

Abstract
Sleep disordered breathing (SDB), which is characterized by intermittent hypoxia (IH) during sleep, causes substantial cardiovascular and neurocognitive complications and has become a growing public health problem. SDB is associated with suppression of growth hormone (GH) secretion, the latter being integrally involved in the growth, development and function of the central nervous system (CNS). Since GH treatment is able to attenuate neurocognitive deficits in a hypoxic-ischemic stroke model, GH, GH receptor (GHR) mRNA expression and GH protein expression were assessed in rat hippocampus after exposures to chronic sustained hypoxia (CH, 10% O2) or intermittent hypoxia (IH, 10% O2 alternating with 21% O2 every 90 sec). In addition, the effect of GH treatment (50 µg/kg, daily s.c. injection) on EPO, VEGF, HO-1 and GLUT-1 mRNA expression and neurobehavioral function was assessed. CH significantly increased GH mRNA and protein expression, as well as IGF-1. In contrast, IH only induced a moderate increase in GH mRNA and a slight elevation in GH protein at day 1, but no increases in IGF-1. CH, but not IH, up-regulated GHR mRNA in the hippocampus. IH induced marked neurocognitive deficits compared to CH or room air (RA). Furthermore, exogenous GH administration increased hippocampal mRNA expression of IGF-1, EPO and VEGF, and not only reduced IH-induced hippocampal injury, but also attenuated IH-induced cognitive deficits. Thus, exogenous GH may provide a viable therapeutic intervention to protect IH-vulnerable brain regions from SDB-associated neuronal loss and associated neurocognitive dysfunction.
Keywords: Sleep disordered breathing, growth hormone, neurocognitive dysfunction
Introduction
Sleep disordered breathing (SDB) has become a prominent and steadily increasing public health problem. In the last 2 decades, SDB has been associated with a variety of morbid consequences, among which the metabolic syndrome, systemic and pulmonary hypertension, ischemic heart disease, cerebrovascular disease, erectile dysfunction, mood disorders, and neurocognitive and behavioral problems have emerged (Mokhlesi and Gozal, 2010;Dempsey et al., 2010). SDB, is a condition characterized by abnormalities in ventilatory homeostasis during sleep that affects all age groups from premature infants, throughout childhood and adolescence and of course middle aged and aged populations. The prevalence revolves around 2% to 4–5% among the various age groups, with major increase in aged populations to up to 15%. Obstructive sleep apnea (OSA), the one of most common form SDB, is characterized by the repetitive collapse of the pharyngeal airway during sleep, sleep fragmentation, intermittent hypoxia, hypercapnia and increased intrathoracic pressure swings (Dempsey et al., 2010). The frequency of IH in clinical settings far exceeds that of sustained chronic hypoxia (CH) which typically occurs during high altitude sojourns. A decade or so ago, we reported that sustained exposures to IH, in the absence of significant sleep deprivation, induces substantial neurocognitive impairments in both adult and developing rodents (Gozal et al., 2001). Such functional alterations were accompanied by evidence of increased oxidative stress, induction and propagation of inflammatory processes, and consequent neuronal cell losses via induction of apoptotic mechanisms in selected brain regions such as the frontal cortex and the CA1 region of the hippocampus (Beebe and Gozal, 2002;Wang et al., 2010). These now repeatedly confirmed findings lend support the notion that IH plays a critical role in SDB-associated neurocognitive deficits (Gozal et al., 2003b;Li et al., 2003;Row et al., 2003;Goldbart et al., 2003;Xu et al., 2004;Li et al., 2004;Row et al., 2004;Kheirandish et al., 2005b;Shan et al., 2007;Douglas et al., 2007;Kheirandish et al., 2005a;Perry et al., 2008;Burckhardt et al., 2008;Hambrecht et al., 2009;Row et al., 2002a;Xie et al., 2010;Hui-guo et al., 2010;Cai et al., 2010;Ward et al., 2009). However, the mechanisms underlying IH-induced neurocognitive deficits remain largely unknown. Furthermore, we are unaware of studies assessing the effects of normobaric CH on cognition.
Growth hormone (GH) is integrally involved in the growth, development and function of the central nervous system (CNS) (van Dam and Aleman, 2004;van Dam et al., 2000;Sartorio et al., 1996;Barta et al., 1981). The expression and localization of growth hormone receptor (GHR) has been characterized in the brain (Lobie et al., 1993), and is altered following brain injury (Scheepens et al., 1999). In addition to a preponderant role of GH in stimulating the postnatal growth of different tissues, GH is also involved in the functional integrity of the CNS in general, and neuronal function in particular, thereby modulating memory and cognitive functions (van Dam and Aleman, 2004;van Dam et al., 2000;Sartorio et al., 1996). Furthermore, GH may localize to the mitochondria where it may serve to regulate cellular metabolism and free radical production (Ardail et al., 2010). Under hypoxic conditions, brain produces neurotrophic factors as an endogenous neuroprotective strategy to mitigate neuronal injury (Shin et al., 2004;Zayour et al., 2003;Zhang and Du, 2000;Xie et al., 2010). In addition, expression of the IGF-I (insulin-growth factor 1) receptor was also up-regulated after hypoxic-ischemic injury (Scheepens et al., 1999;Scheepens et al., 2000), suggesting that IGF-1 is a responsive member of the somatotropic axis, and plays a role in mediating GH functionality in brain. Several studies have demonstrated that GH treatment not only provides some degree of neuroprotection (Scheepens et al., 2001;Gustafson et al., 1999;Scheepens et al., 1999), but also improves neurocognitive outcome following hypoxic-ischemic brain injury (Zhong et al., 2009) and attenuates apoptosis (Shin et al., 2004). IGF-I effectively reduced brain injury in rats subjected to hypoxia-ischemia insults (Gustafson et al., 1999;Liu et al., 2001), suggesting that IGF-1, a downstream component of GH-activated pathways, may afford protection per se. Furthermore, the protective role of GH/GHR in hypoxic injury may be associated with known downstream protective hypoxia-responsive genes, such as EPO, VEGF, HO-1, and GLUT-1. Indeed, the expression of these 4 genes is tightly regulated in the CNS by HIF-1α (Freeman and Barone, 2005;Acker and Acker, 2004), and plays a major protective role in hypoxia and ischemia-induced brain injury (Sharp et al., 2004;Fan et al., 2009). SDB is associated with suppression of growth hormone (GH) secretion (Meston et al., 2003;Cooper et al., 1995;Saini et al., 1993), the presence or absence of cognitive deficits is dependent on the serum levels of IGF-1 at any given level of severity of SDB, and in rodents, physical activity that stimulates IGF-1 is neuroprotective, suggesting that GH suppression may play a role in SDB-associated neurocognitive deficits (Gozal et al., 2009;Gozal et al., 2010). Based on these considerations, the aims of present study were to determine whether similar to IH, CH is associated with spatial learning and memory deficits, and whether exogenous systemic administration of GH will protect the brain from IH-induced neuronal apoptosis and consequent functional deficits.
Experimental Procedures
Hypoxic Exposures
A schematic diagram of all experimental procedures is shown in Figure 1. Male Sprague Dawley rats (175–225 grams, 2 month-old) were purchased from Charles River and used for all experiments. The experimental protocols were approved by the Institutional Animal Use and Care Committee at the University of Louisville, and are in agreement with the NIH guide for the care and use of laboratory animals. All efforts were made to minimize animal suffering. Animals were randomly assigned to 3 experimental groups consisting of (i) chronic sustained hypoxia (CH), 10% O2 for 12 hours during daylight phase leading to nadir oxyhemoglobin levels (SaO2) of ~72–75%; (ii) intermittent hypoxia (IH), alternating 21% O2 and 10% O2 every 90 sec for 12 hours during daylight leading to similar nadir SaO2 levels, and (iii) time-matched normoxic exposures control (RA). Animals were maintained in 4 identical commercially designed chambers (Biospherix, Redfield, NY) operated under a 12 hour light-dark cycle (7:00 am–7:00 pm). Oxygen concentration was continuously measured by an O2 analyzer, and the desired gas profile was administered by a computer controlled system regulating gas outlets. Ambient temperature was kept at 22–24°C.
Figure 1. Experimental scheme for hypoxia exposures and behavioral testing.
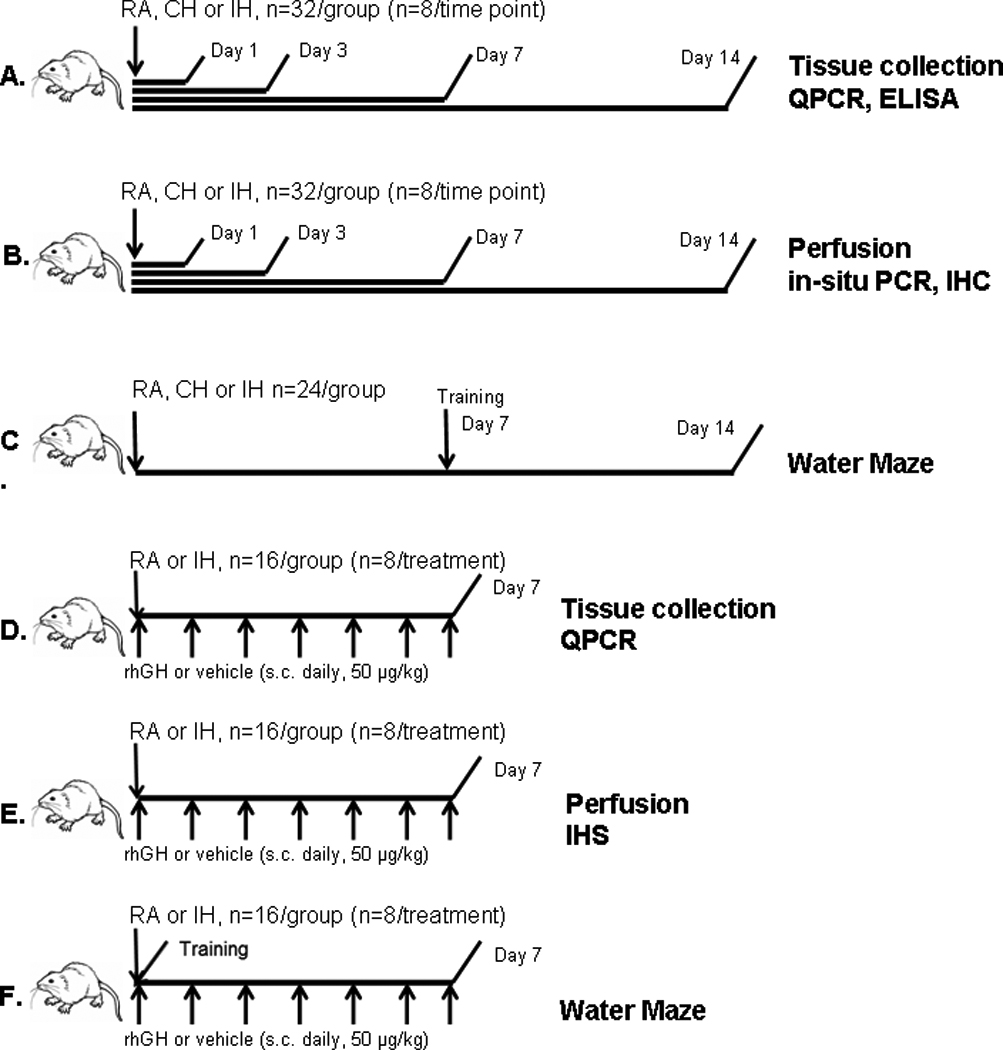
A. Rats were assigned to the designated experimental profiles (IH, CH and RA) for 1, 3, 7 and 14 days. After completion of the respective exposures, rats were anesthetized with pentobarbital (50 mg/kg), and brains were rapidly harvested. The hippocampal tissues were dissected, snap frozen in liquid nitrogen and kept at −80°C. The collected tissue was subjected to ELISA and qPCR analysis. B. After rats were exposed to either IH, CH or RA for 1, 3, 7 and 14 days, rats were deeply anesthetized and perfused intracardially with phosphate-buffered saline (pH 7.4) followed by 4% phosphate-buffered paraformaldehyde. Serial cryosections (7 µm) were cut and stored at −20 °C. The hippocampal sections were subjected to immunohistochemistry and in-situ PCR analysis. C. Rats were exposed to the designated experimental profiles (IH, CH and RA) for 7 days, after which animals underwent water maze training and assessment for an additional 7 days, during which exposures were continued till completion at day 14. D. Rats were exposed to IH or RA and received daily s.c. injections of rhGH (50 µg/kg body weight, Genotropin, Pharmacia & Upjohn, Kalamazoo, MI) or vehicle for 7 days, after which rats were sacrificed, and hippocampal tissues were collected and subject to QPCR analyses. E. Rats were exposed to IH or RA and received daily s.c. injections of rhGH (50 µg/kg body weight) or vehicle for 7 days, after which rats were deeply anesthetized and perfused intracardially with 4 % phosphate-buffered paraformaldehyde. Serial cryosections (7 µm) were cut and stored at −20 °C for immunohistochemistry analysis. F. Rats were exposed to IH or RA and received daily s.c. injections of rhGH (50 µg/kg body weight) or vehicle for 7 days, and underwent water maze training and assessment during that period.
For hypoxic exposure experiments, animals were assigned to the designated experimental profile (IH, CH and RA) for 1, 3, 7 and 14 days. After completion of the respective exposures, animals (n=8/group) were anesthetized with pentobarbital (50 mg/kg), brains were rapidly harvested, and hippocampal tissues were dissected at 4°C, snap frozen in liquid nitrogen, and kept at −80°C until analysis. For histological assessments, rats (n=8/group) were deeply anesthetized and perfused intracardially with phosphate-buffered saline (pH 7.4) followed by 4% phosphate-buffered paraformaldehyde. Serial sections (7 µm) were cut stored at 4°C until use. For the behavioral studies, animals were exposed to the designated experimental profiles (IH, CH and RA) for 14 days, after which animals underwent water maze training and assessment (Figure 1).
Growth hormone administration
For exogenous growth hormone experiments, animals were exposed to IH or RA and received daily s.c. injections of rhGH (50 µg/kg body weight, Genotropin, Pharmacia & Upjohn, Kalamazoo, MI) for 7 days. Rats were randomly assigned to 4 experimental groups (32/group) consisting of: (i) RA exposure and vehicle injection; (ii) RA exposure and GH injection; (iii) IH exposures and vehicle injection; (iiii) IH exposures and GH injection. At day 0, rats received either rhGH or vehicle injection 2 hours before starting RA or IH exposure. Thereafter, rats received daily GH or vehicle injections during the IH exposure phase. In a subset of rats after GH or vehicle treatment (n=8/group), deep anesthesia was induced with pentobarbital (50 mg/kg), brains were rapidly removed, and hippocampal tissues were dissected at 4°C, snap frozen in liquid nitrogen, and stored at −80°C for analysis of IGF-1, EPO, and other genes of interest. For immunohistochemistry assessment, rats (n=8/group) were deeply anesthetized and perfused intracardially with phosphate-buffered saline (pH 7.4) followed by 4% phosphate-buffered paraformaldehyde. Serial sections (40 µm) were cut and stored at 4°C until use. For the behavioral studies, animals (n=16/experimental group) were exposed to IH for 7 days and underwent water maze assessment.
Morris water maze
The Morris water maze was used for the neurobehavioral assessments. An escape platform (10 cm in diameter) was positioned 1 cm below the water surface. Distinctive, geometric, extramaze cues were affixed at specific locations, and were visible to the rats while in the maze. Maze performance was recorded by a video camera suspended above the maze and interfaced with a video tracking system (HVS Imaging, Hampton, UK). Albino rats used in the experiments were temporarily tattooed with a black mark to allow for video tracking.
Animals were handled twice per day 7 days prior to behavior testing to minimize potential bias related to experimenter-induced stress. Animals were initially allowed to acclimate by a 30 s swim in the maze in the absence of the platform and spatial cues prior to behavior testing. They were then given two daily training sessions consisting of eight and four training trials, respectively over a 2-day period. In each training trial, the rat was placed in the maze from a quasirandom start point and allowed a maximum of 90 sec to find the escape platform, where it remained for 15 sec. Rats that failed to locate the platform at the end of 90 sec were manually guided to the platform. The position of the platform remained constant. Training trials were separated by 240 sec. Thirty minutes after completion of the final training session, a 30 sec probe trial, in which the platform was removed, was conducted and the relative proximity of the rat to the target location was used in order to assess spatial bias. After the probe trial, the presence of sensorimotor disturbances was assessed on three cued trials. Each rat was placed into the pool from quasirandom start points and allowed a maximum of 30 sec to escape to a readily visible platform elevated 1 cm above the water surface. The swim distances (pathlengths) derived from the reference memory task (averaged over blocks of four trials) were analyzed using repeated measures analyses of variance (ANOVA) followed by post-hoc tests. The relative average proximity of the rat to the target platform location during the probe trials was analyzed using one-way ANOVA.
Quantitative PCR
Total RNA was prepared from hippocampal tissue samples using TRIzol reagent (Invitrogene) following the manufacturer's instructions. Isolated total RNA was quantified spectrophotometrically. Aliquots of total RNA (1 µg) were reverse transcribed using Superscript II-Reverse Transcriptase (Invitrogene, Carlsbad, CA) according to the manufacturer's protocol. cDNA equivalent to 20 ng of total RNA were subjected to real-time PCR analysis (MX4000, Stratagene, La Jolla, CA) following the manufacturer's protocol. PCR Primers and Taqman probes for GH (Rn01495894_g1), GHR (Rn00567298_m1), IGF-1(Rn00710306_m1), EPO (Rn00566529_m1), VEGF(Rn01511601_m1), HO-1(Rn01536933_m1) and Glut1(Rn01417099_m1) were purchased from ABI (Applied Biosystems) Each reaction (25µl) contained 2.5 µl reaction buffer (10×), 6 mM MgCl2, 0.2 µM dNTP, 0.6 µM each primer, 0.25 µl SureStar Taq DNA Polymerase and 2 µl cDNA dilutions. The cycling condition consisted of 1 cycle at 95°C for 10 min and 40 three-segment cycles (95°C for 30 s, 55°C for 60 s and 72°C for 30 s). Standard curves for gene of interest and housekeeping gene (β-actin) were included in each reaction. We found that the mRNA expression of β-actin was stable and unchanged after either IH or CH exposures. Real-time PCR results were analyzed using MX4000 software (Stratagene, La Jolla, CA).
GH enzyme immunoassay
Tissue concentrations of GH were determined using a commercially available enzyme immunoassay kit (ALPCO Diagnostics, Windham, NH). Hippocampal tissues were homogenized in 0.01 M NaHCO3 and centrifuged at 40,000g for 20 minutes. The supernatant fraction was stored at −80°C until analysis. The protein concentration of tissue samples was determined by Bradford method. Tissue GH measurements were performed according to the manufacture instructions. Briefly, 50 µl of each samples were incubated in duplicate with rat GH antiserum for 20 hours at room temperature. After incubation, 50 µl of acetylcholinesterase (AChE) tracer was added to each well and incubated for another 20 hours. In the next day, samples were incubated with 200 µl of Ellman’s reagent for 30 minutes Finally, GH tissue concentration was determined by measuring optical density at 414 nm using a plate reader (Labsystems Multiskan RC, Helsinki, Finland).
Immunohistochemistry
Serial sections (40 µm) were cut on a microtome and stored at 4°C until use. The free floating sections were blocked with 10% normal goat serum and then incubated with primary cleaved caspase-3 rabbit antibody (1:100, Cell Signaling Technology, Beverly, MA) overnight at 4°C. The binding sites were further detected by goat anti-rabbit antibody conjugated with peroxidase (1:50, KPL,Gaithersburg, MD). Finally, the imunoreactivity was further stained with DAB peroxidase substrate (KPL Gaithersburg, MD). Immunostaining was finally visualized using a Nikon Ellipse E800 microscope (Nikon USA, Melville, NY) and images were captured by a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
In situ RT PCR
Primers and probes: Specific 25-mers oligonucleotide primers, were used after further HPLC-purification (Eurobio, Les Ulis, France). Rat GH sequences used as primers were directed toward selected regions of exons 2 and 5 of the rGH gene as previously described (Binder et al. 1994). The antisense primer (nt 135–160: 5'-CGCAAAGCGGCGACACTTCATGACC-3') is located at the end of the fifth exon and the sense primer (nt 643–668: 5'-CCAGTCTGTTTGCCAATGCTGTGCT-3') is in the second exon. For in situ hybridization, two 30-mers oligonucleotide probes located in the middle of the amplified fragment were synthetized (Eurobio). The sequences of sense probe (nt 407–437) was 5'-AACAGCCTGATGTTTGGTACCTCGGACCGC-3', and antisense probe (nt 248–278) was 5'-TGAGAAGCAGAACGCAGCCTGGGCATTCTG-3'. Probes were 3' end-labeled with digoxigenin-11-dUTP (Roche Diagnostics, Meylan, France).
Antibodies: For in situ RT-PCR revelation, goat anti-digoxigenin and rabbit anti-goat alkaline phosphatase conjugated were from Roche Diagnostics. The streptavidin-avidin-peroxidase complex procedure was employed with diaminobenzidine as chromogen (Dako, Trappes, France).
In situ RT-PCR was performed on 7 µm thick sections mounted on in situ RT-PCR glass slides (Applied Biosystem, Courtaboeuf, France) as described (Recher et al. 2001). Briefly, deparaffinized sections treated with proteinase K (Dako, Trappes, France) were dehydrated and air dried. First, 100 µL of in situ RT reaction mixture containing 50 mM Tris-HCl pH 8.3, 75 mM KCl, 10 mM dithiothreitol, 75 mM MgCl2, 1 mM of dNTP (Promega, Charbonnières, France), 1 µM antisense primer, 100 U RNase inhibitor (Promega), and 200 U M-MLV reverse transcriptase (Life Technologies, Cergy Pontoise, France) was prepared. The sections were then covered with 40 µL of the reaction mixture, sealed with amplicover discs and amplicover clips (Applied Biosystem), and incubated at 42°C for 1 hour. Amplicover discs and clips were removed and sections were washed in 100 mM phosphate buffer for 5 minutes at room temperature, dehydrated, and air-dried. Then, sections were covered with 40 µL of the in situ PCR reaction mixture containing 10 mM Tris/HCl, 50 mM KCl, 3 mM MgCl2, 0.5 mM of each dNTP (Promega), 1 µM of each primers and 20 U Taq DNA polymerase (Eurobio), sealed, and placed on a GeneAmp in situ PCR system 1000 thermal cycler (Perkin-Elmer, Courtaboeuf, France). The hotstart PCR method was used for 25 cycles according to the following program: denaturation at 94°C for 45 sec, annealing at 69°C for 45 sec, extension at 72°C for 1 minute, and final extension at 72°C for 7 minutes. Sections were then washed for 5 minutes in phosphate buffer, fixed 15 minutes in 4% paraformaldehyde at room temperature, rinsed in phosphate buffer for 5 minutes, dehydrated and air-dried. The sections were then hybridized overnight at 40°C with 30 pmoles of sense and antisense labeled probes/mL after 3 minutes denaturation at 94°C. Washing steps were performed sequentially for 1 hour in 2X SSC and 30 minutes in 1X SSC at room temperature and hybrids were revealed using phosphatase alkaline / NBT/BCIP system (Roche Diagnostics). Validation of the in situ RT-PCR reaction was performed as described (Recher et al. 2001) on human pituitary sections as positive control and human liver sections as negative control, and included omission of the reverse transcriptase and omission of the Taq DNA polymerase as supplementary controls. No counterstaining was performed. Qualitative assessment of the slides was performed by investigators who were blinded to the experimental condition. Representative sections of the overall findings for each experimental condition were selected by the blinded investigator (MR) for presentation, such as to qualitatively illustrate the findings.
Data Analysis
Data in text and figures are expressed as mean ± SE. Two group comparisons were evaluated by paired or unpaired t tests, as appropriate. Multiple comparisons were analyzed by ANOVA and Tukey’s or Newman Keuls post-hoc tests. Differences were considered statistically significant for P< 0.05.
Results
CH induces increased GH expression in hippocampus of rat
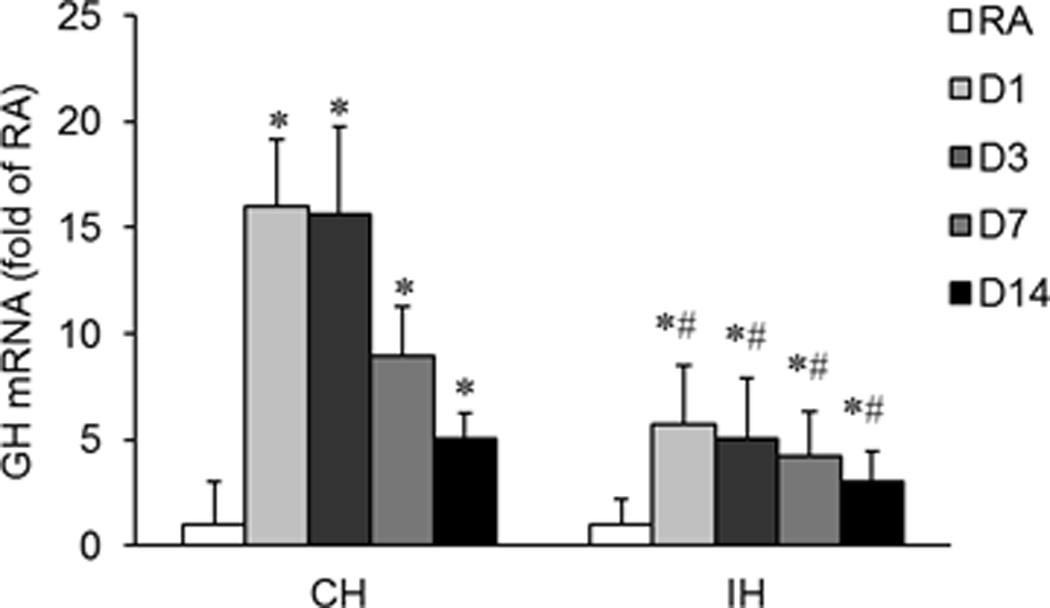
To measure the effect of CH or IH exposures on GH expression in hippocampus, rats were exposed to either CH or IH for 1, 3, 7 and 14 days (n=8/group). CH induced significant increases in GH mRNA expression in the hippocampus of rat brain at all-time points, peaking at day 1, and although decreasing, remained elevated at day 14 (Figure 2A, *P<0.01 vs, RA). In comparison, although GH mRNA expression was also increased in similar pattern after exposure to IH (*P <0.05 vs. RA), the magnitude of change was significantly smaller (#P <0.01 vs. CH). Furthermore, GH protein expression was increased throughout the duration of CH (Figure 2B, *P <0.01 vs. RA). In contrast, increased GH protein expression was found only at day 1 of IH exposures (*P <0.05 vs. RA), subsequently returning to baseline levels. Furthermore, GH protein expression changes during IH were significantly lower than in CH (#P <0.01 IH vs. CH).
Figure 2. Effects of CH or IH exposures on GH mRNA and protein expression in the hippocampus of rat.
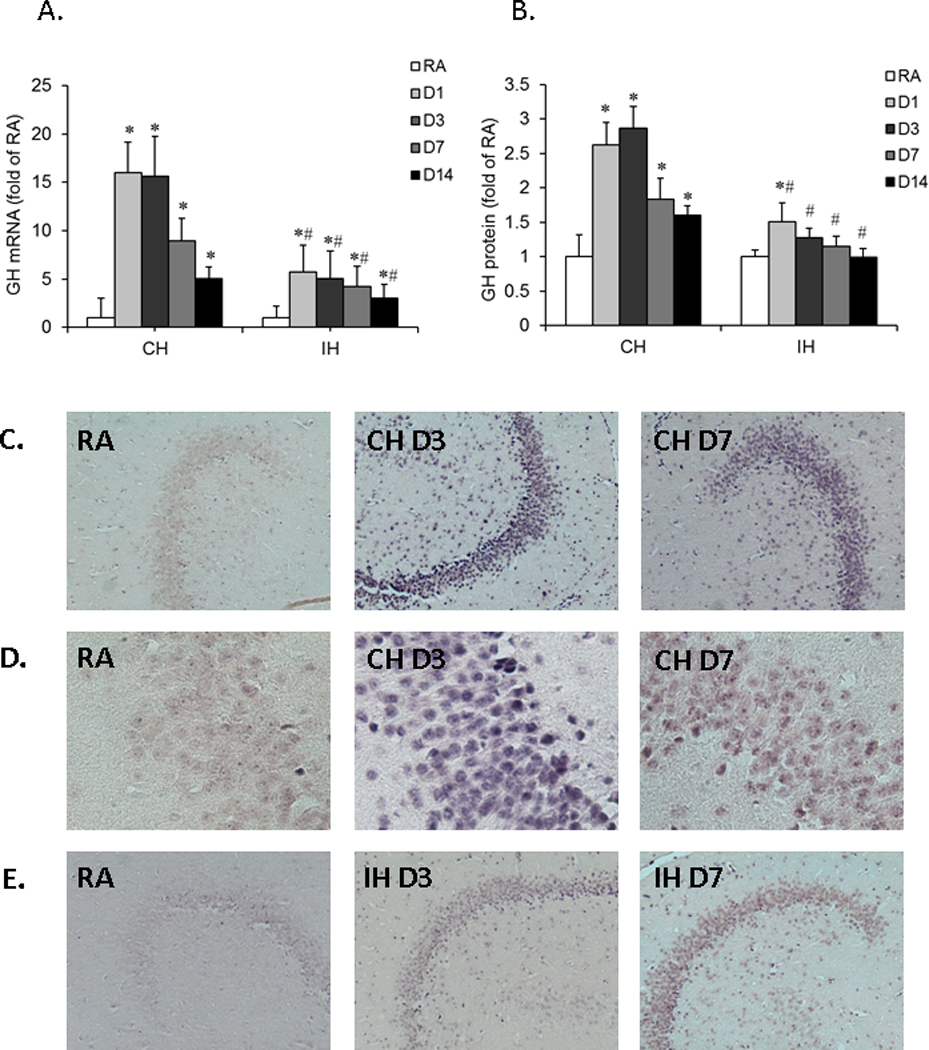
Rats were exposed to either CH or IH for 1, 3, 7 and 14 days (n=8/group). GH mRNA and protein expression were assessed by quantitative real-time RT-PCR or ELISA, respectively. GH mRNA expression was also examined by in-situ RT-PCR. A. Time course of GH mRNA expression in rat hippocampal tissues. Data are expressed as a fold change of RA (mean+SE). Real-time PCR analysis revealed that GH mRNA expression was significantly increased after either CH or IH exposures as compared to GH mRNA expression under RA conditions (*P<0.01 vs. RA, unpaired t tests). IH exposures induced lesser increases in GH mRNA expression as compared with CH (#P<0.01 IH vs. CH, unpaired t tests). B. Time course of GH protein expression in hippocampal tissues. Data are expressed as fold change of RA (mean+SE). GH protein expression was significantly increased after CH, but not after IH exposures (*P<0.01 IH vs. RA, unpaired t tests). C. In-situ RT-PCR analysis for GH mRNA in rat hippocampal sections after CH exposures. An intense positive staining for GH mRNA was detected in hippocampal sections after CH exposures for 3 and 7 days as compared to RA sections, as indicated by the arrow (Scale bar – 50 µ). D. Same as Figure 1C, but in higher magnification, as indicated by the arrow (Scale bar – 5 µm). E. In-situ RT-PCR analysis for GH mRNA in rat hippocampal sections after IH exposures. A slightly enhanced positive staining for GH mRNA was detected in IH sections at 3 and 7 days when compared to RA sections as shown by the arrow (Scale bar – 50 µm)
In a subsequent set of experiments, in-situ RT-PCR was used to further assess topological changes in GH within the hippocampal structure. After CH or IH exposures for 3 and 7 days, the hippocampal sections were prepared for in-situ RT-PCR. In the RA group, the staining for GH mRNA was very weak in the CA1 region of hippocampus, indicating that the basal level of GH mRNA is low. In comparison, enhanced GH mRNA staining was present in the CA1 region of the hippocampus after CH exposures for either 3 or 7 days (CH D3 and CH D7). Most remarkable, enhanced staining was found in the sections of CH at day 3 (Figure 2C). Furthermore, increased GH mRNA was also detected in the CA1 region of the hippocampus after IH exposures for 3 and 7 days, albeit with a markedly less intense pattern when compared CH (Figures 2D and 2E). No staining for GH mRNA was found in negative control sections that were devoid of Taq DNA polymerase (data not shown).
CH but not IH increases the expression of GH receptor
To determine whether hypoxic exposures will also alter expression of GH receptor (GHR) in hippocampus, mRNA expression of GHR was measured by quantitative real-time RT-PCR. After rats were exposed to either CH or IH for 1, 3, 7 and 14 days (n=8/group), mRNA from hippocampal tissues was subjected to real-time PCR analysis and revealed that CH exposures induced significant increases in GHR expression at days 1 and 3 (Figure 3, *P<0.05 vs. RA), and then gradually decreased after day 7 of CH. In contrast, IH exposures did not induce any changes in GHR expression throughout the duration of exposure (P>0.05 vs. RA). GHR mRNA expression after IH exposures was also significantly lower than the levels induced by CH at days 1 and 3 (#P<0.01 vs. CH). These results indicate that CH but not IH lead to increase in GHR expression in the hippocampus of rat brain.
Figure 3. Effects of CH or IH exposure on GHR mRNA expression in the hippocampus of rat.
Rats were exposed to either CH or IH for 1, 3, 7 and 14 days (n=8/group). GHR mRNA expression was assessed by quantitative real-time RT-PCR. Data are expressed as a fold change of RA (mean+SE). Real-time PCR analysis revealed that GHR mRNA expression was significantly increased after exposure to CH when compared to RA exposures (*P<0.05 vs. RA, unpaired t tests); however, such increases were not observed following IH (P>0.05 vs. RA, #P<0.05 vs IH, unpaired t tests).
CH induces increased expression of IGF-1, EPO, VEGF and HO-1 but not of GLUT1 in the rat hippocampus
To examine the effect of CH or IH exposures on the mRNA expression of IGF-1, EPO, VEGF and GLUT1 in hippocampal tissues, rats were exposed to CH or IH for 1, 3, 7 and 14 days (n=8/group). CH induced significant increases in IGF-1, EPO, VEGF and HO-1 mRNA expression at all-time points (Figure 4A–4D, *P<0.01 vs. RA). However, CH did not elicit significant changes in GLUT1 mRNA expression (Figure 4E, P>0.05 vs. RA). IH induced a slight increase in EPO mRNA expression at day 1 (*P<0.05 vs. RA) but not thereafter. Furthermore, IH did not elicit any significant changes in mRNA expression of IGF-1, VEGF, HO-1 and GLUT1 (P>0.05 vs. RA). These findings suggest that CH, but not IH, induces an array of protective gene responses that may mitigate hypoxia-induced injury.
Figure 4. Time course of EPO, VEGF, HO-1, IGF-1, and GLUT1 mRNA expression in rat hippocampal tissue.
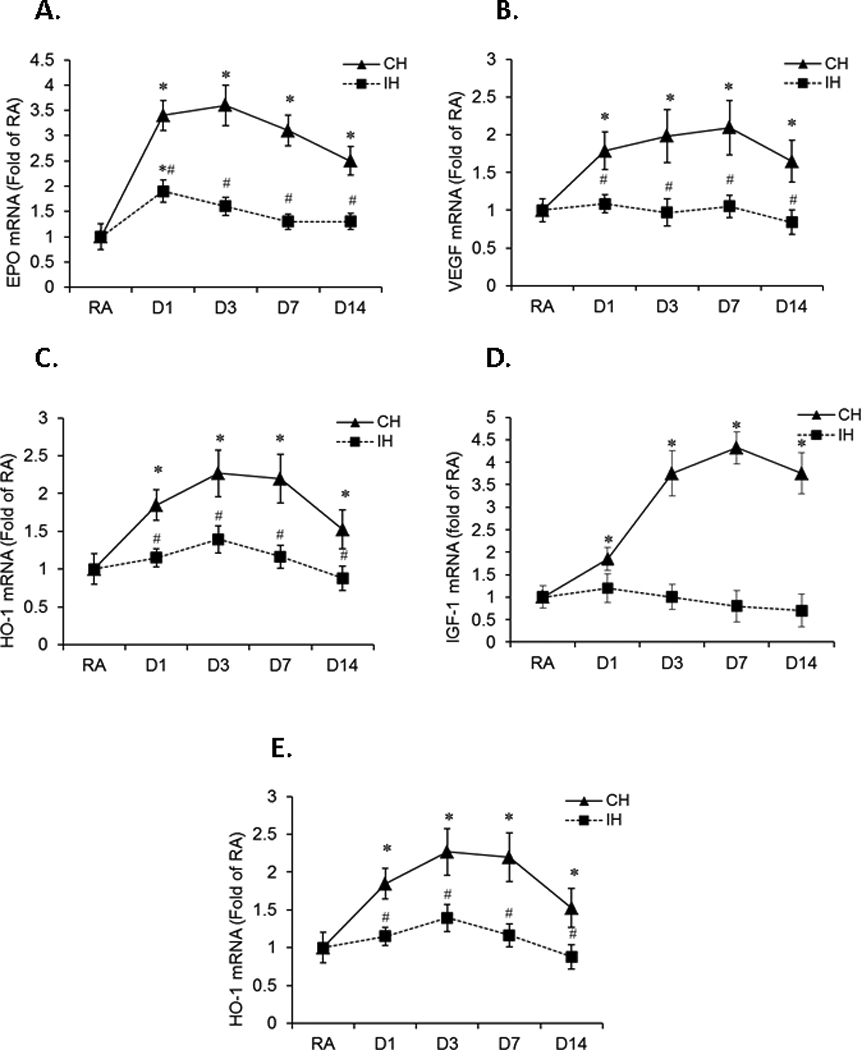
Rats were exposed to either CH or IH for 1, 3, 7 and 14 days (n=8/group). mRNA expression of EPO, VEGF, HO-1, IGF-1, and GLUT-1 was assessed by quantitative real-time RT-PCR. Data are expressed as a fold change of RA (mean+SE). Real-time PCR analyses revealed that CH induced significant increases in hippocampal expression of EPO (Figure 3A), VEGF (Figure 3B), HO-1(Figure 3C), and IGF-1 (Figure 3D) mRNA at all-time points when compared to RA (*P<0.01 vs. RA, #P<0.05 vs. CH, unpaired t tests). However, CH did not change GLUT1 mRNA expression (Figure 3D, P>0.05 vs. RA, unpaired t tests). IH induced a slight increase in EPO mRNA expression at day 1 (*P<0.05 vs. RA, #P<0.05 vs.CH, unpaired t tests) but not thereafter. IH did not change VEGF, HO-1, IGF-1, and GLUT1 gene expression in hippocampal tissues (P>0.05 vs. RA, unpaired t tests).
IH but not CH elicits cognitive deficits in rat
On a standard place discrimination task, rats exposed to 14 days of IH exhibited longer latencies and pathlengths to locate the hidden platform when compared to room air controls RA, and to rats exposed to 14 days sustained hypoxia (CH; n=24 per experimental condition; Figures 5A and 5B). Overall latency analysis for the entire trial blocks revealed significant changes between the different treatment groups, [F= 41.14; P<0.001] and pathlength, [F=16.44; P<0.001] indicating that IH, but not CH, adversely influenced the ability to acquire a spatial task. Significant differences in latencies were observed during block 3 [F=11.38; P<0.001], block 4 [F=12.35; P<0.03], block 5 [F=8.16; P<0.001] and block 6 [F=8.457; P<0.001]. There were no significant changes in Blocks 1 and 2. Repeated measures ANOVA revealed significant differences in pathlengths during blocks 3 [F=6.25; P<0.001], block 4 [F=5.36; P<0.001], block 5 [F=6.83; P<0.001] and block 6 [F=4.99; P<0.001]. There were no significant changes in blocks 1 and 2. In the probe-trial test, one-way ANOVA revealed a significant effect of treatment [F=12.87; P<0.001]. The magnitude of impairment was greatest in IH (Figure 5C). In the reference memory task, IH-exposed rats revealed significant deficits in memory retention in both latency [F=21.61; P<0.001] and pathlength [F=18.84; P<0.001]. However, the CH-exposed rats performed well in the retention task compared to IH and were similar to RA (P>0.05), indicating that only IH treatment elicited deficits in the retention of a spatial task in the water maze.
Figure 5. Effects of CH or IH exposures on water maze performance in rats exposed to either CH or IH for 14 days (n=24/group).
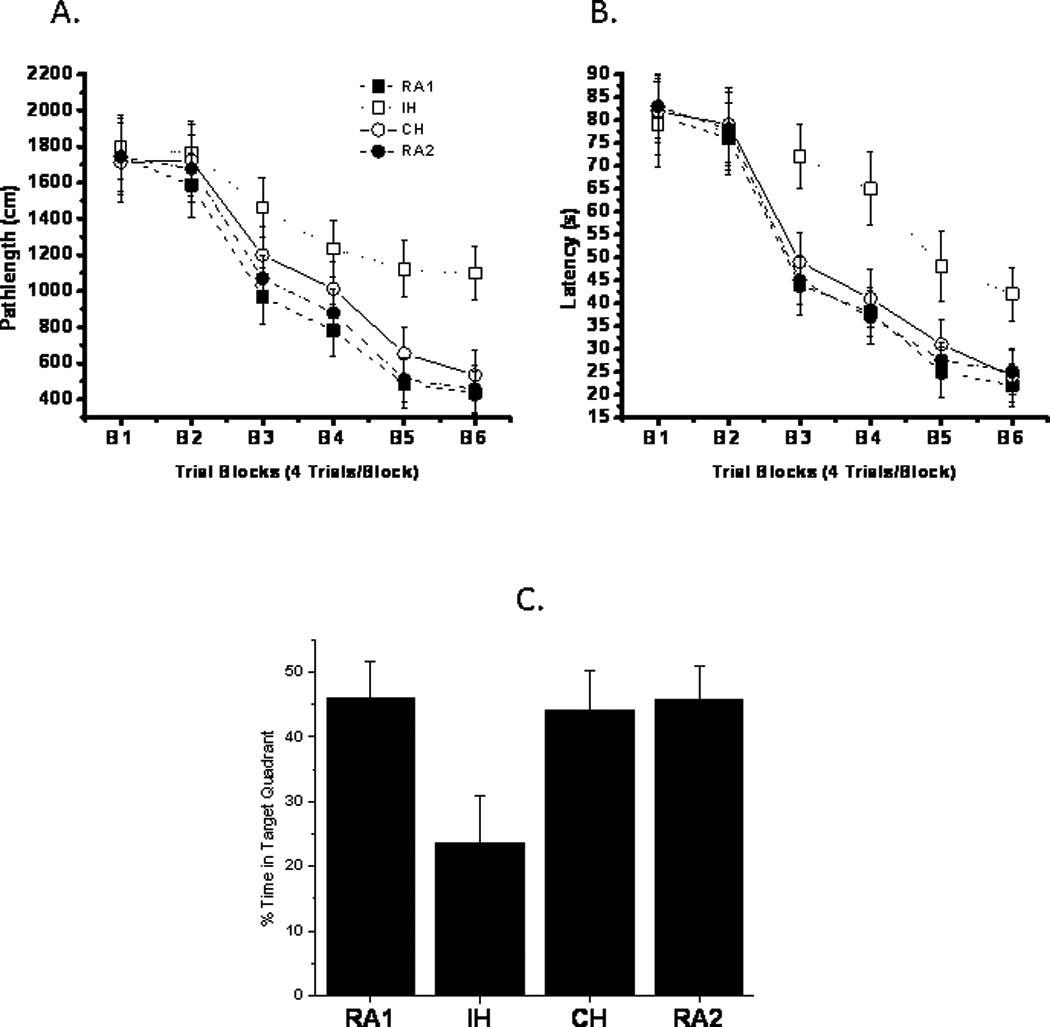
Panels (A) and (B) show mean pathlengths and latencies to locate the target platform during place training in IH and CH exposed rats. Each block represent a total of 4 consecutive trials. RA1 and RA2 (n=24/group) were exposed to normoxic conditions concomitantly with IH and CH, respectively. (see text for more details; IH vs. CH or RA, P<0.001, ANOVA). Panel C shows the mean percentage time that rats spend on target quadrant after completion of training (n=24/group, IH vs. CH or RA1 or RA2: P<0.001; ANOVA).
Administration of GH increases IGF-1 expression in the hippocampus after IH exposures
To assess the effect of exogenous GH on the IGF-1 mRNA expression in the hippocampus of rat brain, rats received daily injection of GH and were exposed to IH or RA for 7 days (n=8/group). After hypoxia exposure and GH administration, hippocampal tissues were collected and mRNA was extracted for measuring IGF-1 mRNA expression. In response to GH administration for 7 days, a significant increase of IGF-1 mRNA expression was detected in the hippocampal tissue as compared to the vehicle treated rat (Figure 6, * P<0.01 vs. vehicle). The increased mRNA expression of IGF-1 was observed in both GH treated IH (IH-GH) and RA (RA-GH) groups, but IGF-1 expression in IH-GH group was significant higher than that in RA-GH group (#P<0.05 vs. RA-GH). These results suggest that peripheral GH administration stimulated a response of GH (hormone) axis in the brain.
Figure 6. Effects of exogenous GH administration on IGF-1 mRNA expression in rat hippocampal tissues.
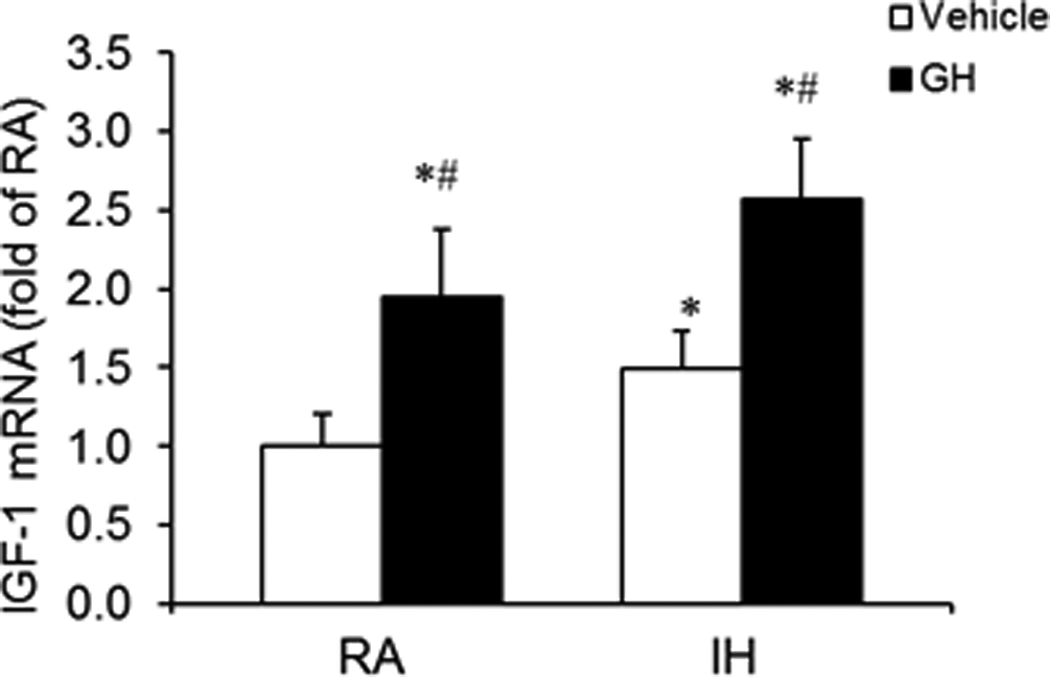
Rats received daily GH or vehicle injections for 7 days and were concomitantly exposed to either IH or RA (n=8/group). IGF-1 mRNA expression was assessed by quantitative real-time RT-PCR. Data are expressed as a fold change of RA-vehicle (mean+SE). GH administration induced significant increases in IGF-1 mRNA expression in both RA-GH and IH-GH conditions when compared to RA-vehicle or IH-vehicle groups (*P<0.01 vs. Vehicle, unpaired t tests). Furthermore, IGF-1 mRNA expression in IH-GH group was significant higher than that in RA-GH group (#P<0.05 vs. RA-GH, unpaired t tests).
GH increases EPO, VEGF and HO-1 but not GLUT1 mRNA expression in the hippocampus during IH
In order to clarify whether GH administration would change the mRNA expression of EPO, VEGF, HO-1 and Glut-1 in hippocampus of rat, rats received daily injections of GH and were exposed to IH or RA for 7 days (n=8/group). EPO, VEGF, HO-1 and Glut-1 mRNA expression were assessed. Administration of GH induced a significant increase in EPO and VEGF, but not in HO-1 and Glut-1 mRNA expression as compared with vehicle treatment in both RA-GH and IH-GH groups (Figure 7A–7D, *P<0.01 vs vehicle). Furthermore, EPO and VEGF mRNA expression in IH-GA group was significantly greater than EPO mRNA expression in RA-GH group (#P<0.01 vs RA-GH). These results suggest that peripheral administration of GH is able to increase EPO and VEFG mRNA expression in the central nervous system, which may in turn play a protective role in hypoxia-induced brain injury.
Figure 7. Effects of GH on EPO, VEGF, HO-1 and GLUT1 mRNA expression in rat hippocampal tissues.
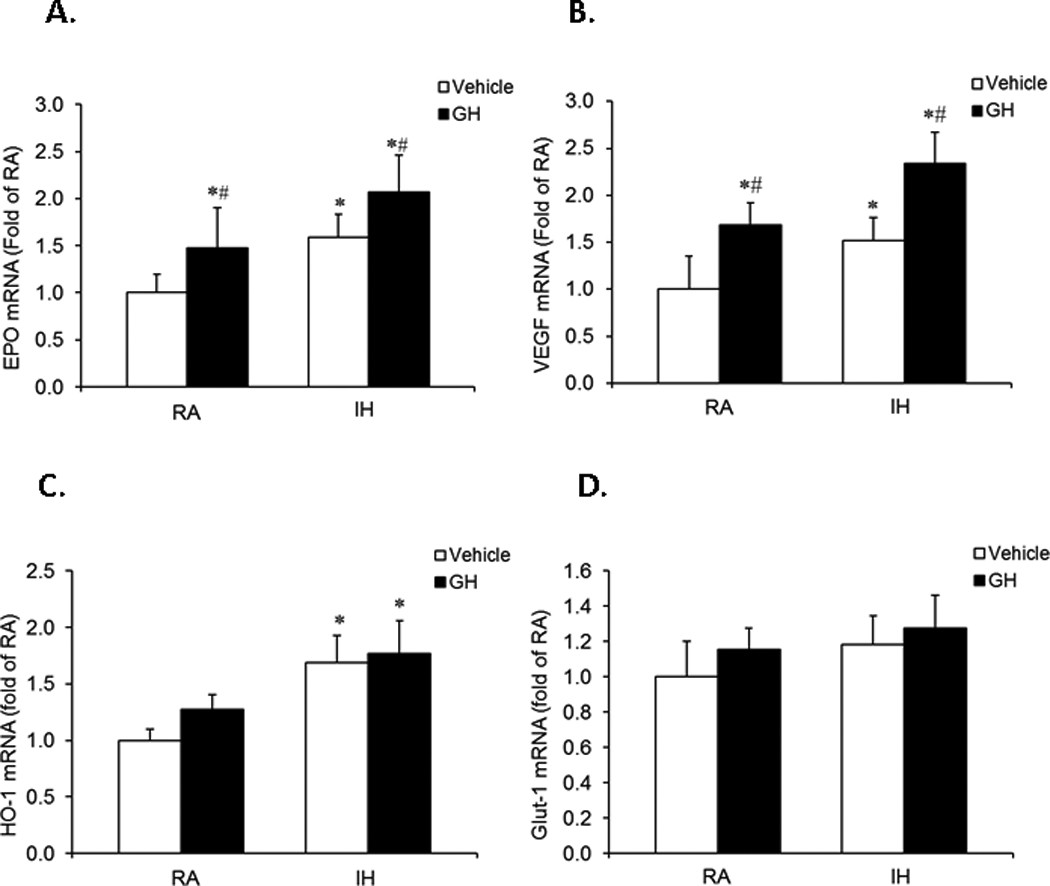
Rats received daily GH or vehicle injections and were exposed to either IH or RA for 7 days (n=8/group). GH administration induced significant increases in EPO (Figure 6A) and VEGF (Figure 6B) gene expression, but not in HO-1(Figure 6C) or GLUT1 (Figure 6D) in both of RA-GH and IH-GH groups as compared to RA-vehicle or IH-vehicle groups (*P<0.01 vs. Vehicle, unpaired t tests). EPO and VEGF mRNA expression in IH-GH treatment group were significantly higher than after RA-GH treatment (#P<0.05 vs. RA-GH, unpaired t tests).
GH decreases IH-induced expression of cleaved caspase 3 in the hippocampus
We have previously shown that IH exposures induce hippocampal neuronal apoptosis and functional losses (Gozal et al., 2001). Therefore, attenuation of hypoxia induced neuronal apoptosis could reduce hypoxic brain injury and improve overall physiological outcomes. The cleaved (i.e., activated) caspase 3 expression in the hippocampus was examined after daily injection of GH and IH exposures for 7 days. Cleaved caspase 3 positive staining was markedly increased in the IH-vehicle sections as compared to the RA-vehicle sections. In addition, IH-induced enhanced staining of cleaved caspase 3 was markedly attenuated by treatment with GH (Figure 8).
Figure 8. Effects of GH on cleaved caspase 3 expression in rat hippocampus.
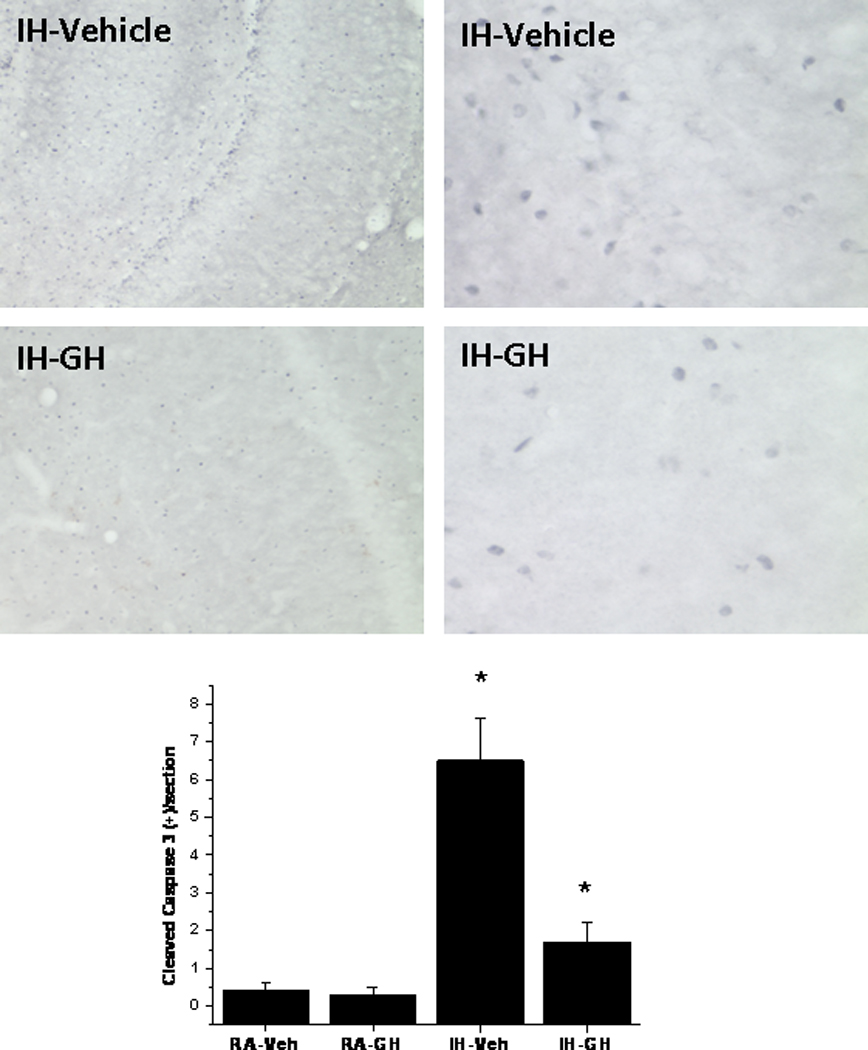
Rats received daily GH or vehicle injections and were exposed to either IH for 7 days (n=8/group). Cleaved caspase 3 expression was assessed by immunohistochemistry in hippocampal sections. For all images, the left panel shows a lower magnification (Scale bar – 50 µ), and the right panel shows a higher magnification of a representative section (Scale bar – 5 µ). Bottom panel shows average counts of cleaved caspase 3 positively labeled cells for each of the 4 experimental conditions (n=10 sections/animal and 5 rats/condition).
GH attenuates IH-induced neurobehavioral deficits
During training trials, vehicle-treated rats exposed to IH for 7 days showed significantly prolonged latencies and pathlengths compared to vehicle-treated RA, GH-treated RA, and GH-treated IH animals (Figure 9A–9B; P<0.002, ANOVA). Similarly, during probe trials, IH-exposed animals treated with vehicle displayed significantly lower spatial bias compared to any of the 3 other experimental groups (P<0.01). Post-hoc analyses further confirmed that IH-exposed animals treated with GH were not significantly different from controls, suggesting that GH attenuated, at least partially, IH-induced deficits in spatial learning and retention. No group differences were observed on the cued task, indicating that the effects of either IH or GH were not due to sensorimotor impairments.
Figure 9. Effects of GH treatment on water maze performance in rats.
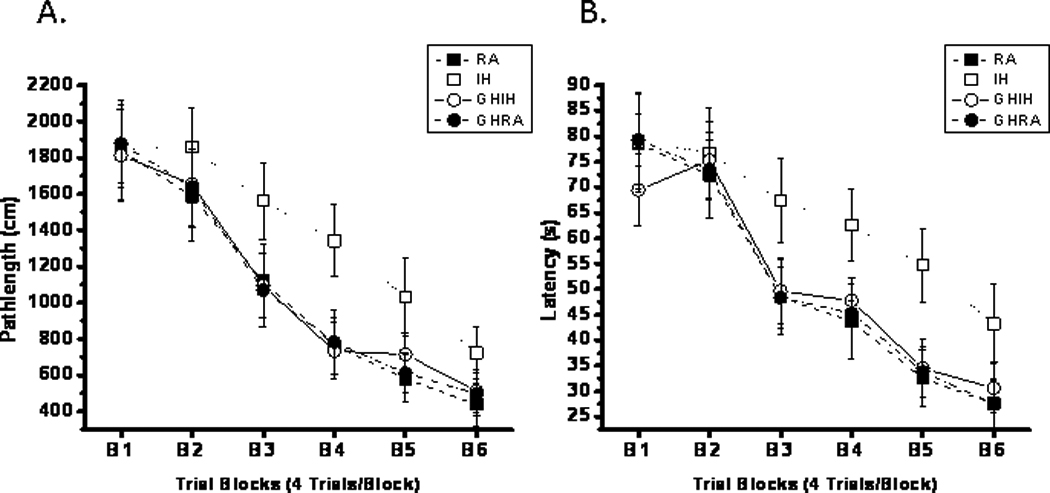
Mean swim distances (cm) (panel A) and latencies (panel B) to locate the target platform during place training in rats exposed to 7 days of IH (IH; open symbols) or RA (RA; filled symbols), receiving either vehicle (RA; square symbols) or GH treatments (circle symbols). (n=16/treatment group; *P<0.001 ANOVA). Data are expressed in cm (mean±SE; n= 16; *P<0.01 vs. RA controls, #P<0.01 vs. IH-GH, ANOVA).
Discussion
In this study, we show for the first time that IH and GH are associated with divergent trajectories in GH pathways within the CNS, and that such divergence is reflected in differential susceptibility to neurocognitive deficits. Indeed, we found increased GH mRNA and protein expression was induced by both CH and IH hypoxic exposures in the hippocampus of rat, although the magnitude of such changes was clearly more prominent and sustained after CH. CH exposures, but not IH exposures, were also associated with increases in EPO, VEGF and HO-1 mRNA expression. Conversely, IH but not CH exposures induced substantial deficits in task acquisition and retention. Treatment with GH led to increases in IGF-1, EPO and VEGF mRNA expression in the hippocampus despite ongoing IH exposures, and not only decreased the expression of cleaved caspase 3 in the hippocampus, but also attenuated IH-induced neurobehavioral deficits, suggesting that exogenous GH administration may serve as an effective therapeutic intervention aiming to mitigate the extent and severity of SDB-associated neuronal cell losses and associated cognitive dysfunction.
In the CNS, GH has emerged as an important player in the formation and maintenance of neuronal plasticity and survival (Scheepens et al., 2001;Donahue et al., 2002;Zearfoss et al., 2008;Baudet et al., 2009). In response to a hypoxia/ischemic injury, the brain initiates a serial endogenous neuroprotective strategies aimed at reducing neuronal cell death (Johansson et al., 1999;Slevin et al., 2005). Several studies suggested that GH may play a complex role in the pathophysiology of brain injury related to ischemia/hypoxia, similar to other neurotrophins in the brain (Shin et al., 2004;Scheepens et al., 2000;Christophidis et al., 2009).
The hypoxic exposures used herein were clearly not associated with ischemia, and our previous experiments have clearly shown that the current IH profile was of a magnitude that not only aimed at mimicking the oxyhemoglobin saturation profiles typically encountered in SDB, but also that such profile induced significant increases in apoptosis within the CA1 region of the hippocampus as well as substantial deficits in water maze performance (Gozal et al., 2001). However, while the field of sustained hypoxia has received much more extensive attention we are only aware of scarce reports on the cognitive consequences of non-oscillatory hypoxic exposures, suggesting that only very severe hypoxia are indeed detrimental (Bahrke and Shukitt-Hale, 1993). Indeed, in a study by Titus colleagues exposures for 7 days to hypobaric hypoxia corresponding to 6,000 meters altitude were needed to induce some deficits in the partially baited radial arm maze task, and were further accompanied by some degree of functional and structural recovery with more extended exposures (Titus et al., 2007;Maiti et al., 2008a;Maiti et al., 2008b). Thus, severe hypoxia, although able to induce temporal endogenous neuroprotection, will eventually cause cell death and behavioral impairments if exposures are carried long enough. Accordingly, the time points we selected for the current experiments (i.e., 14 days after hypoxia) may be only applicable for acute and subacute phases of exposure, such that longer lasting CH may lead to neurodegeneration and cognitive decline, as has been observed in elderly humans with the obstructive sleep apnea syndrome (Cohen-Zion et al., 2001). However, the expected saturation levels from such CH exposures would be much more severe than the experimental conditions in the present study, and as such the absence of measurable cognitive deficits following CH was not unexpected in the time frames studied here. It is well established that a short episodic ischemic/hypoxic event can result in a subsequent resistance to severe ischemic/hypoxic injury, which is so called ischemic/hypoxic preconditioning. Since the preconditioning window is very narrow (<60 min) (Obrenovitch, 2008), it is less likely that the preconditioning protection would occur under the chronic IH or CH exposure. As a methodologic comment, we should point out that IH exposure only provides one aspect of SBD, because sleep fragmentation, episodic hypercapnia, and upper airway collapse are absent in this murine model. Also, the IH profile used here represents a standardization of exposures aiming to reflect severe disease, and as such may not completely represent the variable hypoxic profile characteristics seen in patients with SDB. Also, as we used young rats in our studies, the animal model may not reflect the pathophysiological conditions in other age group, such as aging population. In contradistinction, despite similar nadir oxygenation levels, the presentation of hypoxia in an episodic fashion elicited markedly different effects. One of our leading hypotheses was that the differences between CH and IH could be due to divergent effects on the recruitment of protective mechanisms, such as the GH pathway. Indeed, we found that GH and GHR expression changes diverged in response to CH or IH exposures, and could play a role in differentially activating subsequent signaling events including downstream protective gene expression against hypoxic injury. The local GH in hippocampal tissue may derive from both translation of neuronal GH mRNA and endocrine GH. However, the exact source and distribution of GH under the hypoxia condition is largely unknown. Increased GH and GHR expression or GHR immunoreactivity have been reported after hypoxic-ischemic brain injury (Scheepens et al., 1999;Christophidis et al., 2009). Furthermore, even though it is still controversial whether exogenous GH will cross the blood brain barrier (BBB), we now report on the exogenous GH-mediated effects on the expression of selected genes as well as the mitigation of the functional consequences of IH. Although this effect could be partially due to IH-induced alterations in the permeability of BBB (Natah et al., 2009), there is no evidence to support or dispel this notion, which will need to be investigated in the future. Taken together, our findings support the notion that increased GH and GHR expression in response to CH exposures may play a role in downstream signaling events orchestrating CNS protection and adaptation to hypoxia, and that such responses are maladaptive in the context of IH, thereby leading to the adverse consequences solely found in the latter hypoxic paradigm.
Since hypoxic exposures modified expression of GH and GHR, we therefore proceeded to further explore the potential links between GH/GHR and known downstream protective hypoxia-responsive genes, such as EPO, VEGF, HO-1, and GLUT-1. Indeed, the expression of these 4 genes is tightly regulated in the CNS by HIF-1α (Freeman and Barone, 2005;Acker and Acker, 2004), and plays the protective role in hypoxia and ischemia-induced brain injury (Sharp et al., 2004;Fan et al., 2009). For example, EPO is expressed in the nervous system and involved in normal brain development (Noguchi et al., 2007), and its expression in the brain is modulated by hypoxia and other stresses (Eckardt and Kurtz, 2005) and play an important role in hypoxic/ischemic brain injury (Marti, 2004;Milano and Collomp, 2005). In present study, we found that EPO, VEGF and HO-1, but not GLUT-1 mRNA expression was increased in a sustained manner by CH. In contrast, IH induced only a very modest and transient increase in EPO expression in the hippocampus, but further did not modify VEGF, HO-1 and GLUT-1 mRNA expression. In an in vivo animal model, administration of EPO attenuated global and focal cerebral ischemia-induced brain injury (Junk et al., 2002;Zhang et al., 2006), indicating the EPO is a good candidate gene for hypoxia-ischemia protection. In addition, EPO also show some beneficial effect on ischemia-induced cognitive impairment in adult animals (Kumral et al., 2004), and enhanced expression of EPO has beneficial effects on neuronal survival most likely via activation of EPO receptors (Kilic et al., 2005;Sanchez et al., 2009). Based on the similar favorable effects of increased expression of the HIF-1α-dependent genes, increased expression of these genes in response to CH would confer protection. On the other hand, the relative lack of those protective genes under the IH exposures would be anticipated to contribute to IH-induced neuronal injury and cognitive dysfunction. As mentioned above, the neurocognitive impairments induced by IH in both neonatal and adult rats (Row et al., 2002b;Row et al., 2003;Kheirandish et al., 2005a;Row et al., 2002a;Hui-guo et al., 2010;Cai et al., 2010;Ward et al., 2009) were replicated in the present study, while no such consequences emerged after CH. Although the mechanisms for IH-induced neurocognitive dysfunction are not well understood, increased oxidative stress (Wang et al., 2010;Row et al., 2003;Xu et al., 2004;Shan et al., 2007;Burckhardt et al., 2008), activation of inflammatory pathways (Li et al., 2003), dysregulation of neuronal progenitor cell proliferation (Gozal et al., 2003a), leading to neuronal apoptosis and gliosis. In comparison, CH exposures of such magnitude were apparently not associated with cell injury-promoting processes, thereby suggesting that intrinsic protective mechanisms were operational in the latter but not in the former.
To further establish the potential beneficial effect of GH treatment on hypoxia induced neuronal injury and cognitive impairments, animals were treated daily with recombinant GH while undergoing IH exposures. IGF-1 mRNA expression was up-regulated in the hippocampus following GH treatment, suggesting that the somatotropic axis in the brain was effectively activated by the exogenous subcutaneous GH administration route. GH or IGF-I were effective in reducing brain injury in rats subjected to hypoxia-ischemia insults (Shin et al., 2004;Gustafson et al., 1999;Zhong et al., 2009;Smith, 2003;Liu et al., 2001), suggesting that IGF-1, a downstream component of GH-activated pathways, may afford protection per se. Similar to GHR, IGF-I receptor was also up-regulated after hypoxic-ischemic injury (Scheepens et al., 1999;Scheepens et al., 2000), suggesting that IGF-1 is a responsive member of the somatotropic axis and plays a role in mediating GH functionality in brain. After GH treatment, we also found that EPO and VEGF were up-regulated in the hippocampus of the brain, suggesting that GH treatment may activate some of hypoxia-protective gene expression, which may in turn attenuate IH-induced injury. IH-induced spatial learning deficits were manifest after 7 days of IH exposures, and GH treatment greatly attenuated such deficits, as well as reduced the extent of apoptosis. Therefore, anti-apoptotic mechanisms induced by GH, most likely via induction of protective gene expression appear to ultimately improve overall neurological outcomes in this model of SDB.
Conclusion
Dysregulation of recruitment of GH and associated downstream protective pathways underlies components of hippocampal injury and dysfunction following exposures to the episodic hypoxia during sleep. Furthermore, exogenous GH therapy promotes enhanced expression of protective genes, thereby reducing IH-induced neuronal injury in hippocampus and neurobehavioral deficits. GH-based interventions may provide the basis for potential clinical therapeutic interventions aimed at palliating SDB-associated end-organ morbidity.
Highlights.
Intermittent hypoxia that characterizes sleep-disordered breathing leads to neurocognitive deficits.
Dysregulation of recruitment of GH and associated downstream protective pathways occurs after intermittent hypoxia, but not sustained hypoxia.
Divergent GH pathways underlies components of hippocampal vulnerability and ultimately injury and dysfunction following exposures to the episodic hypoxia.
Exogenous GH therapy promotes enhanced expression of protective genes, thereby reducing IH-induced neuronal apoptosis in hippocampus and functional deficits.
GH-based interventions may provide the basis for potential clinical therapeutic interventions aimed at palliating SDB-associated end-organ morbidity.
Acknowledgements
This study was supported by National Institutes of Health grants HL-086662 (to DG) and American Heart Association grant AHA-0930129N (to RCL).
Abbreviations
- CH
chronic sustained hypoxia
- CNS
central nervous system
- EPO
erythropoietin
- GH
growth hormone
- GHR
growth hormone receptor
- Glut-1
glucose transporter-1
- HO-1
heme oxygenase-1
- IGF-1
insulin-like growth factor-1
- IH
intermittent hypoxia
- RA
room air
- SDB
Sleep disordered breathing
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207:3171–3188. doi: 10.1242/jeb.01075. [DOI] [PubMed] [Google Scholar]
- Ardail D, Debon A, Perret-Vivancos C, Biol-N'Garagba MC, Krantic S, Lobie PE, Morel G. Growth hormone internalization in mitochondria decreases respiratory chain activity. Neuroendocrinology. 2010;91:16–26. doi: 10.1159/000268289. [DOI] [PubMed] [Google Scholar]
- Bahrke MS, Shukitt-Hale B. Effects of altitude on mood, behaviour and cognitive functioning. A review. Sports Med. 1993;16:97–125. doi: 10.2165/00007256-199316020-00003. [DOI] [PubMed] [Google Scholar]
- Barta A, Richards RI, Baxter JD, Shine J. Primary structure and evolution of rat growth hormone gene. Proc Natl Acad Sci U S A. 1981;78:4867–4871. doi: 10.1073/pnas.78.8.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudet ML, Rattray D, Martin BT, Harvey S. Growth hormone promotes axon growth in the developing nervous system. Endocrinology. 2009;150:2758–2766. doi: 10.1210/en.2008-1242. [DOI] [PubMed] [Google Scholar]
- Beebe DW, Gozal D. Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res. 2002;11:1–16. doi: 10.1046/j.1365-2869.2002.00289.x. [DOI] [PubMed] [Google Scholar]
- Burckhardt IC, Gozal D, Dayyat E, Cheng Y, Li RC, Goldbart AD, Row BW. Green tea catechin polyphenols attenuate behavioral and oxidative responses to intermittent hypoxia. Am J Respir Crit Care Med. 2008;177:1135–1141. doi: 10.1164/rccm.200701-110OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai XH, Zhou YH, Zhang CX, Hu LG, Fan XF, Li CC, Zheng GQ, Gong YS. Chronic intermittent hypoxia exposure induces memory impairment in growing rats. Acta Neurobiol Exp (Wars) 2010;70:279–287. doi: 10.55782/ane-2010-1799. [DOI] [PubMed] [Google Scholar]
- Christophidis LJ, Gorba T, Gustavsson M, Williams CE, Werther GA, Russo VC, Scheepens A. Growth hormone receptor immunoreactivity is increased in the subventricular zone of juvenile rat brain after focal ischemia: a potential role for growth hormone in injury-induced neurogenesis. Growth Horm IGF Res. 2009;19:497–506. doi: 10.1016/j.ghir.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Cohen-Zion M, Stepnowsky C, Marler, Shochat T, Kripke DF, Ancoli-Israel S. Changes in cognitive function associated with sleep disordered breathing in older people. J Am Geriatr Soc. 2001;49:1622–1627. doi: 10.1046/j.1532-5415.2001.t01-1-49270.x. [DOI] [PubMed] [Google Scholar]
- Cooper BG, White JE, Ashworth LA, Alberti KG, Gibson GJ. Hormonal and metabolic profiles in subjects with obstructive sleep apnea syndrome and the acute effects of nasal continuous positive airway pressure (CPAP) treatment. Sleep. 1995;18:172–179. [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue CP, Jensen RV, Ochiishi T, Eisenstein I, Zhao M, Shors T, Kosik KS. Transcriptional profiling reveals regulated genes in the hippocampus during memory formation. Hippocampus. 2002;12:821–833. doi: 10.1002/hipo.10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RM, Miyasaka N, Takahashi K, Latuszek-Barrantes A, Haddad GG, Hetherington HP. Chronic intermittent but not constant hypoxia decreases NAA/Cr ratios in neonatal mouse hippocampus and thalamus. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1254–R1259. doi: 10.1152/ajpregu.00404.2006. [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest. 2005;35 Suppl 3:13–19. doi: 10.1111/j.1365-2362.2005.01525.x. 13–19. [DOI] [PubMed] [Google Scholar]
- Fan X, Heijnen CJ, van der KOOIJ MA, Groenendaal F, van BF. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. 2009;62:99–108. doi: 10.1016/j.brainresrev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Barone MC. Targeting hypoxia-inducible factor (HIF) as a therapeutic strategy for CNS disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:85–92. doi: 10.2174/1568007053005154. [DOI] [PubMed] [Google Scholar]
- Goldbart A, Row BW, Kheirandish L, Schurr A, Gozal E, Guo SZ, Payne RS, Cheng Z, Brittian KR, Gozal D. Intermittent hypoxic exposure during light phase induces changes in cAMP response element binding protein activity in the rat CA1 hippocampal region: water maze performance correlates. Neuroscience. 2003;122:585–590. doi: 10.1016/j.neuroscience.2003.08.054. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Nair D, Goldbart AD. Physical activity attenuates intermittent hypoxia-induced spatial learning deficits and oxidative stress. Am J Respir Crit Care Med. 2010;182:104–112. doi: 10.1164/rccm.201001-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Row BW, Gozal E, Kheirandish L, Neville JJ, Brittian KR, Sachleben LR, Jr, Guo SZ. Temporal aspects of spatial task performance during intermittent hypoxia in the rat: evidence for neurogenesis. Eur J Neurosci. 2003a;18:2335–2342. doi: 10.1046/j.1460-9568.2003.02947.x. [DOI] [PubMed] [Google Scholar]
- Gozal D, Row BW, Kheirandish L, Liu R, Guo SZ, Qiang F, Brittian KR. Increased susceptibility to intermittent hypoxia in aging rats: changes in proteasomal activity, neuronal apoptosis and spatial function. J Neurochem. 2003b;86:1545–1552. doi: 10.1046/j.1471-4159.2003.01973.x. [DOI] [PubMed] [Google Scholar]
- Gozal D, Sans CO, McLaughlin CV, Serpero LD, Witcher LA, Kheirandish-Gozal L. Plasma IGF-1 levels and cognitive dysfunction in children with obstructive sleep apnea. Sleep Med. 2009;10:167–173. doi: 10.1016/j.sleep.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Gustafson K, Hagberg H, Bengtsson BA, Brantsing C, Isgaard J. Possible protective role of growth hormone in hypoxia-ischemia in neonatal rats. Pediatr Res. 1999;45:318–323. doi: 10.1203/00006450-199903000-00005. [DOI] [PubMed] [Google Scholar]
- Hambrecht VS, Vlisides PE, Row BW, Gozal D, Baghdoyan HA, Lydic R. G proteins in rat prefrontal cortex (PFC) are differentially activated as a function of oxygen status and PFC region. J Chem Neuroanat. 2009;37:112–117. doi: 10.1016/j.jchemneu.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui-guo L, Kui L, Yan-ning Z, Yong-jian X. Apocynin attenuate spatial learning deficits and oxidative responses to intermittent hypoxia. Sleep Med. 2010;11:205–212. doi: 10.1016/j.sleep.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Johansson BB, Zhao L, Mattsson B. Environmental influence on gene expression and recovery from cerebral ischemia. Acta Neurochir Suppl. 1999;73:51–55. doi: 10.1007/978-3-7091-6391-7_8. [DOI] [PubMed] [Google Scholar]
- Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirandish L, Gozal D, Pequignot JM, Pequignot J, Row BW. Intermittent hypoxia during development induces long-term alterations in spatial working memory, monoamines, and dendritic branching in rat frontal cortex. Pediatr Res. 2005a;58:594–599. doi: 10.1203/01.pdr.0000176915.19287.e2. [DOI] [PubMed] [Google Scholar]
- Kheirandish L, Row BW, Li RC, Brittian KR, Gozal D. Apolipoprotein E-deficient mice exhibit increased vulnerability to intermittent hypoxia-induced spatial learning deficits. Sleep. 2005b;28:1412–1417. doi: 10.1093/sleep/28.11.1412. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Kumral A, Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, Kiray M, Genc S, Duman N, Koroglu TF, Ozkan H, Genc K. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res. 2004;153:77–86. doi: 10.1016/j.bbr.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Gozal E, Kheirandish L, Fan Q, Brittian KR, Guo SZ, Sachleben LR, Jr, Gozal D. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med. 2003;168:469–475. doi: 10.1164/rccm.200211-1264OC. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, Sachleben LR, Jr, Gozal D. Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol Dis. 2004;17:44–53. doi: 10.1016/j.nbd.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Thorne RG, DeFor TA, Frey WH. Intranasal administration of insulin-like growth factor-I bypasses the blood-brain barrier and protects against focal cerebral ischemic damage. J Neurol Sci. 2001;187:91–97. doi: 10.1016/s0022-510x(01)00532-9. [DOI] [PubMed] [Google Scholar]
- Lobie PE, Garcia-Aragon J, Lincoln DT, Barnard R, Wilcox JN, Waters MJ. Localization and ontogeny of growth hormone receptor gene expression in the central nervous system. Brain Res Dev Brain Res. 1993;74:225–233. doi: 10.1016/0165-3806(93)90008-x. [DOI] [PubMed] [Google Scholar]
- Maiti P, Muthuraju S, Ilavazhagan G, Singh SB. Hypobaric hypoxia induces dendritic plasticity in cortical and hippocampal pyramidal neurons in rat brain. Behav Brain Res. 2008a;189:233–243. doi: 10.1016/j.bbr.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Maiti P, Singh SB, Mallick B, Muthuraju S, Ilavazhagan G. High altitude memory impairment is due to neuronal apoptosis in hippocampus, cortex and striatum. J Chem Neuroanat. 2008b;36:227–238. doi: 10.1016/j.jchemneu.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207:3233–3242. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- Meston N, Davies RJ, Mullins R, Jenkinson C, Wass JA, Stradling JR. Endocrine effects of nasal continuous positive airway pressure in male patients with obstructive sleep apnoea. J Intern Med. 2003;254:447–454. doi: 10.1046/j.1365-2796.2003.01212.x. [DOI] [PubMed] [Google Scholar]
- Milano M, Collomp R. Erythropoietin and neuroprotection: a therapeutic perspective. J Oncol Pharm Pract. 2005;11:145–149. doi: 10.1191/1078155205jp162oa. [DOI] [PubMed] [Google Scholar]
- Mokhlesi B, Gozal D. Update in sleep medicine 2009. Am J Respir Crit Care Med. 2010;181:545–549. doi: 10.1164/rccm.200912-1948UP. [DOI] [PubMed] [Google Scholar]
- Natah SS, Srinivasan S, Pittman Q, Zhao Z, Dunn JF. Effects of acute hypoxia and hyperthermia on the permeability of the blood-brain barrier in adult rats. J Appl Physiol. 2009;107:1348–1356. doi: 10.1152/japplphysiol.91484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi CT, Asavaritikrai P, Teng R, Jia Y. Role of erythropoietin in the brain. Crit Rev Oncol Hematol. 2007;64:159–171. doi: 10.1016/j.critrevonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- Perry JC, D'Almeida V, Lima MM, Godoi FR, Vital MA, Oliveira MG, Tufik S. Intermittent hypoxia and sleep restriction: motor, cognitive and neurochemical alterations in rats. Behav Brain Res. 2008;189:373–380. doi: 10.1016/j.bbr.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Row BW, Kheirandish L, Li RC, Guo SZ, Brittian KR, Hardy M, Bazan NG, Gozal D. Platelet-activating factor receptor-deficient mice are protected from experimental sleep apnea-induced learning deficits. J Neurochem. 2004;89:189–196. doi: 10.1111/j.1471-4159.2004.02352.x. [DOI] [PubMed] [Google Scholar]
- Row BW, Kheirandish L, Neville JJ, Gozal D. Impaired spatial learning and hyperactivity in developing rats exposed to intermittent hypoxia. Pediatr Res. 2002a;52:449–453. doi: 10.1203/00006450-200209000-00024. [DOI] [PubMed] [Google Scholar]
- Row BW, Kheirandish L, Neville JJ, Gozal D. Impaired spatial learning and hyperactivity in developing rats exposed to intermittent hypoxia. Pediatr Res. 2002b;52:449–453. doi: 10.1203/00006450-200209000-00024. [DOI] [PubMed] [Google Scholar]
- Row BW, Liu R, Xu W, Kheirandish L, Gozal D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med. 2003;167:1548–1553. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- Saini J, Krieger J, Brandenberger G, Wittersheim G, Simon C, Follenius M. Continuous positive airway pressure treatment. Effects on growth hormone, insulin and glucose profiles in obstructive sleep apnea patients. Horm Metab Res. 1993;25:375–381. doi: 10.1055/s-2007-1002123. [DOI] [PubMed] [Google Scholar]
- Sanchez PE, Fares RP, Risso JJ, Bonnet C, Bouvard S, Le-Cavorsin M, Georges B, Moulin C, Belmeguenai A, Bodennec J, Morales A, Pequignot JM, Baulieu EE, Levine RA, Bezin L. Optimal neuroprotection by erythropoietin requires elevated expression of its receptor in neurons. Proc Natl Acad Sci U S A. 2009;106:9848–9853. doi: 10.1073/pnas.0901840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorio A, Conti A, Molinari E, Riva G, Morabito F, Faglia G. Growth, growth hormone and cognitive functions. Horm Res. 1996;45:23–29. doi: 10.1159/000184754. [DOI] [PubMed] [Google Scholar]
- Scheepens A, Sirimanne E, Beilharz E, Breier BH, Waters MJ, Gluckman PD, Williams CE. Alterations in the neural growth hormone axis following hypoxic-ischemic brain injury. Brain Res Mol Brain Res. 1999;68:88–100. doi: 10.1016/s0169-328x(99)00051-0. [DOI] [PubMed] [Google Scholar]
- Scheepens A, Sirimanne ES, Breier BH, Clark RG, Gluckman PD, Williams CE. Growth hormone as a neuronal rescue factor during recovery from CNS injury. Neuroscience. 2001;104:677–687. doi: 10.1016/s0306-4522(01)00109-9. [DOI] [PubMed] [Google Scholar]
- Scheepens A, Williams CE, Breier BH, Guan J, Gluckman PD. A role for the somatotropic axis in neural development, injury and disease. J Pediatr Endocrinol Metab. 2000;13 Suppl 6:1483–1491. doi: 10.1515/jpem-2000-s623. [DOI] [PubMed] [Google Scholar]
- Shan X, Chi L, Ke Y, Luo C, Qian S, Gozal D, Liu R. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol Dis. 2007;28:206–215. doi: 10.1016/j.nbd.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1:26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DH, Lee E, Kim JW, Kwon BS, Jung MK, Jee YH, Kim J, Bae SR, Chang YP. Protective effect of growth hormone on neuronal apoptosis after hypoxia-ischemia in the neonatal rat brain. Neurosci Lett. 2004;354:64–68. doi: 10.1016/j.neulet.2003.09.070. [DOI] [PubMed] [Google Scholar]
- Slevin M, Krupinski J, Kumar P, Gaffney J, Kumar S. Gene activation and protein expression following ischaemic stroke: strategies towards neuroprotection. J Cell Mol Med. 2005;9:85–102. doi: 10.1111/j.1582-4934.2005.tb00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PF. Neuroprotection against hypoxia-ischemia by insulin-like growth factor-I (IGF-I) IDrugs. 2003;6:1173–1177. [PubMed] [Google Scholar]
- Titus AD, Shankaranarayana Rao BS, Harsha HN, Ramkumar K, Srikumar BN, Singh SB, Chattarji S, Raju TR. Hypobaric hypoxia-induced dendritic atrophy of hippocampal neurons is associated with cognitive impairment in adult rats. Neuroscience. 2007;145:265–278. doi: 10.1016/j.neuroscience.2006.11.037. [DOI] [PubMed] [Google Scholar]
- van Dam PS, Aleman A. Insulin-like growth factor-I, cognition and brain aging. Eur J Pharmacol % 2004;19:87–95. doi: 10.1016/j.ejphar.2004.02.047. 490. [DOI] [PubMed] [Google Scholar]
- van Dam PS, Aleman A, de Vries WR, Deijen JB, van der Veen EA, de Haan EH, Koppeschaar HP. Growth hormone, insulin-like growth factor I and cognitive function in adults. Growth Horm IGF Res. 2000;10 Suppl B:S69–S73. doi: 10.1016/s1096-6374(00)80013-1. S69–S73. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang SX, Gozal D. Reactive oxygen species and the brain in sleep apnea. Respir Physiol Neurobiol. 2010;174:307–316. doi: 10.1016/j.resp.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CP, McCoy JG, McKenna JT, Connolly NP, McCarley RW, Strecker RE. Spatial learning and memory deficits following exposure to 24 h of sleep fragmentation or intermittent hypoxia in a rat model of obstructive sleep apnea. Brain Res. 2009;1294:128–137. doi: 10.1016/j.brainres.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Leung KL, Chen L, Chan YS, Ng PC, Fok TF, Wing YK, Ke Y, Li AM, Yung WH. Brain-derived neurotrophic factor rescues and prevents chronic intermittent hypoxia-induced impairment of hippocampal long-term synaptic plasticity. Neurobiol Dis. 2010;40:155–162. doi: 10.1016/j.nbd.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Zayour D, Azar ST, Azar N, Nasser M, Obeid M, Mroueh S, Dbaibo GS, Bitar FF. Endocrine changes in a rat model of chronic hypoxia mimicking cyanotic heart disease. Endocr Res. 2003;29:191–200. doi: 10.1081/erc-120022301. [DOI] [PubMed] [Google Scholar]
- Zearfoss NR, Alarcon JM, Trifilieff P, Kandel E, Richter JD. A molecular circuit composed of CPEB-1 and c-Jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus. J Neurosci. 2008;28:8502–8509. doi: 10.1523/JNEUROSCI.1756-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Signore AP, Zhou Z, Wang S, Cao G, Chen J. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: potential signaling mechanisms. J Neurosci Res. 2006;83:1241–1251. doi: 10.1002/jnr.20816. [DOI] [PubMed] [Google Scholar]
- Zhang YS, Du JZ. The response of growth hormone and prolactin of rats to hypoxia. Neurosci Lett. 2000;279:137–140. doi: 10.1016/s0304-3940(99)00968-4. [DOI] [PubMed] [Google Scholar]
- Zhong J, Zhao L, Du Y, Wei G, Yao WG, Lee WH. Delayed IGF-1 treatment reduced long-term hypoxia-ischemia-induced brain damage and improved behavior recovery of immature rats. Neurol Res. 2009;31:483–489. doi: 10.1179/174313208X338133. [DOI] [PubMed] [Google Scholar]