

Abstract
Spinal muscular atrophy is characterized by degeneration of α motor neurons in the anterior horns of the spinal cord, which leads to progressive symmetrical muscle weakness and atrophy. Spinal muscular atrophy is the leading fatal autosomal recessive disorder in infancy, and genetic counseling is an essential component of the care of families of these patients. However, little guidance is available in the published literature regarding the process and benefit of genetic counseling for families. Accordingly, the authors designed a questionnaire to assess parents’ knowledge of the disease, gauge their access to genetic counseling, and determine how parents use information gained from counseling to guide choices for future pregnancies. The questionnaire specifically targeted when genetic counseling was received, from whom, parental knowledge regarding spinal muscular atrophy genetics, parental choices regarding spinal muscular atrophy and their child, frequency of prenatal testing, perceived relevance of newborn screening, and opinions regarding the disease. Most families clearly received some type of genetic counseling. Yet how and from whom they received the information varied greatly, as did their genetic knowledge of spinal muscular atrophy. The highest percentage of families received counseling from neurologists, who may not be appropriately prepared to provide formal genetic counseling. Many respondents reported having a negative experience with genetic counseling, possibly because it occurred at the time of diagnosis or shortly afterward, a period of great emotional turmoil. These data suggest that a consistent approach for facilitating how and when genetic counseling is received is greatly needed.
Keywords: spinal muscular atrophy, genetic counseling, genetic testing, carrier testing, parents’ perspectives
Spinal muscular atrophy is caused by a deletion or mutation in the survival motor neuron 1 (SMN1)gene. SMN1 was discovered in 1995 and is located on chromosome 5q11-q13.1 SMN1 contains 9 exons that encode a 294 amino acid protein. Two copies of SMN lie in tandem on each chromosome 5: telomeric SMN1 and centromeric SMN2. SMN1 and SMN2 differ in the coding sequence by a single nucleotide (840C→T).2 SMN2 produces approximately 10% of the full-length SMN protein.1,3,4 SMN2 copy numbers are a demonstrated phenotypic modifier in the spinal muscular atrophy population; milder forms of the disease are associated with an increased copy number of SMN2.5,6
Spinal muscular atrophy is characterized by degeneration of α motor neurons in the anterior horns of the spinal cord, which leads to progressive symmetrical muscle weakness and atrophy. It affects approximately 1 in 6000 to 10 000 white newborns and previously was the second most common fatal autosomal recessive disorder after cystic fibrosis.7 Advances in the treatment of cystic fibrosis have resulted in a marked decrease in childhood mortality in that disorder, such that spinal muscular atrophy is now the leading fatal autosomal recessive disorder of infancy. Spinal muscular atrophy is clinically subdivided into 3 types.8 Type 1 (Werdnig-Hoffmann) is characterized by muscle weakness prior to 6 months of age. Affected infants are unable to sit without support. Spinal muscular atrophy type 1 infants account for 60% to 70% of cases. Prognosis is grave, with most not surviving beyond their second birthday without substantial respiratory support.9 Type 2 is characterized by symptoms of muscle weakness prior to 18 months of age. Affected patients are able to sit unsupported at some point in their clinical course but often suffer significant respiratory morbidity.9 Prognosis is widely variable. Type 3 is characterized by the ability to achieve independent ambulation9 and a normal life expectancy.10 These patients suffer from proximal muscle weakness, frequent falls, and significant fatigue, yet 40 years after onset, 59% remain ambulatory.11 Often, these individuals go on to start families of their own.
Spinal muscular atrophy is a common and frequently life-threatening condition. Many family planning options are available to parents (eg, egg or sperm donation, termination of pregnancy, adoption, and preimplantation genetic diagnosis). Early diagnosis can be beneficial in implementing a proactive care plan for the associated respiratory and nutritional issues in affected children. Genetic counseling is an essential component of support for families of patients with spinal muscular atrophy.8 There is a lack of literature regarding the process and benefit of genetic counseling for families affected by spinal muscular atrophy. In addition, there appears to be no consistent approach to the genetic counseling process for parents of children with spinal muscular atrophy, nor is this service provided in a consistent setting.
To add to the body of knowledge regarding genetic counseling for spinal muscular atrophy, we devised a descriptive and exploratory investigation to assess parents’ knowledge regarding the disease, gauge their access to genetic counseling, and determine how they use the information gained from genetic counseling in future pregnancies.
Materials and Methods
A questionnaire composed of 48 quantitative questions and 5 concluding qualitative questions was developed and used to obtain information from parents of children with spinal muscular atrophy. The questionnaire specifically targeted when genetic counseling was received and from whom, parental knowledge regarding spinal muscular atrophy genetics, parental choices regarding spinal muscular atrophy and their child, frequency of prenatal testing, perceived relevance of newborn screening, and opinions regarding the disease. In our study, genetic counseling was defined as any health professional speaking with a patient about how spinal muscular atrophy occurs. This definition included counseling received by a variety of professionals, including neurologists, geneticists, genetic counselors, or other health care practitioners.
Quantitative and qualitative methods were used, including in-person and telephone questionnaires to obtain a descriptive and exploratory investigation to assess parents’ knowledge and access to genetic counseling. This study was approved by the University of Utah Institutional Review Board.
Study Participants
Study participants (n = 103) were parents of children with spinal muscular atrophy types 1 to 3 enrolled in a natural history study at the University of Utah. They included 99 Americans from 27 of the 50 states and 4 from Argentina. Consent was initiated by the first author, Ms Candice Meldrum, either in person, via electronic mail, or by telephone. The response rate for in-person interviews was 94.5%, whereas the telephone interview response rate was 46.1%. The telephone interview response rate included messages left on answering machines that were not responded to by participants. Parents who were contacted but elected not to participate most often cited time constraints as the reason for declining. Some were unavailable when we attempted to contact them later.
Data Collection
Data were collected by Ms Meldrum, who personally administered the questionnaire to all study participants and recorded all answers. Fifty-two questionnaires were administered, either at the University of Utah General Clinical Research Center or at the Primary Children’s Medical Center outpatient clinics in Salt Lake City, Utah. Fifty-one questionnaires were administered over the telephone. Ms Meldrum was available to clarify questions during the questionnaire process and to explain answers after completion of the questionnaire. All responses were recorded using a digital voice recorder. Ms Meldrum documented the answers given by parents during administration in an effort to achieve full completion of each questionnaire.
Data Analysis
There were 103 survey respondents: 43 single responses from a household and 30 couples. The data were separated into couples and single respondents because of the correlated nature of couple data. Mothers were selected as representatives from the couples to include with the single respondents to create an analysis set of 1 respondent per household. Analyses of 2 × 2 contingency tables were performed using the Fisher exact test. Contingency tables with greater than 2 response categories were analyzed using the Pearson χ2 test. All reported P values are 2-sided, and statistical significance was set at .05.
All qualitative question responses were fully transcribed and coded manually according to mutually agreed-on themes. Qualitative software was deemed unnecessary because of the relatively small sample and the diminutive fraction of questions in relation to the full interview schedule. Coding was completed by a graduate-level research assistant, and a subsample of 20 interviews was double-coded by Ms Meldrum to ensure reliability. Multiple themes were identified for each of the 5 questions, and coding for each question was not mutually exclusive; many responses expressed multiple themes and were subsequently coded as such.
Results
Table 1 shows demographics of parents answering questions with regard to parental age, ethnicity, relationship, and perceived current health of their child. The typical respondent was a white female. Age range varied from 20 to 55 years, with the majority between 30 and 44 years of age. Most families that participated had children with spinal muscular atrophy type 2. This reflects the general prevalence of this population and ability to travel to a research center. It may also be a result of the greater medical needs of children with spinal muscular atrophy type 2 compared with those who have spinal muscular atrophy type 3.
Table 1.
Demographics of Questionnaire Respondents
| % | |
|---|---|
| Parent age | |
| 20-24 | 4 |
| 25-29 | 12 |
| 30-34 | 18 |
| 35-39 | 33 |
| 40-44 | 15 |
| 45-49 | 11 |
| 50-54 | 6 |
| 55+ | 1 |
| Ethnicity | |
| White | 86 |
| Hispanic | 8 |
| Asian | 6 |
| Relationship | |
| Mothera | 89 |
| Father | 11 |
| Perceived current health of child with spinal muscular atrophy | |
| Healthyb | 85 |
| Health problems | 10 |
| Not applicable | 5 |
Most often, 1 parent accompanied the child for visits. Mothers were much more likely to have flexibility in schedule than fathers.
Questionnaires administered during outpatient visits when children were well.
Table 2 shows genetic counseling experiences and demographics. Neurologists conducted 49% of genetic counseling. However, neurologists were perceived as having the poorest understanding of what families wanted compared with other genetic counseling providers (P = .021), shown in Table 4. Somewhat surprisingly, parents found that genetic counselors were the least helpful in expanding their understanding of spinal muscular atrophy, but there was no statistical difference, with a P value of .068 among the professionals shown in Table 3.
Table 2.
Genetic Counseling Experiences and Demographics
| Topics | % |
|---|---|
| Parents received genetic counselinga | 92 |
| Timing of genetic counseling | |
| After birth/before diagnosis | 1 |
| Coincident with diagnosis | 22 |
| After diagnosis | 68 |
| No counseling | 8 |
| Professional conducting genetic counseling | |
| Geneticist | 18 |
| Genetic counselor | 28 |
| Neurologist | 46 |
| Nurse | 1.5 |
| Other | 1.5 |
| Geneticist + neurologist | 1.5 |
| Genetic counselor + neurologist | 1.5 |
| Genetic counselor + other | 1.5 |
| Perceived need for genetic counseling | 89 |
| Parents who pursued carrier testing | 37 |
| Parents who pursued carrier testing in nonsymptomatic children | 16 |
| Demographics of other relatives who received carrier testing | |
| Siblings of parents | 12 |
| Otherb | 12 |
According to the definition provided in the questionnaire.
In-laws.
Table 4.
Parents’ Perceptions Regarding Health Care Provider Understanding of Wants
| Health Care Professional | Helpful | %a | Not Helpful | No Answer | Total |
|---|---|---|---|---|---|
| Geneticist | 11 | 92 | 0 | 1 | 12 |
| Genetic counselor | 17 | 89 | 2 | 0 | 19 |
| Neurologist | 21 | 68 | 10 | 0 | 31 |
| Nurse | 1 | 100 | 0 | 0 | 1 |
| Other | 1 | 100 | 0 | 0 | 1 |
| Geneticist + neurologist | 1 | 100 | 0 | 0 | 1 |
| Genetic counselor + neurologist | 1 | 100 | 0 | 0 | 1 |
| Genetic counselor + other | 1 | 100 | 0 | 0 | 1 |
| Geneticist + genetic counselor + neurologist | 1 | 100 | 0 | 0 | 1 |
| Total | 55 | 12 | 1 | 68 |
Percentages represent total of each profession, not total of health care professionals.
Table 3.
Parents’ Perceptions Regarding Helpfulness of Health Care Provider
| Health Care Professional | Helpful | %a | Not Helpful | No Answer | Total |
|---|---|---|---|---|---|
| Geneticist | 10 | 83 | 1 | 1 | 12 |
| Genetic counselor | 13 | 68 | 6 | 0 | 19 |
| Neurologist | 28 | 90 | 3 | 0 | 31 |
| Nurse | 1 | 100 | 0 | 0 | 1 |
| Other | 1 | 100 | 0 | 0 | 1 |
| Geneticist + neurologist | 1 | 100 | 0 | 0 | 1 |
| Genetic counselor + neurologist | 1 | 100 | 0 | 0 | 1 |
| Genetic counselor + other | 1 | 100 | 0 | 0 | 1 |
| Geneticist + genetic counselor + neurologist | 1 | 100 | 0 | 0 | 1 |
| Total | 68 |
Percentages represent total of each profession, not total of health care professionals.
With regard to genetic knowledge and analysis, 4 questions required a correct response. From these questions, genetic knowledge was assessed. Table 5 shows genetic knowledge in parents of children with spinal muscular atrophy. Most parents were aware of the potential effect of SMN2 on disease severity. Parents were generally accurate in their perception of recurrence risk but may not have completely understood what constitutes affected genotypes. Most parents reported that knowing about the genetics of spinal muscular atrophy helped them understand the disease. A parent’s decision to have carrier testing was not dependent on genetic knowledge, as shown by answers to these questions. Families receiving counseling had a greater percentage of perfect scores, but the overall distribution was no different between families (P = .206). The level of knowledge was the same for men and women within a couple. There was no strong relationship between the time passed between genetic counseling and administration of the survey or the number of answers correctly given.
Table 5.
Genetic Knowledge of Parents of Children Affected With Spinal Muscular Atrophy (SMA)
| Topics | % |
|---|---|
| Parents who identify causative gene as SMN1 | 70 |
| Parents who know SMN is on a chromosome | 85 |
| Parents who identify affected genotype | 56 |
| Parents who identify carrier genotype | 73 |
| Parents who identify recurrence risk | 93 |
| Parents who think genetic information is helpful | 88 |
| Parents who understand that a negative carrier test does not entirely rule out SMA recurrence | 59 |
| Parents who believe SMA can occur when both parents are carriers | 99 |
| Parents who believe SMA can occur when 1 parent is a carrier | 27 |
| Parents who believe SMA can occur with 0 carriers | 68 |
| Parents who infer a second child with SMA will have the same type as the firsta | 63 |
| Parents who perceive that SMN2 copy number predicts severity | 77 |
| Parents who know their child’s SMN2 copy number | 51 |
Data refer only to type 2 families.
Table 6 shows parents’ perceptions of spinal muscular atrophy. Parents were clearly unsure of what is most predictive of the future level of disability. All 3 answers with regard to future level of disability received relatively equal responses. A significant percentage of parents rated the current level of disability as being equal in importance to SMN2 copy number in determining their child’s future level of disability. This would seem to indicate that parents overestimate the importance of SMN2 in providing additional information in the setting of an already established phenotype. With regard to future therapies, most parents idealized stem cell research and underestimated the impact of currently available therapies on their child’s health and future prognosis.
Table 6.
Parents’ Perceptions About Spinal Muscular Atrophy (SMA)
| % | |
|---|---|
| Factors considered most predictive of child’s future disability | |
| SMA type | 22 |
| Current disability | 38 |
| SMN2 copy number | 38 |
| No answer | 1 |
| Parents’ perceptions regarding most important future therapy | |
| Gene therapy | 22 |
| Stem cell therapy | 56 |
| Medication | 12 |
| Physical therapy | 10 |
Table 7 shows parents’ perceptions regarding reproductive decision making. In this population, most children with spinal muscular atrophy were the first symptomatic individuals identified in their families. Fifty-three percent of families were worried about having another child with the disease. Many families clearly choose to limit subsequent pregnancies once they have had a child with spinal muscular atrophy. Genetic counseling was only reported to be helpful in guiding such decisions in just over half of the respondents.
Table 7.
Parents’ Perceptions Regarding Reproductive Decision Making
| % | |
|---|---|
| Perception of 25% recurrence risk | |
| Risk considered high | 77 |
| Risk considered low | 11 |
| Risk considered information of no value | 12 |
| Parents who acknowledged benefit of genetic counseling in family planning | 56 |
| Parents who worry about possible recurrence | 53 |
| Parents who want another child with spinal muscular atrophy (SMA) | 3 |
| Parents who preplanned number of children prior to SMA | 81 |
| Parents who chose to limit subsequent pregnancies due to SMA | 53 |
| Subsequent pregnancies in which parents chose prenatal diagnosis | 21 |
| Prenatal diagnostic choice in those undergoing prenatal diagnosis | |
| Chorionic villus sampling (CVS) | 33 |
| Amniocentesis | 40 |
| Preimplanation genetic diagnosis (PGD) | 13 |
| CVS and PGD | 13 |
Table 8 shows parents’ perceptions about newborn screening for spinal muscular atrophy. The vast majority of parents were supportive of newborn screening for spinal muscular atrophy. Many parents indicated in retrospect that they wished their child had been diagnosed sooner.
Table 8.
Parents’ Perceptions About Newborn Screening for Spinal Muscular Atrophy (SMA)
| Topics | % |
|---|---|
| Parents who are aware of newborn screening (NBS) | 96 |
| Parents who believe SMA testing would be helpful if included in NBS | 93 |
| Parents who would choose to pursue diagnostic testing in unaffected children | 68 |
| Parents who would choose NBS if symptom onset delayed 6 months | 92 |
| Parents who would choose NBS if symptom onset delayed 10 years | 77 |
Qualitative Section
The last 5 questions of the questionnaire were qualitative in nature, addressing perceptions of genetic diagnosis and genetic counseling and assessing how parents might proceed differently if spinal muscular atrophy were diagnosed in a subsequent child. A last question provided an opportunity for open-ended advice to health care professionals. Coding each response generated themes regarding parents’ opinions. Parents seemed unable to distinguish between genetic diagnosis and genetic counseling when answering the qualitative questions; thus, we combined the first 2 questions for analysis.
Genetic diagnosis and genetic counseling were perceived to help families connect with resources, understand and help their child, better understand the nature of the disease, feel empowered, and make decisions regarding family planning. Some families thought it helped them inform extended family and absolved guilt. In families who perceived that genetic diagnosis and genetic counseling were not helpful, it was deemed too late to make a difference, their interactions with health care professionals were negative, the material was inadequate, and the proximity of counseling to the initial diagnosis was emotionally overwhelming. Some families also reported not having or needing genetic diagnosis or counseling.
Families had mixed perceptions about having another child with spinal muscular atrophy. They understood the statistical likelihood that they would have another child with the same type. They reported they would be better prepared emotionally and more proactive in seeking medical interventions for the next child. Other major themes included concerns regarding quality of life, emotional reactions to a recurrence, family planning, and acceptance of recurrence. Some parents indicated they would pursue an altered parenting style should they have a second child with the disease.
If families already had more than 1 child with spinal muscular atrophy, they were asked what choices they had made differently with the second child. Some families reported a proactive approach to diagnosis and preventive care; however, other families reported no change or simple acceptance. Advice for health care professionals included the request that they become better informed about spinal muscular atrophy and provide more proactive treatment interventions when children are ill. Parents reported needing more information on treatment options and wanted more research on spinal muscular atrophy. Parents emphasized a desire for an early diagnosis and more effective proven treatment interventions and, ultimately, a cure. Parents requested more resources, more support and direction, and clear explanations of medical options. Parents indicated a desire that their choices and opinions be respected by health care professionals. In addition, they asked that health care professionals be more compassionate when presenting the initial diagnosis to families and emphasize the positive quality of life possible. An additional theme included a need for increased community awareness.
Discussion
Most families in our study received some type of genetic counseling. Yet how and from whom they received the information varied greatly. Families that reported receiving counseling had a greater percentage of perfect scores with regard to assessment of genetic knowledge, but the overall distribution was no different between families. This suggests families that acquire genetic knowledge from support group sites, other Internet sources, or personal study have comparatively the same genetic knowledge as those that receive genetic counseling from their health care professional. Genetic information about spinal muscular atrophy is readily available on the Internet, but the quality of information varies greatly with the source. Our survey indicates that most families receive genetic counseling solely from their child’s neurologist, who may not be appropriately prepared to provide formal genetic counseling. This is problematic because neurologists were also perceived as having the poorest understanding of parents’ needs. Because receiving an initial diagnosis is often an emotionally traumatic event for families, limiting genetic information at the time of the diagnostic visit and focusing instead on a compassionate and supportive message for parents may be more beneficial for families.
Surprisingly, genetic counselors were perceived the least helpful health care professionals, for reasons that were unclear. This may primarily be because many parents perceive that formal genetic counseling is only needed in the setting of prenatal testing. Many families choose to forgo having additional children and, in doing so, do not see a need to meet with a genetic counselor. The fact that many families choose not to pursue formal genetic counseling may explain why very few extended family members have received carrier testing. The low number of relatives reported to have received carrier testing implies that many extended family members are not receiving genetic counseling, which could be helpful to them. Genetic counselors generally discuss carrier testing much more extensively than neurologists, whose focus is primarily on diagnosis and management of the disease in their patient. Our data support the premise that parents are interested and willing to pursue formal genetic counseling given the opportunity, with potential benefit to themselves as well as their extended family.
Many respondents had a negative experience with genetic counseling, possibly because most receive counseling at the time of diagnosis or shortly afterward. Parents are often overwhelmed at the time of the initial diagnosis and may not have adequately processed the information they received. Alternatively, they may not have received appropriate or complete genetic counseling regarding the complex genetics of spinal muscular atrophy. At the time of diagnosis, they are much more likely to be focused on the present medical needs of their child and not future pregnancies. It may be helpful to identify the most appropriate time for addressing genetic counseling issues.
Because of the variety of health professionals conducting genetic counseling, their variable genetic knowledge, and where such counseling was received, it is clear parents did not receive consistent counseling. Inconsistent counseling may lead to confusion between parents as well as variable genetic knowledge. This may affect parental decision making for family planning and health care management.
Many parents automatically assume they are carriers, which may or may not reflect true recurrence risk. This is likely because of insufficient counseling on this issue. The possibilities of false negatives and spontaneous mutations should ideally be addressed within a counseling session. However, in a time-limited setting, discussion of such concepts can lead to significant confusion.
Parents clearly appreciate professionals who educate themselves when they initially have little knowledge concerning a specific disorder. Parents report a desire for their health care professional to focus on all aspects of the child’s care. They want to be provided with additional resources they can pursue on their own. In addition, parents stressed the need for a message of hope despite the terrible realities of spinal muscular atrophy. Most families indicate that presently, they view stem cell therapy as their greatest hope for a cure for their child’s disease. Health care professionals could greatly benefit families by refocusing them on present treatment interventions, such as supportive nutritional and respiratory therapies that could greatly influence their child’s future health and well-being.
The parents in this study were extremely supportive of newborn screening for spinal muscular atrophy, even in the event that their child might receive a diagnosis many years before the onset of symptoms. However, this is a special population clearly in favor of participating in research studies. Thus, they may have more proactive views than a general population of parents of children with spinal muscular atrophy. The issue of the occasional patient with type 3 spinal muscular atrophy who may present years before the onset of symptoms needs to be discussed in more detail as we consider newborn screening of spinal muscular atrophy. The large majority of parents with children with type 3 spinal muscular atrophy believe there would be a benefit to early diagnosis because it would give them more time to adjust to having a child with the disease before having to make crucial life decisions about health care management.
Because of the inherent characteristics of the study design, limitations are expected. The data were self-reported by respondents. The study was retrospective and nonrandom. Order effects may have been present in the questionnaire. The possibility of selection bias exists. The inability to probe all responses is also limiting. By virtue of this population participating in an existing research study, the participants may be more likely to elect participation in additional research studies. We would expect that these participants may be better informed than those encountered in assessment in clinical practice. Despite these limitations, the population sampled was diverse in geographic location, adding to the strength of this study. In addition, the number of participants helped overcome some of the limitations.
Conclusions and Recommendations
It would be helpful to develop a consistent paradigm by which families receive genetic counseling. An example of one such paradigm is illustrated in Figure 2. During the first 2 visits to the neurologist, the primary focus remains on the diagnosis and management of the child, but a brief introduction to the genetics of spinal muscular atrophy could also be provided. The benefit of seeking a separate appointment for a more detailed discussion of genetics could be introduced at this time; the family may or may not be ready to pursue scheduling for such a visit. At each subsequent visit, reassessment of genetic knowledge and referral for formal genetic counseling should be offered. Ideally, a genetic counselor would be available at the next clinical visit to formally meet with the family. Currently, most neurology departments do not have a genetic counselor available for such services. However, some neurology specialty or multi-disciplinary clinics have begun to incorporate such services into the clinical setting. Aside from prenatal testing, accessing formal genetic counseling services can be problematic. Most genetic counselors are employed by perinatal or pediatric genetics clinics, thus limiting access to patients with spinal muscular atrophy who are typically followed in the neurology clinic. Neurologists may benefit from increasing collaboration with genetics colleagues to provide their patients with more access for formal genetic counseling or to contract genetic counselors for specific indications.
Figure 2.
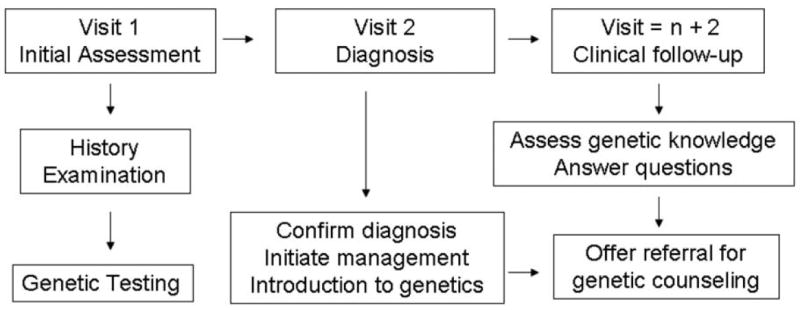
Incorporating genetic counseling into the current health care paradigm. Visits pertain to neurologist consultation.
More emphasis on outreach to relatives is needed, particularly in light of the relatively high carrier status in the general population. Although privacy concerns and regulations prevent direct contact of extended family members concerning genetic carrier testing, health care professionals should put an increased emphasis on the value of carrier testing for adults of reproductive age and try to facilitate opportunities for relatives to receive such testing.
Health care professionals ideally need to supplement or improve upon existing information to maximize the benefit of genetic counseling for families. Many families may not have the time or ability to easily access Internet-related resources. Therefore, providing written materials during the counseling process and periodically verbally reassessing genetic knowledge during subsequent clinical visits would improve significantly on current practice.
If newborn screening for spinal muscular atrophy becomes available, parents will need anticipatory guidance and proactive management strategies to maximize health benefit. The more widespread implementation of standards of care management will greatly facilitate this process, but the incorporation of neurologists into the current newborn screening algorithms for care and monitoring of symptomatic as well as presymptomatic newborns and infants will clearly be needed.
Figure 1.
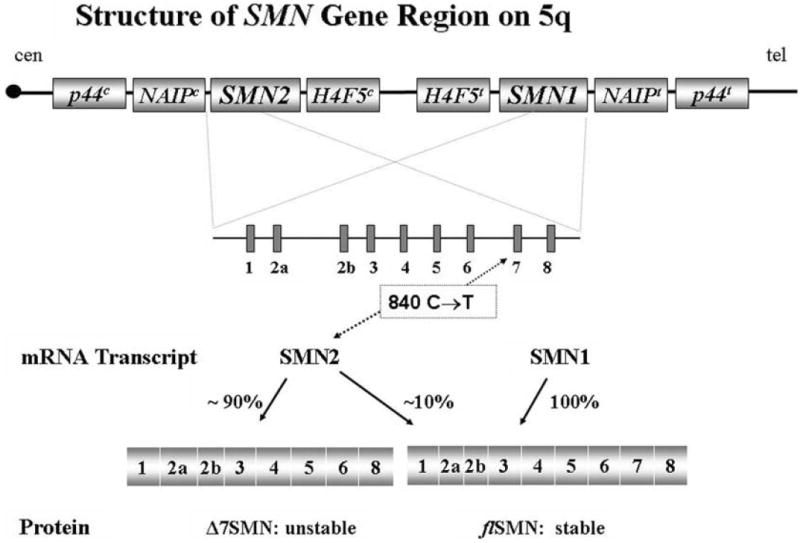
SMN genes and transcripts. SMN2 is alternatively spliced, creating a 90% unstable transcript lacking exon 7. Adapted from a drawing by Livija Medne, MS.
Acknowledgments
We acknowledge funding support from Families of Spinal Muscular Atrophy. This investigation was also supported in part by the Department of Health and Human Services research grant M01-RR00064 from the National Center for Research Resources. Additional funding was provided by the Muscular Dystrophy America and by the Spinal Muscular Atrophy and American Academy of Neurology Foundations (Young Investigator Award). We thank the families and patients for their assistance and observations. We also gratefully acknowledge support from Nicola Longo, MD, PhD, Brent Hafen, MS, Janalee Hobson, MS, Livija Medne, MS, and Bonnie J. Baty, MS.
Footnotes
Presented at the Neurobiology of Disease in Children: Symposium on Spinal Muscular Atrophy, in conjunction with the 35th annual meeting of the Child Neurology Society, Pittsburgh, Pennsylvania, October 18-21, 2006. The work was completed at the University of Utah General Clinical Research Center, Primary Children’s Medical Center outpatient clinics, Salt Lake City, Utah, and at the University of Utah School of Medicine.
References
- 1.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Burglen L, Lefebvre S, Clermont O, et al. Structure and organization of the human survival motor neuron (SMN) gene. Genomics. 1996;32:479–482. doi: 10.1006/geno.1996.0147. [DOI] [PubMed] [Google Scholar]
- 3.Gennarelli M, Lucarelli M, Capon F, et al. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 4.Helmken C, Hofmann Y, Schoenen F, et al. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114:11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 5.Wirth B, Brichta L, Schrank B, et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006;119:422–428. doi: 10.1007/s00439-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 6.Prior TW, Swoboda KJ, Scott HD, et al. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet. 2004;130A:307–310. doi: 10.1002/ajmg.a.30251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA) Hum Genet. 2002;111:477–500. doi: 10.1007/s00439-002-0828-x. [DOI] [PubMed] [Google Scholar]
- 9.Swoboda K, Prior T, Scott C, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 11.Zerres K, Rudnik-Schoneborn S, Forrest E. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146:67–72. doi: 10.1016/s0022-510x(96)00284-5. [DOI] [PubMed] [Google Scholar]