

Abstract
We evaluated a new concept in cancer therapy, coiled-coil mediated induction of apoptosis in Raji B cells, for treatment of human B-cell lymphoma in a preclinical animal model. The system is composed of a pair of complementary coiled-coil peptides, CCE and CCK, forming antiparallel heterodimers; Fab’ fragment of the 1F5 anti-CD20 antibody; and N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer. One peptide is conjugated to the Fab’ fragment (Fab’-CCE), the other is conjugated in multiple grafts to polyHPMA (CCK-P; P is the HPMA copolymer backbone). Intravenous administration of Fab’-CCE conjugate, followed by the administration of CCK-P produced long-term survivors in SCID (C.B.-17) mice bearing human B-lymphoma xenografts. The rationale of the design is the absence of low molecular weight drugs and the fact that crosslinking of CD20 at B-cell surface results in apoptosis. This approach creates a new paradigm for manipulating molecular recognition principles in the design of improved cancer treatment.
Keywords: Apoptosis, CD20, coiled-coils, B-cell lymphoma, polymer-peptide conjugates
1. Introduction
Non-Hodgkin’s lymphoma (NHL) is a prevalent cancer with over a half-million individuals having a history in the United States [1]. About 85% of NHLs are malignancies originating from B cells while the remaining malignancies are of T cell origin [2]. B-cell NHLs include Burkitt’s, diffuse large B-cell, follicular, immunoblastic large cell, precursor B-lymphoblastic, and mantle cell lymphomas. These cancers are generally classified as either indolent or aggressive, which then dictates the type of therapy the patient may receive. In general aggressive NHLs are more treatable than indolent NHLs [3]. Immunotherapeutic approaches targeted to CD20 have revolutionized the treatment of NHL [2]. However, large refractory populations exist that are not responsive to these therapies [4]. Thus, although treatments for NHLs have made great improvements, refractive malignancies still occur that are nonresponsive to current therapies in about half of all patients, indicating that improved treatment strategies are needed [4].
As with any well-designed drug delivery system, the biological target is of great importance. CD20 is one of the most reliable cell surface markers of B lymphocytes [5–8]. Since it remains on cell surface after antibody ligation [5,9] it has become an attractive target for immunotherapies intended to deplete B-cells [9]. CD20 is expressed on most NHL malignant cells as well as on normal B cells. However, it is not expressed on stem cells and mature plasma cells. Consequently, normal numbers of B cells can be restored after treatment [10]. CD20 is a cell cycle regulatory protein [8,11] that either controls or functions as a store operated calcium channel [12]. The protein forms dimers and tetramers [7,8] constitutively associated in lipid rafts of the cell membrane [13]. The fact that crosslinking of CD20 leads to apoptosis is well established [14–17]. Clinical success was reached when Rituximab, a human-murine antibody (Ab) chimera, received FDA approval for the treatment of NHL [2,4,18–20]. It has since been approved for a number of immune disorders and transplant proliferative disorders that have been linked to B cells [21]. Rituximab’s ability to clear B cell populations has been attributed to antibody dependent cellular cytotoxicity, complement dependent cytotoxicity, and apoptosis [14,15,22,23]. To a limited extent, Rituximab can cause homoaggregation of CD20, and induce apoptosis. This response is magnified by crosslinking ligated Rituximab with a secondary antibody or with effector cells [14]. Covalently crosslinked Rituximab preparations enhanced induction of apoptosis and tumor clearance when compared with Rituximab alone [16,17].
The self-assembly of hybrid materials, composed of synthetic and biological macromolecules, is mediated by the biorecognition of biological motifs [24–27]. We have previously designed self-assembling hybrid hydrogel systems composed of a synthetic N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer backbone and coiled-coil peptide motifs; the results showed that it is possible to impose properties of a well-defined coiled-coil peptide onto a whole hybrid hydrogel [28]. Recently, we designed a pair of oppositely charged pentaheptad peptides (CCE and CCK) that formed antiparallel coiled-coil heterodimers (see Figure 1a for explanation of coiled-coil structure) and served as physical crosslinkers [29]. HPMA graft copolymers, CCE-P and CCK-P (P is the HPMA copolymer backbone), self-assembled into hybrid hydrogels with a high degree of biorecognition [29,30].
Figure 1.

Drug-free macromolecular therapeutics. (a) Cartoon of overall design and possible mechanism of treatment of NHL with conjugates of antiparallel coiled-coil forming peptides, CCE and CCK. The induction of apoptosis in human Burkitt’s NHL Raji B cells was triggered by crosslinking of the surface CD20 antigen due to biorecognition processes of antigen-antibody and complementary peptides. Raji B cells were first exposed to Fab’ conjugate with unfolded CCE. Binding to (non-internalizing) CD20 receptor decorated the cell surface with CCE peptide. Subsequent exposure of decorated B cells to the HPMA copolymer containing multiple CCK grafts resulted in the formation of antiparallel coiled-coil heterodimers and apoptosis. The diagram is not drawn to scale. Inset: Helical wheel representation of two-stranded, antiparallel α-helical coiled coils formed by the dimerization of CCE and CCK. The primary sequence of a typical coiled-coil is composed of 7-residue repeats, designated as heptads. The amino acid residues in a heptad are conventionally denoted as “a, b, c, d, e, f, g”. Hydrophobic residues at positions “a” and “d” form an inter-helical hydrophobic core, providing a stabilizing interface between the helices. Charged residues at positions “e” and “g” form electrostatic interactions, which contribute to coiled-coil stability and mediate specific association among helices. The view is shown looking down the superhelical axis from the N-terminus of CCE and from the C-terminus of the CCK. CC denotes the coiled-coil peptides. E and K denote peptides in which most of the e and g positions are occupied by either glutamic acid or lysine, respectively. The sequences are written in the one-letter amino acid code. (b) Schematic outline of preparations of peptide-conjugates, Fab’-CCE and CCK-P. (c) Circular dichroism spectrum of the equimolar mixture of CCE and CCK in PBS. (d) Confocal fluorescence images of Raji B cells after exposure to the mixture of Fab’-CCE + CCK-P showing biorecognition of CCE/CCK on the surface of Raji B cells within 4 h.
In our previous report [31] we hypothesized that this unique biorecognition of CCK and CCE peptide motifs could be expanded past biomaterials design and be applied to a living system and mediate a biological process. This would provide a bridge between the designs of biomaterials and macromolecular therapeutics. We presented a new paradigm for apoptosis induction in B cells [31] based on the biorecognition of peptide motifs at cell surface and formation of antiparallel coiled-coils with concomitant crosslinking of CD20 receptors (Figure 1a). The new model for treating NHL is based on the biorecognition of peptide motifs at cell surface. This binary system consists of a pair of complementary peptides (CCE and CCK) [29]; one attached to Fab’ fragment of 1F5 Ab [9] (Fab’-CCE) and the other as grafts to HPMA–based graft copolymer (CCK-P) (Figure 1b). CCE and CCK form antiparallel coiled-coil heterodimers in saline (Figure 1c). It is important to note that, in contrast to Rituximab, ligated 1F5 requires crosslinking to generate a biological response of apoptosis [14]. Exposure of CD20+ Raji B cells to Fab’-CCE results in decoration of the cell surface with multiple copies of CCE via antigen-antibody fragment biorecognition (CD20 is a non-internalizing receptor; thus decoration of Raji B cell surface with peptides holds for hours as demonstrated in Figure 1d). Subsequent exposure of the decorated cells to CCK-P produces CCE/CCK coiled-coil heterodimers at the cell surface. This second biorecognition event causes crosslinking of CD20 receptors and triggers apoptosis of Raji B cells in vitro as identified by caspase 3, TUNEL, and Annexin V apoptotic assays [31].
Here, we used an in vivo model of NHL, SCID (C.B.-17) mice bearing human Raji cell B-lymphoma xenografts, with the aim to evaluate if the biorecognition of CCE/CCK sequences results in tumor treatment.
2. Materials and methods
2.1 Preparation of conjugates Fab’-CCE and CCK-P
The structures of coiled-coil forming peptides, CCE and CCK, are shown below; they were prepared by solid phase peptide synthesis and their structure validated with mass spectrometry and reversed phase HPLC, as previously reported [29–31].

Fab’-CCE and CCK-P were synthesized [31] following the schemes shown in Figure 1b. The details are briefly described below:
Fab’-CCE conjugate
Anti-CD20 mAb 1F5, prepared from the anti-CD20 hybridoma clone 1F5 (ATTC, Bethesda, MD) in a CellMax© bioreactor (Spectrum Laboratories, Rancho Dominguez, CA) as previously described [32], was digested into F(ab’)2 with lysyl endopeptidase (Wako Chemicals USA, Richmond, VA). F(ab’)2 was then labeled with Rhodamine Red-X succinimidyl ester (Invitrogen, Carlsbad, CA) to introduce a fluorophore that facilitated the fluorescence microscopy investigation. Briefly, 40 µl of Rhodamine Red-X succinimidyl ester solution (5 mM) in DMF was added into 3 ml of F(ab’)2 solution (3 mg/ml) in PBS (pH 7.4). The pH of the mixture was gradually adjusted to 8.3 using 0.1 N NaOH under constant stirring. Additional 30 min were allowed to complete the labeling reaction. The labeled F(ab’)2 was then purified twice using a PD10 column (GE Healthcare, Buckinghamshire, UK) to remove the unreacted labeling agent.
Fab’-CCE was obtained by conjugation of Fab’ with CCE using maleimide-thiol chemistry. Immediately prior to use, the labeled F(ab′)2 was reduced to Fab’ with 10 mM tris(2-carboxyethyl) phosphine hydrochloride (TCEP) in PBS (pH 7.4) containing 5 mM EDTA for 1 h at 37 °C in the dark. CCE (1.5× in excess to Fab’) was added and the coupling reaction proceeded at 4 °C in the dark overnight. The crude product was then purified twice using a PD10 column.
CCK-P conjugate
To synthesize HPMA copolymer grafted with peptide CCK, HPMA was copolymerized with N-(3-aminopropyl)methacrylamide (MA-NH2) in methanol at 50 °C using AIBN as the initiator as previously described [29]. N-methacryloylaminopropyl fluorescein thiourea (MA-FITC) was optionally used when a fluorescent probe in polymer was needed. The molecular weight and molecular weight distribution of the copolymer were measured on the ÄKTA FPLC system (Amersham Pharmacia Biotech, Piscataway, NJ) equipped with UV and RI detectors using a Superose 6HR10/30 column with PBS (pH=7.4) as the mobile phase. Linear polyHPMA fractions were employed for calibration of the column. The copolymer (P-NH2) yield was 82% with an amine content of 397 nmol/mg (5.8 mole%) as determined by ninhydrin assay. The number average molecular weight was 99.6 kDa (Mw/Mn = 2.26).
P-NH2 was further reacted with succinimidyl trans-4-(maleimidylmethyl)cyclohexane-1-carboxylate (SMCC) (molar ratio [NH2]:[SMCC] = 1:1.5) in DMF in the presence of triethylamine (3× [NH2]) at room temperature overnight. After precipitation into acetone/ether (3:2), the product was re-dissolved in methanol and precipitated into acetone twice to remove unreacted SMCC. The HPMA copolymer with maleimide side chain (P-mal) was obtained. The maleimide content was 285 nmol/mg (4.35 mole %) as measured by the modified Ellman’s assay.
Attachment of CCK to P-mal was accomplished via thioether bond formed by the reaction of maleimide with cysteine at the N-terminus of CCK ([Mal]:[CCK] = 1:1). The coupling reaction proceeded at room temperature overnight with analytical RP-HPLC monitoring the progress. The conjugate was then dialyzed against DI water using a dialysis tube with molecular weight cutoff 6–8 kDa (to remove unreacted CCK) and then lyophilized.
The graft content in the resulting copolymer was determined by amino acid analysis. The copolymer (2.08 mg) was hydrolyzed in 0.2 ml 6 N HCl at 120 °C for 16 h in a sealed ampoule and then dried under vacuum. Precolumn derivatization with ophthaldialdehyde was used and the samples were analyzed by HPLC with fluorescence detector (excitation 229 nm and emission 450 nm) using gradient elution from buffer A to buffer B, where buffer A: 0.05 M sodium acetate, pH 6.0, and buffer B: 70% methanol in buffer A [31]. The average number of CCK grafts per macromolecule was determined as 8.94. Thus, we denoted the HPMA copolymer grafted with CCK as (CCK)9-P.
2.2 Cell line and systemic NHL SCID mouse model
Human Burkitt’s B-cell NHL Raji cells (ATCC, Manassas VA) were cultured as described before [31]. The cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere of 5% CO2. Cells at the exponential growth phase were used for mice inoculation.
Female C.B-17 SCID mice (~6 weeks old) were purchased from Charles River Laboratories (Wilmington, MA) and maintained following the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Utah. Following the animal models described by Ghetie et al. [33] and Griffiths et al. [34], mice with body weight around 20 g were intravenously injected with 4×106 Raji cells in 200 µl normal saline (N.S.) via the tail vein. The onset of hind limb paralysis time is the end point of this animal model [34].
2.3 In vivo efficacy studies
Inoculated mice were divided into five groups: Control, group without treatment; CS, consecutive administration of single dose; PS, premixed administration of single dose; CM, consecutive administration of multiple (3) doses; and PM, premixed administration of multiple (3) doses. Here consecutive administration involved the i.v. injection of Fab’-CCE first and 1 h later the (CCK)9-P conjugate were applied via the same route; while in the premixed administration, the two conjugates were first mixed outside the body for 1 h, and then injected via the tail vein. For single-dose groups, conjugates were administered 24 h post inoculation; for multiple-dose groups, conjugates were applied 24, 72, 120 h post inoculation.
The doses in all treatments were 50 µg/20 g Fab’-CCE and 324 µg/20 g (CCK)9-P. This combination ensured the molar ratio between CCE and CCK to be 1:25, a proved efficient ratio to allow for maximum apoptosis induction in vitro [31].
Post-operation monitoring was exercised at least once a day. Major aspects of the mice closely assessed included body weight, physical appearance (e.g. hunched back, piloerection), food intake and hind limb paralysis. Body weight of mice was monitored every day. Animals were sacrificed at signs of sickness, such as the onset of hind-limb paralysis or when body weight loss was >20%. Animals without signs of paralysis were sacrificed at 100 days. Statistical comparison between groups was performed with the log-rank test using Prism (Windows version 5.01, GraphPad) software.
2.4 Residual Raji cell analysis
Bone marrow cells were collected by purging fresh femurs with cold PBS and then 5 volumes of cold red blood lysing solution was added into 1 volume bone marrow cell suspension. The mixture was incubated for 2–3 min at room temperature and centrifuged to remove cell debris. After washing with cold washing buffer once, the bone marrow cells were divided into three tubes (~50 µl each) and suspended again in cold washing buffer. One tube was spared as control, and the other two tubes were stained by Raji cell specific primary antibodies [35]: R-phycoerythrin (PE) mouse anti-human CD10 (mouse IgG1, κ isotype) and allophycocyanin (APC) mouse anti-human CD19 (mouse IgG1, κ isotype). Both antibodies were purchased from BD Biosciences (San Jose, CA). Twenty microliter fluorescently labeled antibody (CD10 or CD19) was added and incubated with processed bone marrow cells for 40 min at 4 °C in the dark. Finally, flow cytometry was employed to identify the residual Raji tumor cells.
2.5 Pathological examinations and histopathological evaluation
Immediately after the sacrifice, organs were harvested: brain, heart, lung, liver, spleen, kidneys, and femurs. Except femurs, all other organs were fixed and preserved in formalin at 4 °C and then sent for pathological examinations; the organs were mounted 4-µm sections on glass slides and stained by hematoxylin and eosin. Harvested organs were evaluated by a veterinary pathologist.
2.6 Macrophage activation in response to peptides CCE, CCK, the mixture of CCE/CCK, and HPMA copolymer - peptide conjugate
Murine macrophage RAW 264.7 cell line [36] was maintained in DMEM medium supplemented with 10% fetal bovine serum. RAW 264.7 cells were plated at 8×104 cells in 200 µl per well in 96-well tissue culture plates. After 48 h, cells were incubated with peptides and HPMA copolymer-peptide conjugate for 1 and 7 days, respectively in the presence of 5 µg/ml polymyxin B. Polymyxin B was used to inhibit LPS contamination. Cells incubated with solvent used for dissolving conjugates were also used as the negative control. After incubation, supernatants in 4 replicate wells were collected and the release of TNF-α was measured by ELISA (R&D Systems, Minneapolis, MN).
3. Results and Discussion
The translational potential of hybrid macromolecular systems, composed from both natural and synthetic molecules, is immense [26]. The design of drug free macromolecular therapeutics was inspired by our previous work on the self-assembly of hybrid materials composed of synthetic and biological macromolecules. Individually, the oppositely charged pentaheptad (35 amino acid) peptides, CCE and CCK, are random coils, but their equimolar mixture formed antiparallel coiled-coil heterodimers [29]. Equimolar mixtures of HPMA graft copolymers, CCE-P and CCK-P, self-assembled into hybrid hydrogels with a high degree of biorecognition [29,30]. In vitro exposure of Raji B cells to Fab’-CCE followed by subsequent exposure to (CCK)9-P resulted in high degree of apoptosis in the presence of 10% of fetal calf serum [31].
Here, we examined the biorecognition of coiled-coil forming peptides CCE and CCK in an in vivo situation. To this end, the therapeutic efficacy of our new strategy in a preclinical animal model was evaluated. We used the human Raji B cell line and female SCID (C.B.-17) mice (6–7 weeks of age) bearing human B-lymphoma xenografts [33,34].
Post-operation, the mice were monitored daily for hind limb paralysis and general physical appearance such as weight changes, food intake, piloerection, and hunched back. The mice were sacrificed upon onset of paralysis; otherwise the mice were kept until 100 days and considered long-term survivors.
Preliminary experiment with three mice per group was conducted for dose selection (Figure 2). For each treatment, 50 µg/20 g of Fab’-CCE and 324 µg/20 g of CCK-P were administered, respectively. This value was based on in vitro apoptosis induction experiments: the molar ratio between CCE and CCK 1:25 allowed for maximum apoptosis induction [31]. One mouse out of three in PM group and two out of three in CM group survived for 100 days without paralysis (Figure 2), which indicated the dose selected was appropriate.
Figure 2.
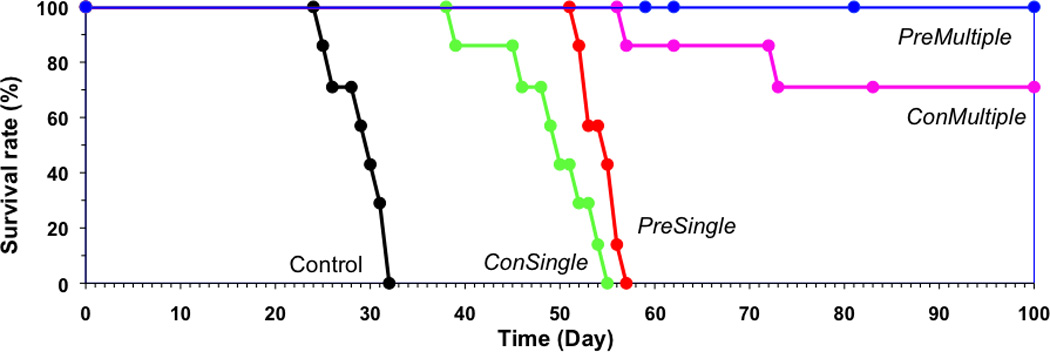
Preliminary evaluation of therapeutic efficacy of drug-free macromolecular therapeutics against systemically disseminated Raji B cell lymphoma in C.B.-17 SCID mice (3 mice per group). Four million Raji B cells were injected into the tail vein on day 0 to initiate the disseminated disease. The incidence of hind-limb paralysis or survival of mice was monitored up to 100 days. Five groups of animals were evaluated: i) untreated (Control); ii) consecutive administration of single dose (CS); iii) premixed administration of single dose (PS); iv) consecutive administration of three doses at days 1, 3, and 5 (CM); and v) premixed administration of three doses at days 1, 3, and 5 (PM). Consecutive administration involved the i.v. injection of 50 µg/20 g Fab’-(CCE)1 first and 1 h later the i.v. administration of 324 µg/20 g (CCK)9-P conjugate; in the premixed administration, the two conjugates were first mixed outside the body, kept for 1 h, and then injected via the tail vein.
We repeated the experiment on a larger group of animals (seven mice per groups). The results are shown in Figure 3a. Untreated (control) mice developed hind-limb paralysis within 24–32 days. This observation was in good agreement with literature [34,35,37]. Single dose groups had a substantially longer survival time (38–57 days) than controls; whereas the group that received three doses produced 5 out of 7 long-term survivors (100 days) for consecutive administration, and 7 out of 7 long-term survivors for premixed administration group.
Figure 3.

Therapeutic efficacy of drug-free macromolecular therapeutics against systemically disseminated Raji B cell lymphoma in C.B.-17 SCID mice (7 mice per group). (a) Top panel shows timeline for the in vivo efficacy study. Four million Raji B cells were injected into the tail vein on day 0 to initiate the disseminated disease. The incidence of hind-limb paralysis or survival of mice was monitored until day 100. Five groups of animals were evaluated: untreated controls; consecutive administration of single dose (CS); premixed administration of single dose (PS); consecutive administration of three doses at days 1, 3, and 5 (CM); and v) premixed administration of three doses at days 1, 3, and 5 (PM). Consecutive administration involved the i.v. injection of 50 µg/20 g Fab’-CCE first and 1 h later the i.v. administration of 324 µg/20 g CCK-P conjugate; For premixed administration, the two conjugates were mixed together 1 h before injection via the tail vein. Bottom panel shows survival rate of tumor-bearing mice that received above treatments. The curve was presented in a Kaplan-Meier plot with indication of numbers of long-term survivors (7 mice per group); (b) Estimation of residual Raji B lymphoma cells in the bone marrow. Shown are results from representative mice that received the indicated treatment. Revealed are histograms of bone marrow cells isolated from mice (as indicated) followed by staining with PE mouse anti-human CD10 and APC mouse anti-human CD19. (c) Preliminary evaluation of immunogenicity. TNF-α released from RAW 264.7 cells upon exposure to peptides (1 day) and HPMA copolymer-peptide conjugate (7 days) in vitro. The values are shown as averages (n = 4) ± S.D.
Statistical analyses showed that all administration modes of the drug-free macromolecular therapeutic system produced statistically significant (P<0.002) enhancement of treatment efficacy compared with the control group. In addition, multiple treatments resulted in significantly (P<0.001) higher effectiveness than the single dose treatment. However, the differences in treatment efficacy of consecutive and premixture treatments were not statistically significant.
To further assess that the surviving mice in the therapy groups (specially PM and CM) were tumor free, we sacrificed the mice to detect if there was any residual Raji cells in the bone marrow [35]. Two fluorescently labeled mouse anti-human antibodies, PE labeled mouse anti-human CD10 and APC labeled mouse anti-human CD19, have been used for flow cytometry analysis. As shown in Figure 3b, the presence of residual Raji B cells was confirmed in control group, and also in mice that received single-dose treatment (and developed paralysis); however, no residual malignant B cells were detected in mice treated with three doses that survived for 100 days without sign of hind-limb paralysis.
Harvested organs (lung, kidney, spleen, liver, heart and brain) were evaluated by a veterinary pathologist. Results revealed that there was acute congestion in all tissues (lung, kidney, liver, spleen, heart) with minimal variation in lesions between the individuals. Smooth muscle hypertrophy of the lung tissues was also observed. Prominent extramedullary hematopoiesis occurred in the spleens, which demonstrated the reserved ability of blood cell reestablishment after treatments. Importantly, no toxicity of the treatment was suggested in any of the tissues evaluated.
Immunogenicity is a major biocompatibility concern when peptide/protein-based therapeutic agents are used for treatment of human diseases. We have evaluated the potential of coiled-coil forming peptides to activate RAW 264.7 macrophages in vitro. The release of the inflammatory cytokine, TNF-α (tumor necrosis factor-α) has been frequently used as a first line of biocompatibility evaluation [36]. We incubated the macrophages with free peptides CCK and CCE; with CCK peptide grafted in multiple copies to HPMA copolymer, CCK-P (to increase the molecular weight and evaluate if the peptide may act as a hapten), and with a premixed equimolar mixture of CCE/CCK (to evaluate the impact of peptide conformation). The RAW 264.7 macrophages were incubated with 1, 10, 100, and 1000 nM of peptides. The results bode well for the biocompatibility of the approach – minimal TNF-α release was observed even at the highest concentrations used. The maximum release was only 2.5 times larger than non-treated controls (Figure 3c). We are aware that numerous tests need to be done before a conclusion on the immunogenicity of peptides can be made. We have performed such tests in the past with shorter oligopeptide sequences attached to HPMA copolymers [38–42]. HPMA copolymers with oligopeptide side chains behaved as thymus independent antigens with low immunogenicity [39]. The intensity of response depended on the structure of the side-chains, dose, and genetic background of the mice [38]. Neither the homopolymer nor the HPMA copolymers possessed mitogenic activity. The immune response towards oligopeptides attached to HPMA copolymers was about 4 orders of magnitude lower when compared with bovine gamma globulin [42]. Generally, attachment of peptides and/or antibodies to HPMA copolymers results in a decrease in their immunogenicity [39,41].
4. Conclusions
In summary, we presented a new paradigm for NHL treatment, drug-free macromolecular therapeutics, based on the biorecognition of peptide sequences at the cell surface, formation of antiparallel coiled-coil heterodimers, and crosslinking of CD20 receptors with concomitant induction of apoptosis. This approach produced a large number of long-term survivors in a disseminated NHL model in SCID mice. The difference between consecutive and pre-mixture treatment were statistically not significant. However, we believe that the two-step (consecutive) delivery possesses an advantage for the translation into the clinics. As shown by other groups [43–46] maximal tumor concentration of antibody (in our case antibody fragment-CCE conjugate) is achieved after 12 – 24 hours. Finding the optimal time when to administer the second part of the delivery system (graft copolymer CCK-P) will result in enhanced, specific tumor accumulation with favorable tumor to tissue ratio. The two-step pre-targeting system has the potential to optimize the delivery based on the individual patient condition. Thus our approach, in addition to providing an effective alternative to NHL treatment, may potentially serve as a model for other biocompatible drug-free macromolecular therapeutics.
Acknowledgements
This study was supported in part by NIH grants EB00588 and GM95606 (to JK). KW acknowledges support from the American Foundation for Pharmaceutical Education (AFPE). We thank Y. Zhou and M. Sima for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J. Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin's lymphoma. N. Engl. J. Med. 2008;359:613–626. doi: 10.1056/NEJMra0708875. [DOI] [PubMed] [Google Scholar]
- 3.Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J. Clin. Oncol. 1998;16:2780–2795. doi: 10.1200/JCO.1998.16.8.2780. [DOI] [PubMed] [Google Scholar]
- 4.Molina A. A decade of Rituximab: Improving survival outcomes in non-Hodgkin’s lymphoma. Annu. Rev. Med. 2008;59:237–250. doi: 10.1146/annurev.med.59.060906.220345. [DOI] [PubMed] [Google Scholar]
- 5.Press OW, Farr AG, Borroz KI, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting human B-cell malignancies. Cancer Res. 1989;49:4906–4912. [PubMed] [Google Scholar]
- 6.Riaz W, Hernandez-Ilizaliturri FJ, Czuczman MS. Strategies to enhance rituximab anti-tumor activity in the treatment of CD20-positive B-cell neoplasms. Immunol. Res. 2010;46:192–205. doi: 10.1007/s12026-009-8121-x. [DOI] [PubMed] [Google Scholar]
- 7.Tedder TF, McIntyre G, Schlossman SF. Heterogeneity in the B1 (CD20) cell surface molecule expressed by human B-lymphocytes. Mol. Immunol. 1988;25:1321–1330. doi: 10.1016/0161-5890(88)90047-8. [DOI] [PubMed] [Google Scholar]
- 8.Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol. 1994;15:450–454. doi: 10.1016/0167-5699(94)90276-3. Today. [DOI] [PubMed] [Google Scholar]
- 9.Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, Thomas ED. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987;69:584–591. [PubMed] [Google Scholar]
- 10.Anderson KC, Bates MP, Slaugenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63:1424–1433. [PubMed] [Google Scholar]
- 11.Golay JT, Clark EA, Beverley PC. The CD20 (Bp35) antigen is involved in activation of B cells from the G0 to the G1 phase of the cell cycle. J. Immunol. 1985;135:3795–3801. [PubMed] [Google Scholar]
- 12.Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF. Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in B lymphocytes. J. Cell Biol. 1993;121:1121–1132. doi: 10.1083/jcb.121.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deans JP, Li H, Polyak MJ. CD20-mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176–182. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–1652. [PubMed] [Google Scholar]
- 15.Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol. Dis. 2000;26:133–143. doi: 10.1006/bcmd.2000.0287. [DOI] [PubMed] [Google Scholar]
- 16.Ghetie MA, Bright H, Vitetta ES. Homodimers but not monomers of Rituxan (chimeric anti-CD20) induce apoptosis in human B-lymphoma cells and synergize with a chemotherapeutic agent and an immunotoxin. Blood. 2001;97:1392–1398. doi: 10.1182/blood.v97.5.1392. [DOI] [PubMed] [Google Scholar]
- 17.Zhang N, Khawli LA, Hu P, Epstein AL. Generation of rituximab polymer may cause hyper-cross-linking-induced apoptosis in non-Hodgkin's lymphomas. Clin. Cancer Res. 2005;11:5971–5980. doi: 10.1158/1078-0432.CCR-05-0554. [DOI] [PubMed] [Google Scholar]
- 18.Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JF, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. [PubMed] [Google Scholar]
- 19.McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 20.McLaughlin P, White CA, Grillo-López AJ, Maloney DG. Clinical status and optimal use of rituximab for B-cell lymphomas. Oncology (Williston Park) 1998;12:1763–1769. discussion 1769–1770, 1775–1777. [PubMed] [Google Scholar]
- 21.Boye J, Elter T, Engert A. An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann. Oncol. 2003;14:520–535. doi: 10.1093/annonc/mdg175. [DOI] [PubMed] [Google Scholar]
- 22.Maloney DG. Mechanism of action of Rituximab. Anticancer Drugs. 2001;12 Suppl 2:S1–S4. [PubMed] [Google Scholar]
- 23.Johnson P, Glenie M. The mechanism of action of rituximab in the elimination of tumor cells. Semin. Oncol. 2003;30(1 Suppl. 2):3–8. doi: 10.1053/sonc.2003.50025. [DOI] [PubMed] [Google Scholar]
- 24.Kopeček J. Smart and genetically engineered biomaterials and drug delivery systems. Eur. J. Pharm. Sci. 2003;20:1–16. doi: 10.1016/s0928-0987(03)00164-7. [DOI] [PubMed] [Google Scholar]
- 25.Kopeček J Hydrogels. From Soft Contact Lenses and Implants to Self-Assembled Nanomaterials. J. Polym. Sci: Part A: Polym. Chem. 2009;47:5929–5946. doi: 10.1002/pola.23607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopeček J, Yang J. Peptide-directed self-assembly of hydrogels. Acta Biomaterialia. 2009;5:805–816. doi: 10.1016/j.actbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths PC, Paul A, Apostolovic B, Klok H-A, de Luca E, King SM, Heenan RK. Conformational consequences of cooperative binding of a coiled-coil peptide motif to poly(N-(2-hydroxypropyl) methacrylamide) HPMA copolymers. J. Control. Release. 2011 doi: 10.1016/j.jconrel.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Stewart RJ, Kopeček J. Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature. 1999;397:417–420. doi: 10.1038/17092. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Xu C, Wang C, Kopeček J. Refolding hydrogels self-assembled from HPMA graft copolymers by antiparallel coiled-coil formation. Biomacromolecules. 2006;7:1187–1195. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Wu K, Koňák Č, Kopeček J. Dynamic light scattering study of the self-assembly of HPMA hybrid graft copolymers. Biomacromolecules. 2008;9:510–517. doi: 10.1021/bm701001f. [DOI] [PubMed] [Google Scholar]
- 31.Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Drug-free macromolecular therapeutics: Induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew. Chem. Int. Ed. 2010;49:1451–1455. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RN, Kopečková P, Kopeček J. Synthesis and evaluation of multivalent branched HPMA copolymer-Fab’ conjugates targeted to B-cell antigen CD20. Bioconjugate Chem. 2009;20:129–137. doi: 10.1021/bc800351m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghetie MA, Tucker K, Richardson J, Uhr JW, Vitteta ES. The antitumor activity of an anti-CD22 Immunotoxin in SCID mice with disseminated Daudi lymphoma is enhanced by either an anti-CD19 antibody or an anti-CD19 immunotoxin. Blood. 1992;80:2315–2320. [PubMed] [Google Scholar]
- 34.Griffiths GL, Mattes MJ, Stein R, Govindan SV, Horak ID, Hansen HJ, Goldenberg DM. Cure of SCID mice bearing human B-lymphoma xenografts by an anti-CD74 antibody-anthracycline conjugate. Clin. Cancer Res. 2003;9:6567–6571. [PubMed] [Google Scholar]
- 35.Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood. 2010;115:4778–4786. doi: 10.1182/blood-2009-12-257386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Schutte RJ, Abu-Shakra A, Reichert WM. Protein array method for assessing in vitro biomaterial-induced cytokine expression. Biomaterials. 2005;26:1081–1085. doi: 10.1016/j.biomaterials.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 37.Hernandez-Ilizaliturri FJ, Jupudy V, Ostberg J, Oflazoglu E, Huberman A, Repasky E, Czuczman MS. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin. Cancer Res. 2003;9:5866–5873. [PubMed] [Google Scholar]
- 38.Říhová B, Kopeček J, Ulbrich K, Pospíšil J, Mančal P. Effect of the chemical structure of N-(2-hydroxypropyl)methacrylamide copolymers on their ability to induce antibody formation in inbred strains of mice. Biomaterials. 1984;5:143–148. doi: 10.1016/0142-9612(84)90048-6. [DOI] [PubMed] [Google Scholar]
- 39.Říhová B, Kopeček J, Ulbrich K, Chytrý V. Immunogenicity of N-(2-hydroxypropyl)methacrylamide copolymers. Makromol. Chem. 1985 Suppl. 9:13–24. [Google Scholar]
- 40.Šimečková J, Říhová B, Plocová D, Kopeček J. Activity of complement in the presence of N-(2-hydroxypropyl)methacrylamide copolymers. J. Bioact. Comp. Polym. 1986;1:20–31. [Google Scholar]
- 41.Hart P, Kopečková P, Omelyanenko V, Enoutina E, Kopeček J. HPMA copolymer-modified avidin: Immune response. J. Biomat.Sci. Polym. Ed. 2000;11:1–12. doi: 10.1163/156856200743454. [DOI] [PubMed] [Google Scholar]
- 42.Kopeček J. Controlled biodegradability of polymers - a key to drug delivery systems. Biomaterials. 1984;5:19–25. doi: 10.1016/0142-9612(84)90062-0. [DOI] [PubMed] [Google Scholar]
- 43.Goodwin DA, Meares CF. Pretargeting, Cancer. 1997;80:2675–2680. doi: 10.1002/(sici)1097-0142(19971215)80:12+<2675::aid-cncr45>3.3.co;2-x. [DOI] [PubMed] [Google Scholar]
- 44.Goodwin DA, Meares CF. Advances in pretargeting technology. Biotechnology Advances. 2001;19:435–450. doi: 10.1016/s0734-9750(01)00065-9. [DOI] [PubMed] [Google Scholar]
- 45.He J, Liu G, Gupta S, Zhang Y, Rusckowski M, Hnatowich DJ. Amplification targeting: A modified pretargeting approach with potential for signal amplification – proof of a concept. J. Nucl. Med. 2004;45:1087–1095. [PubMed] [Google Scholar]
- 46.Sharkey RM, Karacay H, Litwin S, Rossi EA, McBride WJ, Chang C-H, Goldenberg DM. Improved therapeutic results by pretargeted radioimmunotherapy of non-Hodgkin’s lymphoma with a new recombinant, trivalent, anti-CD20, bispecific antibody. Cancer Res. 2008;68:5282–5290. doi: 10.1158/0008-5472.CAN-08-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]