyvinylidene difluoride membrane (PerkinElmer Life Sciences). After transfer, membranes were blocked in 5% (wt/vol) nonfat dry milk in PBS-0.1% Tween 20 and probed with the antibodies specified. Horseradish peroxidase-conjugated secondary antibodies (Sigma) and enhanced chemiluminescence substrate kit (PerkinElmer Life Science) were used in the detection of specific proteins.
Statistical analysis
All data were expressed as means ± SD. Statistical analysis was performed using a one-way ANOVA and was analyzed further by Newman-Keuls test for statistical difference. Differences between treatments were considered to be statistically significant at P < 0.05.
Results
Excessive alcohol intake causes fat accumulation in the liver and liver injury
The pathological alterations in livers from both groups were evaluated by the measurements of hepatic triglyceride contents and circulating liver enzyme levels. As expected, long-term excessive alcohol diet feeding resulted in increased triglyceride accumulation in the liver (Fig. 1A) and liver injury in rats, as judged by increased plasma ALT and AST levels in alcohol-fed animals (Fig. 1B & C).
Fig. 1.

Chronic alcohol feeding caused hepatic fat accumulation and liver injury. Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Liver triglyceride contents (A), Plasma ALT levels (B), and Plasma AST levels (C) were measured with corresponding assay kit. Data are means ± SD (n = 8). * P < 0.05. PF: pair-fed; AF: alcohol-fed.
Increase of hepatic cholesterol content in alcohol-fed rats is associated with enhanced cholesterol biosynthesis pathway
The effects of chronic alcohol feeding on hepatic cholesterol content were examined. As shown in Fig. 2A, alcohol feeding increased hepatic cholesterol levels. Cholesterol biosynthesis in hepatocytes is essentially regulated by transcription factor, sterol regulatory element-binding protein (SREBP)-2. To examine the effect of alcohol on cholesterol biosynthesis pathway, we measured SREBP-2 activation by detecting mature form of SREBP-2 protein abundance in the liver. As shown in Fig. 2B & C, increased mature form of SREBP-2 protein was observed in both whole tissue lysates and nuclei from alcohol fed rats. Furthermore, we examined the effects of alcohol feeding on gene expression of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase, one of SREBP-2 targets and the rate-limiting enzyme for cholesterol synthesis. We showed that HMG-CoA reductase gene expression was significantly increased by chronic alcohol exposure (Fig. 2D), suggesting that chronic alcohol exposure enhanced de novo cholesterol synthesis in the liver.
Fig. 2.
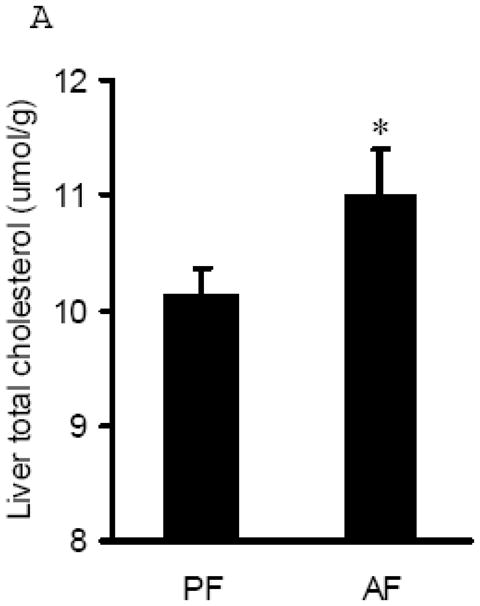
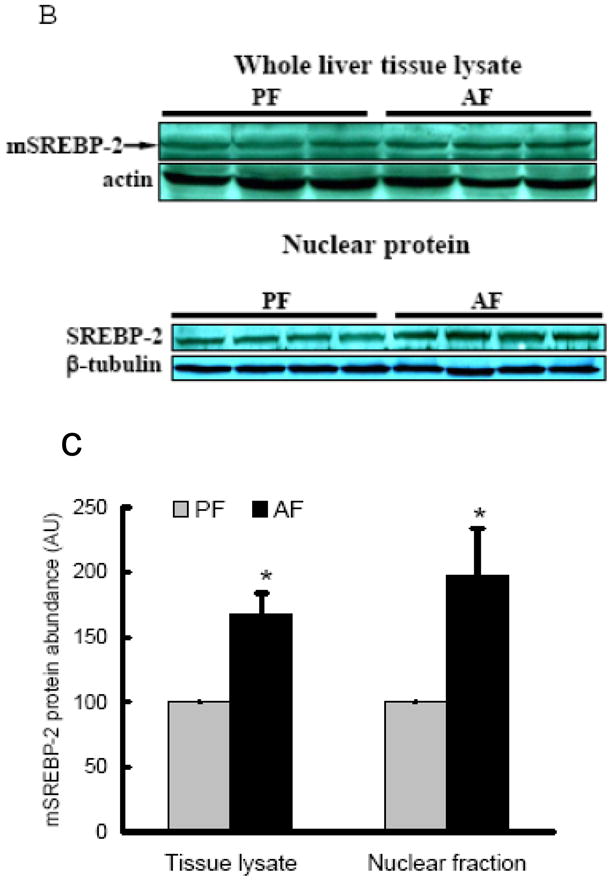
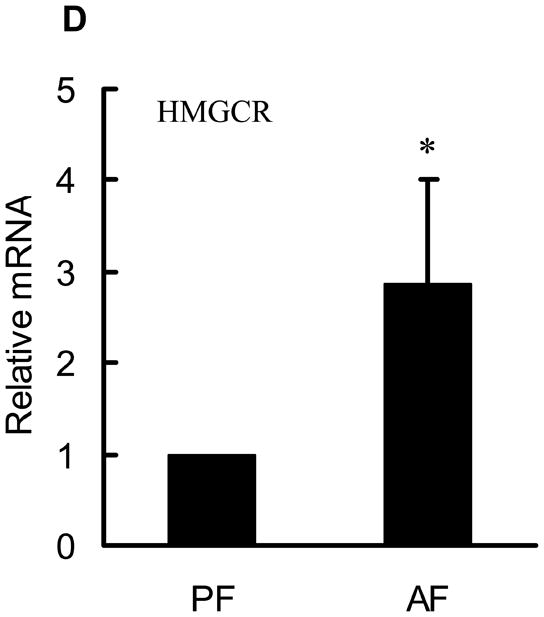
Alcohol-induced increases in hepatic total cholesterol levels were associated with enhanced cholesterol biosynthesis pathway. A. Liver total cholesterol levels; B. Representative Western blots of mature form SREBP-2 proteins in both whole liver and nuclei lysates; C. Quantitation of Western blots of mature form SREBP-2 proteins in both whole liver and nuclei lysates (n=8). Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Equal amounts of protein were loaded onto the gel for comparison of pair-fed and alcohol-fed animals. PF: pair-fed; AF: alcohol-fed. SREBP-2: sterol regulatory element binding proteins-2; D. Gene expression analysis of HMGCR by real-time RT-PCR. Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Total RNA from liver tissues were isolated thereafter and subjected to real-time RT-PCR assay to quantitate mRNA for HMGCR. Data are means ± SD (n = 8). * P < 0.05. PF: pair-fed; AF: alcohol-fed. HMGCR: 3-hydroxy-3-methyl-glutaryl-CoA reductase.
Alcohol feeding induced hypercholesterolemia and reduced hepatic LDL receptor (LDLr)
Plasma total cholesterol, HDL cholesterol, and LDL cholesterol levels in both pair-fed and alcohol-fed rats were measured. As shown in Fig. 3, alcohol-fed rats showed significantly elevated plasma total cholesterol levels (Fig. 3A) in comparison to pair-fed animals, and this was associated with markedly increased cholesterol enrichments in both HDL and LDL (Fig. 3B & C). LDLr plays a central role in the regulation of hepatic LDL clearance and plasma cholesterol homeostasis. To examine the effect of chronic alcohol feeding on LDLr production, we measured hepatic LDLr mRNA levels and protein abundance in both groups. As shown in Fig. 3D, long-term alcohol exposure suppressed hepatic LDLr gene expression. Correspondingly, a lower LDLr protein abundance was observed in livers of alcohol-fed rats in comparison to pair-fed animals (Fig. 3E & F).
Fig. 3.
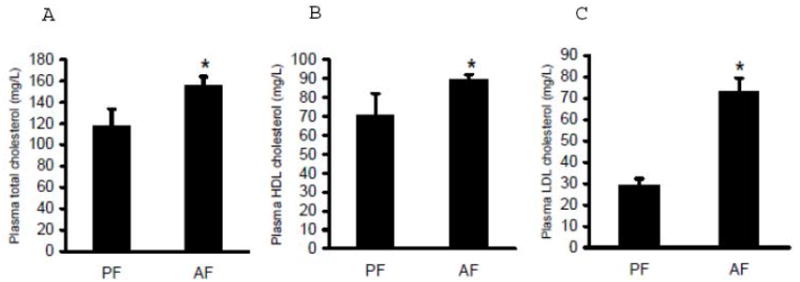
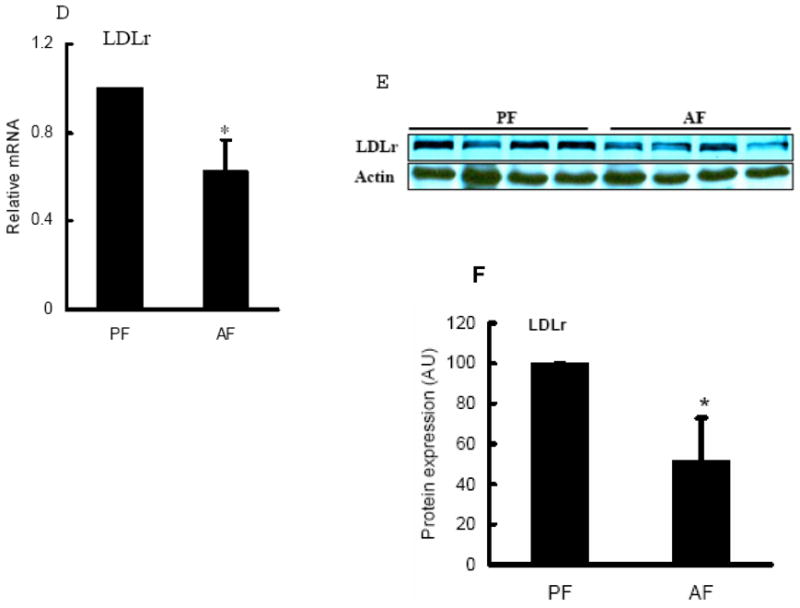
Alcohol-induced hypercholesterolemia was associated with reduced hepatic low-density lipoprotein receptor (LDLr) levels. Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Plasma total cholesterol (A), HDL-cholesterol (B), and LDL-cholesterol (C) levels were measured using commercially available kits. Data are means ± SD (n = 8). * P < 0.05. PF: pair-fed; AF: alcohol-fed; D. Gene expression analysis of LDLr by real-time RT-PCR. Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Total RNA from liver tissues were isolated thereafter and subjected to real-time RT-PCR assay to quantitate mRNA for LDLr. Data are means ± SD (n = 8). * P < 0.05. PF: pair-fed; AF: alcohol-fed; E. Representative Western blots of LDLr proteins in the liver; F. Quantitation of Western blots of LDLr proteins in the liver (n=8). Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Equal amounts of protein were loaded onto the gel for comparison of pair-fed and alcohol-fed animals. PF: pair-fed; AF: alcohol-fed.
Chronic alcohol exposure decreased proprotein convertase subtilisin/kexin type 9 (PCSK9) levels in the liver
PCSK9 is a recently discovered enzyme capable of degrading LDLr protein through post-translational mechanisms (Lopez, 2008; Seidah, 2009). In present study, we examined hepatic PCSK9 levels in both groups and our data showed that both mRNA levels and cleaved form of PCSK9 protein levels were increased by alcohol feeding (Fig. 4).
Fig. 4.
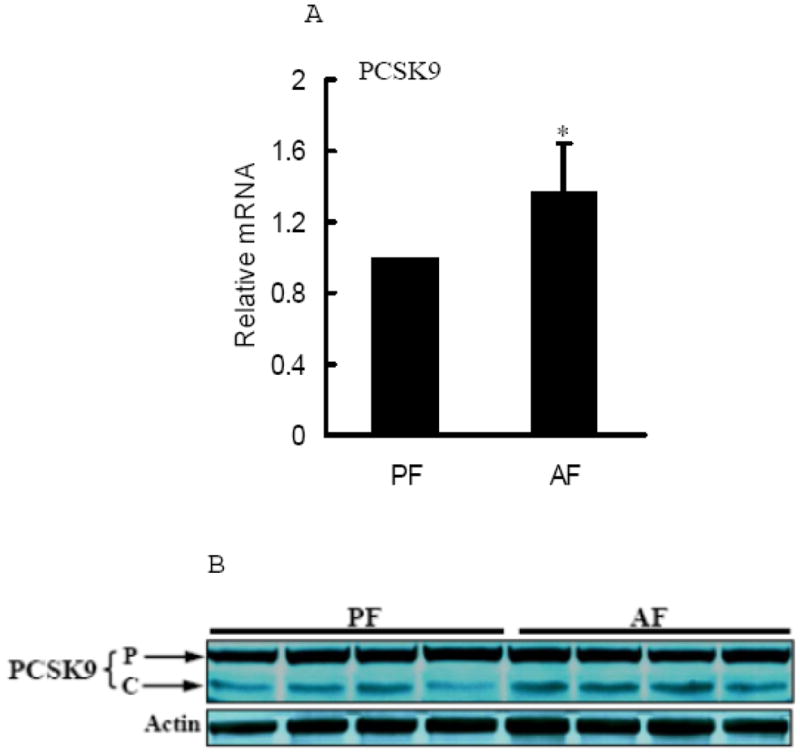
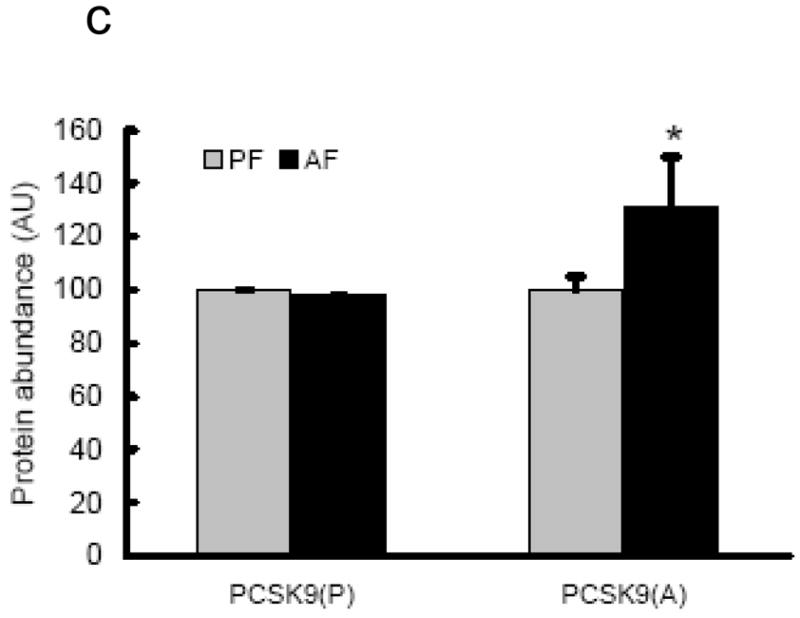
Effects of chronic alcohol feeding on hepatic PCSK9 levels. A. Gene expression analysis of PCSK9 by real-time RT-PCR. Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Total RNA from liver tissues were isolated thereafter and subjected to real-time RT-PCR assay to quantitate mRNA for PCSK9. Data are means ± SD (n = 8). * P < 0.05. PF: pair-fed; AF: alcohol-fed. PCSK9: proprotein convertase subtilisin/kexin type 9; B. Representative Western blots of PCSK9 proteins in the liver; C. Quantitation of Western blots of precursor and cleaved form of PCSK9 proteins in the liver (n=8). Male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Equal amounts of protein were loaded onto the gel for comparison of pair-fed and alcohol-fed animals. PF: pair-fed; AF: alcohol-fed. PCSK9: proprotein convertase subtilisin/kexin type 9. p: precursor. c/a: cleaved (active) form.
Involvement of MEK/ERK pathway in the regulation of LDLr production
To assess the possible involvement of ERK1/2 activation in the reducing effects of alcohol on hepatic LDLr contents, we first examined ERK1/2 activation in livers of alcohol-fed rats by Western blot analysis. As shown in Fig. 5A & B, alcohol exposure reduced ERK1/2 protein phosphorylation in the liver. To further determine if the reduced MEK/ERK activation is associated with decreased LDLr production, we next examined the effect of U0126, a specific MEK/ERK inhibitor, on LDLr production in HepG2 cells. As shown in Fig. 5C & D, addition of U0126 decreased LDLr levels in HepG2 cells, while SREBP-2 levels were not affected, suggesting that inhibition of the MEK/ERK pathway contribute to LDLr reduction by alcohol.
Fig. 5.
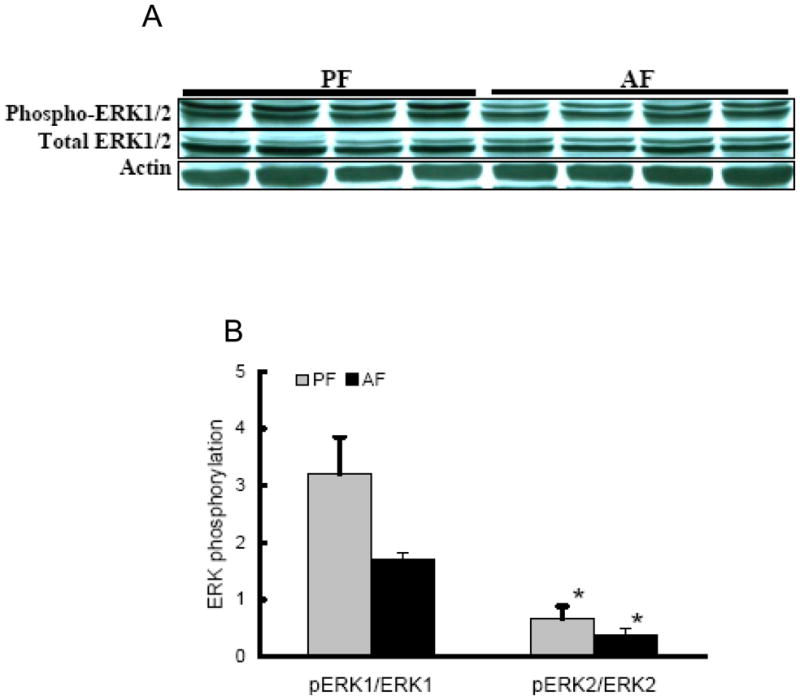

Involvement of ERK activities on LDLr production. In the in vivo studies, male Sprague-Dawley rats were fed with either control or Lieber-De Carli ethanol-containing diet for 4 weeks. Total protein extracts from the liver were prepared thereafter. Forty micrograms of protein were subjected to Western blot analysis for ERK1/2 phosphorylation using specific antibodies. A. Representative Western blots for phosphorylated and total ERK1/2 in the liver; B. Quantitation of Western blots of ERK1/2 phosphorylation in the liver (n=8). PF: pair-fed; AF: alcohol-fed. In the in vitro investigation, HepG2 cells were cultured in DMEM medium overnight and incubated with DMEM containing U0126, an ERK inhibitor, for 12 hours. Total cell extracts were subjected to Western blot analysis with specific antibodies. C: Representative Western blots for phosphorylated and total ERK1/2, LDLr, and mSREBP-2 in whole cell lysate of HepG2 cells; D. Quantitation of Western blots of phosphorylated and total ERK1/2, LDLr, and mSREBP-2. Data represent a mean of at least 3 experiments ± SD, *P < 0.05. SREBP-2: sterol regulatory element binding proteins-2. LDLr: low-density lipoprotein receptor.
Discussion
In present study, we demonstrated that chronic alcohol exposure for 4 wk resulted in disturbed cholesterol homeostasis in rats. Chronic alcohol feeding constantly activated SREBP-2 in the liver and increased gene expression of HMG-CoA reductase, the rate-limiting enzyme for cholesterol de novo synthesis, indicating that cholesterol biosynthesis was enhanced. This may account, at least partially, for increased hepatic total cholesterol contents in alcohol fed rats. Plasma total cholesterol levels were also elevated, which was associated with decreased LDL receptor gene expression and protein abundance in the liver. Additional investigations revealed that chronic alcohol exposure activated PCSK9, a proprotein convertase to downregulate LDLr via post-translational mechanisms. Moreover, alcohol feeding suppressed ERK activation in the liver. In vitro investigations demonstrated that inhibition of ERK activation was associated with decreased LDLr expression in HepG2 cells, suggesting that multiple mechanisms are involved in alcohol-induced downregulation of LDLr.
Both exogenous uptake and endogenous de novo synthesis contribute to intracellular cholesterol content. The LDL is the major cholesterol-carrying lipoprotein in the blood and the liver is the major organ for LDL clearance via mechanisms essentially mediated by LDL receptor. LDL-LDLr interaction is responsible for ~70% of LDL removed from the circulation and LDLr deficiency is the major cause for familial hypercholesterolemia (Brown and Goldstein, 1986; Goldstein and Brown, 2009; Hobbs et al., 1992). Therefore, in this study, observed decreases in hepatic LDLr abundance in alcohol-fed rats suggested that suppressive effects of alcohol on LDL receptor may contribute to its hypercholesterolemic effects. On the other hand, LDLr deficiency may also cause decreased exogenous uptake of cholesterol by hepatocytes. Surprisingly, in this study, alcohol-induced reduction of LDLr in the liver was concomitant with increased hepatic total cholesterol levels, suggesting that increased intracellular de novo synthesis may compensate, or even surmount, the insufficient uptake of exogenous cholesterol due to LDLr reduction. Biosynthesis of cholesterol involves the actions of numerous enzymes and SREBP-2 is a major transcription factor controlling gene expressions of many enzymes involved in this pathway (Eberle et al., 2004; Webber et al., 2004). Synthesized as a 125 kDa protein and subsequently localized to the endoplasmic reticulum, SREBP-2 precursor is transported to the Golgi by SCAP (SREBP-cleavage activating protein), a chaperone protein, and then cleaved by two proteases to release the mature, transcriptionally active 68 kDa amino terminal domain, which enters the nucleus and up-regulates transcription of many genes for the cholesterol synthesis, including HMG-CoA reductase, the rate-limiting enzyme for intracellular cholesterol synthesis (Caballero et al., 2009; Konig et al., 2006). Our present study demonstrated that chronic alcohol feeding resulted in enhanced SREBP-2 activation, manifested by increased mature form of SREBP-2 proteins in the liver of alcohol-fed rats, and this was associated with increased HMG-CoA gene expression, indicating that alcohol enhanced intracellular cholesterol biosynthesis pathway. Since the liver is the only organ for cholesterol disposition via bile acids synthesis pathway, therefore, the potential effects of chronic alcohol intake on bile acids synthesis/excretion may also contribute to observed alterations in hepatic cholesterol contents. In present study, we did not examine whether or not alcohol altered bile acids synthesis or excretion in our animal model, however, previous studies using the same model demonstrated that chronic alcohol feeding decreased bile acids excretion (Lefevre et al., 1972), suggesting the both pathways may contribute to elevated hepatic cholesterol levels.
Apart from controlling cholesterol biosynthesis, SREBP-2 also plays a critical role in the regulation of LDLr gene expression. The transport of the proSREBP-2-SCAP complex to the Golgi is inhibited by sterols and activated in response to depleted intracellular cholesterol levels (Caballero et al., 2009; Konig et al., 2006). Cholesterol-lowering therapy with statins, a group of inhibitors for cholesterol biosynthesis, represents a paradigm for this regulation. Inhibition of cholesterol synthesis by statin treatment activates SREBP-2 and subsequently induces LDLr gene expression, thereby enhancing hepatic clearance of LDL (Attie and Seidah, 2005). Interestingly, in present study, although SREBP-2 activations were observed in alcohol-fed rats, hepatic LDLr gene expressions in these animals were suppressed. These observations implied that other regulatory pathways than SREBP-2 activation may be involved. As a matter of fact, the involvement of the sterol-independent mechanisms in LDLr gene expression has been reported. Among these, extracellular signal-regulated kinase (ERK) pathway activation has been demonstrated to up-regulate LDLr gene expression in hepatocyte by enhancing LDLr mRNA stability through sequences present in the proximal section of the LDLr mRNA 3′ untranslated region. This pathway has been reported to attribute to the LDL-lowering properties of bile acids and several herbal medicines, including berberine (Abidi et al., 2005; Kong et al., 2004; Nakahara et al., 2002). To test the potential involvement of this pathway in alcohol-induced reduction of LDLr gene expression, we first examined the effects of alcohol exposure on MEK/ERK activation in the liver. Our data clearly showed that alcohol feeding suppressed ERK1/2 activation. Studies using HepG2 cells confirmed that inhibition of ERK activation decreased LDLr production without affecting SREBP-2 levels, suggesting that suppressed ERK pathway activation contribute to the inhibitory effects of alcohol on hepatic LDLr gene expression.
Another interesting finding of this study is that PCSK9 activations were observed in alcohol-fed rats. PCSK9 is a recently discovered molecule which plays a critical role in post-translational degradation of LDLr. PCSK9 is highly expressed in hepatocytes and small intestine and is a sterol-responsive gene, upregulated by statins through SREBP-2 activation (Dubuc et al., 2004; Maxwell et al., 2004; Rashid et al., 2005). This may explain why many patients undergoing statin treatment (to increase LDLr gene expression) can not always attain their therapeutic goals. Adenovirus-mediated overexpression of PCSK9 in mice resulted in dramatic reduction of LDLr protein in the liver and elevated circulating LDL levels (Benjannet et al., 2004; Maxwell et al., 2003; Park et al., 2004). Conversely, PCSK9-siRNA studies in HepG2 cells revealed the reverse effect (Benjannet et al., 2004). Furthermore, PCSK9-knockout mice exhibit ~50% reduction in total cholesterol and ~3-fold increase in hepatic LDLr (Rashid et al., 2005). That statins dramatically upregulated the LDLr and potently increased LDL clearance in the absence of PCSK9 (Rashid et al., 2005) convincingly demonstrated that PCSK9 was responsible for the inability of these drugs to increase the level of LDLr protein. Since PCSK9 gene expression is upregulated by SREBP-2, it is rationale to postulate that alcohol feeding should increase its expression. As we expected, increased mRNA levels of PCSK9 were observed in alcohol-fed animals. Importantly, we also observed that alcohol feeding increased active form of PCSK9 protein abundance in livers of alcohol-fed rats, indicating that PCSK9 activation was induced in response to alcohol feeding. This observation provided further evidence to explain observed hepatic LDLr reduction in alcohol-fed rats. Together with decreased gene expression of LDLr by alcohol, our results suggested that both transcriptional and post-translational mechanisms participate in the suppressive effects of chronic alcohol consumption on LDL receptor.
Upregulation of intracellular cholesterol levels in the liver by alcohol exposure plays an important role in the development of alcoholic liver disease. For instance, acetaldehyde, a major metabolite of hepatic ethanol metabolism, elevates intracellular cholesterol production through inducing endoplasmic reticulum stress and sensitizes hepatocytes to TNF-α induced cytotoxicity (Lluis et al., 2003). Using a heavy drinking animal model, our data showed that chronic alcohol exposure enhanced cholesterol biosynthesis and caused hypercholesterolemia, which may result from decreased LDL receptor abundance in the liver. Our study provides the first evidence that both PCSK9 activation and ERK deactivation contributes to alcohol-induced hypercholesterolemia in rats.
Acknowledgments
Supported by the National Institutes of Health grants K01 AA015344 and R01 AA017442 (Z Song).
References
- Abidi P, Zhou Y, Jiang JD, Liu J. Extracellular signal-regulated kinase-dependent stabilization of hepatic low-density lipoprotein receptor mRNA by herbal medicine berberine. Arterioscler Thromb Vasc Biol. 2005;25:2170–2176. doi: 10.1161/01.ATV.0000181761.16341.2b. [DOI] [PubMed] [Google Scholar]
- Attie AD, Seidah NG. Dual regulation of the LDL receptor--some clarity and new questions. Cell Metab. 2005;1:290–292. doi: 10.1016/j.cmet.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chrétien M, Prat A, Seidah NG. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Caballero F, Fernández A, De Lacy AM, Fernández-Checa JC, Caballería J, García-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. 2009;50:789–796. doi: 10.1016/j.jhep.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Chait A, Mancini M, February AW, Lewis B. Clinical and metabolic study of alcoholic hyperlipidaemia. Lancet. 1972;2:62–64. doi: 10.1016/s0140-6736(72)91552-8. [DOI] [PubMed] [Google Scholar]
- Chiang JY. Bile acid regulation of hepatic physiology: III. Bile acids and nuclear receptors. Am J Physiol Gastrointest Liver Physiol. 2003;284:G349–536. doi: 10.1152/ajpgi.00417.2002. [DOI] [PubMed] [Google Scholar]
- Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- Donohue TM., Jr Alcohol-induced steatosis in liver cells. World J Gastroenterol. 2007;13:4974–4978. doi: 10.3748/wjg.v13.i37.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Fernández A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Cholesterol and sphingolipids in alcohol-induced liver injury. J Gastroenterol Hepatol Suppl. 2008;1:S9–15. doi: 10.1111/j.1440-1746.2007.05280.x. [DOI] [PubMed] [Google Scholar]
- Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- Kang S, Davis RA. Cholesterol and hepatic lipoprotein assembly and secretion. Biochim Biophys Acta. 2000;1529:223–230. doi: 10.1016/s1388-1981(00)00151-7. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Todero SL, Ward BW, Cannella JJ, 3rd, Tuma DJ. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem. 2009;327:75–78. doi: 10.1007/s11010-009-0044-2. [DOI] [PubMed] [Google Scholar]
- Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, Wang Y, Wang Z, Si S, Pan H, Wang S, Wu J, Wang Y, Li Z, Liu J, Jiang JD. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10:1344–1351. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- König B, Koch A, Spielmann J, Hilgenfeld C, Stangl GI, Eder K. Activation of PPARalpha lowers synthesis and concentration of cholesterol by reduction of nuclear SREBP-2. Biochem Pharmacol. 2006;73:574–585. doi: 10.1016/j.bcp.2006.10.027. [DOI] [PubMed] [Google Scholar]
- Lefevre AF, DeCarli LM, Lieber CS. Effect of ethanol on cholesterol and bile acid metabolism. J Lipid Res. 1972;13:48–55. [PubMed] [Google Scholar]
- Lieber CS, De Carli LM. Ethanol dependence and tolerance: a nutritionally controlled experimental model in the rat. Res Commun Chem Pathol Pharmacol. 1973;6:983–991. [PubMed] [Google Scholar]
- Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- Lopez D. PCSK9: an enigmatic protease. Biochim Biophys Acta. 2008;1781:184–186. doi: 10.1016/j.bbalip.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci USA. 2004;101:7100–7105. doi: 10.1073/pnas.0409736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109–2119. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- Menon KV, Gores GJ, Shah VH. Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin Proc. 2001;76:1021–1029. doi: 10.4065/76.10.1021. [DOI] [PubMed] [Google Scholar]
- Nakahara M, Fujii H, Maloney PR, Shimizu M, Sato R. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J Biol Chem. 2002;277:37229–37234. doi: 10.1074/jbc.M206749200. [DOI] [PubMed] [Google Scholar]
- Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- Seidah NG. PCSK9 as a therapeutic target of dyslipidemia. Expert Opin Ther Targets. 2009;13:19–28. doi: 10.1517/14728220802600715. [DOI] [PubMed] [Google Scholar]
- Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Joshi-Barve S, Barve S, McClain CJ. Advances in alcoholic liver disease. Curr Gastroenterol Rep. 2004;6:71–76. doi: 10.1007/s11894-004-0029-y. [DOI] [PubMed] [Google Scholar]
- You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- Weber LW, Boll M, Stampfl A. Maintaining cholesterol homeostasis: sterol regulatory element-binding proteins. World J Gastroenterol. 2004;10:3081–3087. doi: 10.3748/wjg.v10.i21.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lütjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]