

Abstract
Background
Notch signaling in vascular smooth muscle precursors is required for smooth muscle differentiation. Jagged1 expression on endothelium activates Notch in vascular smooth muscle precursors including those of neural crest origin to initiate the formation of a smooth muscle layer in a maturing blood vessel.
Methods and Results
Here, we show that Jagged1 is a direct Notch target in smooth muscle, resulting in a positive feedback loop and lateral induction that propagates a wave of smooth muscle differentiation during aortic arch artery development. In vivo, we show that Notch inhibition in cardiac neural crest impairs Jagged1 mRNA expression and results in deficient smooth muscle differentiation and resultant aortic arch artery defects. Ex vivo, Jagged1 ligand activates Notch in neural crest explants and results in activation of Jagged1 mRNA, a response that is blocked by Notch inhibition. We examine 15 evolutionary conserved regions within the Jagged1 genomic locus and identify a single Notch response element within the second intron. This element contains a functional Rbp-J binding site demonstrated by luciferase reporter and chromatin immunoprecipitation assays and is sufficient to recapitulate aortic arch artery expression of Jagged1 in transgenic mice. Loss of Jagged1 in neural crest impairs vascular smooth muscle differentiation and results in aortic arch artery defects.
Conclusions
Taken together, these results provide a mechanism for lateral induction that allows for a multilayered smooth muscle wall to form around a nascent arterial endothelial tube and identify Jagged1 as a direct Notch target.
Keywords: Muscle, smooth; Heart defects, congenital; Vasculature
Vascular smooth muscle is derived from multiple embryonic sources including neural crest and lateral plate mesoderm. 1, 2 Notch signaling in vascular smooth muscle precursors is required for smooth muscle differentiation. The Notch ligand, Jagged1, is expressed by endothelium and activates Notch in vascular smooth muscle precursors to initiate the formation of a smooth muscle layer in a maturing blood vessel. 3 Thus, inhibition of Notch signaling in neural crest leads to impaired smooth muscle differentiation and aortic arch artery defects, although neural crest migration is unaffected. 4 In vitro studies suggest that Notch directly regulates smooth muscle α-actin expression in vascular smooth muscle cells. 5, 6
The importance of the Notch signaling in the endothelium and vascular smooth muscle is further highlighted by the spectrum of cardiovascular defects associated with mutations of Notch ligands or receptors. Mutations in NOTCH3, encoding a Notch receptor found in vascular smooth muscle, cause the autosomal dominant disorder CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) syndrome. 7, 8 Notch3 knockout mice, though viable, show diminished expression of some vascular smooth muscle markers in a subset of arteries, indicating that Notch may promote some aspects of smooth muscle differentiation or maturation in vivo. 9 Mutations in NOTCH2 or the Notch ligand JAGGED1 are associated with Alagille syndrome (AGS). 10, 11 AGS is a multifaceted disorder including congenital heart defects and vascular pathologies. 10–12 Mice lacking Jagged1 die early in development due to defective remodeling of both the embryonic and yolk sac vasculature. 13 Similar vascular defects are observed when Jagged1 is specifically deleted in the endothelium. 3 Jagged1 is also expressed by smooth muscle, and deletion using SM22-Cre resulted in the absence of intrahepatic bile ducts 14, a feature of Alagille syndrome. Deletion of Jagged1 in murine smooth muscle, again utilizing SM22-Cre, is also associated with early postnatal mortality due to patent ductus arteriosus (PDA), a common human congenital heart defect. 15 Normally, the ductus arteriosus closes at birth, but defective smooth muscle differentiation following Jagged1 deletion prevents proper vessel remodeling. 15
Notch is a highly-conserved signaling pathway. In mammals, association of one of four Notch receptors (Notch 1–4) with one of five Notch ligands (Jagged1, Jagged2, Delta-like 1 (Dll1), Delta-like 3 (Dll3) and Delta-like 4 (Dll4)) initiates juxtacrine signaling. Following ligand-receptor association, proteolytic cleavage releases the Notch intracellular domain (NICD) which translocates to the nucleus. NICD then forms an active transcriptional complex with the DNA-binding protein Rbp-J and the coactivator Mastermind-like (MAML) and target genes are transcribed. 16 Classically, Notch signaling has been thought to function through lateral inhibition in which a stochastic decision by one cell prevents adjacent cells from adopting the same cell fate. 16, 17 This asymmetry in cell fate is typically associated with a decrease of Notch ligand expression in neighboring cells imparting a selective advantage to the single differentiating cell. 16, 17 Alternatively, Notch can function as part of a positive feedback loop in which Notch receptor activation promotes Notch ligand expression in surrounding cells thus relaying a signal – a process known as lateral induction. 18
Lateral induction of Notch signaling has been documented in diverse physiological systems. 19 In the developing inner ear, for example, over expression of Notch both induces Jagged1 expression and sensory specification from nonsensory epithelium. 20 Additional examples of Notch/Jagged lateral induction can be found both in macrophages and the ocular lens. 21, 22 In studies most relevant to vascular development, endothelial Jagged1 expression activates Notch3 in mural cells resulting in increased Jagged1 expression, and Jagged1 protein is decreased in retinal blood vessels of Notch3 knockout animals. 23
In this report, we demonstrate that smooth muscle precursors derived from neural crest up-regulate Jagged1 mRNA upon Notch activation. We show that Jagged1 is a direct Notch target and we identify a Notch response element in the Jagged1 genomic locus that mediates vascular smooth muscle lateral induction. These results help to explain how a multilayered vascular smooth muscle wall forms around a nascent endothelial tube.
Methods
Mice
All mice were maintained on a mixed genetic background. Pax3Cre 24, DNMAML 25 and Jagged1flox/+ 26 mice were genotyped as previously described. ECR6-hsp68-LacZ stable mice were genotyped using the following primers:
ECR6Tg Forward: 5′ CGGACCTACAACCACTAACAG 3′
ECR6Tg Reverse: 5′ GGTAACGCCAGGGTTTTCCCAGTC 3′
All animal protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Histology, immunohistochemistry and in situ hybridization
Samples were harvested, fixed overnight in 4% paraformaldehyde and subsequently dehydrated through an ethanol series. All samples were then paraffin embedded and sectioned. Antibodies used for immunohistochemistry were anti-Jag1 rabbit polyclonal H-114 (sc-8303, Santa Cruz Biotechnology, Inc.) and anti-αSMA mouse monoclonal 1A4 (Sigma-Aldrich). The Jagged1 and HRT1 in situ probes utilized were previously published. 4 Radioactive in situ hybridizations were completed as detailed in a previous protocol. 27
Primary vascular smooth muscle cell culture
To isolate neural crest-derived smooth muscle, the aortic arch to the origin of the left subclavian artery and the proximal regions of the major arch vessels were dissected from E17.5 or E18.5 embryos. Primary smooth muscle cell cultures were performed according to previously published protocols for aortic vascular smooth muscle cells. 28 Jagged1-Fc and control-Fc constructs were kindly provided by Tom Kadesch and have been described elsewhere. 29 Smooth muscle cells were stimulated by plating on Jagged1-Fc or control-Fc coated plates at a density of 2.5 × 105 per 10 cm plate. Cells were harvested 24 hours following stimulation and RNA was extracted using Trizol reagent (Invitrogen). Semi-quantitative RT-PCR was performed using standard protocols and the following primers:
Jagged1 forward: 5′-GCTTCCACTGGCACTGGTAGTTTC-3′
Jagged1 reverse: 5′-TGCTGACATCAAATCCCCCCTC-3′
GAPDH forward: 5′-ACCACAGTCCATGCCATCAC-3′
GAPDH reverse: 5′-GAAGTCACAGGAGACAACCTGGTC-3′
Sequence analysis
A comparative sequence alignment was completed using the NCBI DCODE resource (www.dcode.org) to ascertain evolutionarily conserved regions (ECRs) within and surrounding the Jagged1 genomic locus. This analysis encompassed approximately 30 kb upstream, all intronic regions (~40 kb) and 10 kb downstream of the Jagged1 locus on mouse chromosome 2. The ECRs were identified as elements of at least 200 bp bearing a minimum sequence identity of 70% between mouse, human, dog and opposum. This analysis identified 15 ECRs. All conserved regions were amplified with primers containing a 5′ attB1 site and a 3′ attB2 site to allow for Gateway recombination into pDONR221 (Invitrogen). Genomic locations of all ECRs are listed in Supplemental Table 1 while primer sequences can be found in Supplemental Table 2. All ECRs were sequence verified following insertion into pDONR221. Conserved elements were then recombined into a Gateway compatible pGL3-Promoter vector for use in luciferase assays (Promega, vector courtesy of R. Addis and J. Gearhart, University of Pennsylvania).
Luciferase assay
Twenty-four hours prior to transfection, HeLa cells were plated in 12 well plates. Similarly, in experiments utilizing the Jagged1-Fc and control-Fc constructs, HeLa cells were plated on the appropriate conditions 24 hours prior to transfection. HeLa cells were maintained at 37 °C with 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum, penicillin and streptomycin. Transfections were completed using FuGene6 (Roche) with 300 ng of the ECRx-pGL3-Promoter DNA, 300 ng of mouse Notch Intracellular Domain (mNICD) or empty vector and 1.5 ng CMV-renilla luciferase (Promega). In luciferase experiments utilizing the Jagged1-Fc and control-Fc constructs, no mNICD was transfected. Cellular extracts were collected 48 hours post-transfection for use in a dual-luciferase assay (Promega). Twenty μl of cellular extract was used to assess first firefly luciferase activity, followed immediately by the measurement of renilla luciferase activity. Firefly luciferase activity from the ECRx-pGL3-Promoter vector was normalized to the renilla luciferase activity. All experiments were performed in duplicate on at least three separate occasions. Statistical differences between conditions were analyzed using a two-tailed, paired t test.
Deletion Series
ECR6 deletion constructs were PCR amplified from full-length ECR6-pGL3-Promoter. All deletion constructs were amplified with primers containing a 5′ attB1 site and a 3′ attB2 site to allow for Gateway recombination into pDONR221 (Invitrogen) and all primer sequences can be found in Supplemental Table 2. All ECR6 deletions were sequence verified following insertion into pDONR221. Deletion constructs were then recombined into a Gateway compatible pGL3-Promoter vector for use in a luciferase assay.
ECR Mutagenesis
The predicted Rbp-J binding site within ECR6 was mutated using QuikChange Mutagenesis (Stratagene). All mutagenic primer sequences can be found in Supplemental Table 2 with underlined nucleotides indicating mutagenic sequences. M1 (**) primers modify the wildtype Rbp-J site sequence from 5′-TTTCCCACAGT-3′ to 5′-AATCCCACAGT-3′. M2 (§§) primers modify the wildtype Rbp-J site sequence from 5′-TTTCCCACAGT-3′ to 5′-TTTCGGACAGT-3′. M3 (XXXX) primers modify the wildtype Rbp-J site sequence from 5′-TTTCCCACAGT-3′ to 5′-TGCAGCACAGT-3′. The mutations were created in ECR6-pDONR221 vector and were fully sequenced to verify the presence of the intended mutations and exclude other polymerase errors. ECR6 mutants were then recombined into a Gateway compatible pGL3-Promoter vector for use in luciferase assays.
Generation of transgenic mice
Wildtype and mutant (M3) ECR6 were inserted upstream of the gene with an hsp68 minimal promoter. The mutation in ECR6 was identical to “M3” used for luciferase assays and described above. Transgenic mice were generated by injection of linearized DNA into the male pronucleus of C57BL6xSJL-F1/J zygotes. Embryos were harvested at E11.5 for analysis of β-galactosidase expression and genotyping. Genotyping for lacZ was performed as previously described. 30 Embryos were fixed with 2% paraformaldehyde in PBS for 20 minutes at 4 °C. Following fixation, samples were washed twice for 10 minutes in PBS at 4 °C. Embryos were stained in 1 mg/ml X-gal, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 0.01% NP-40, 0.01% sodium deoxycholate, in PBS. Staining reactions were incubated overnight at 37 °C. For optical projection tomography analysis, embryos were embedded in 1% low-melt agarose, dehydrated in methanol, and then cleared in 1:2 (v/v) benzyl alcohol and benzyl benzoate. 31 Embryos were scanned using the Bioptonics OPT Scanner (3001M). Image stacks were reconstructed using OsiriX software.
Chromatin immunoprecipitation (ChIP)
Twenty-four hours prior to transfection, 293T or HeLa cells were plated in 100mm plates. 3–100mm plates were prepared per condition. Cells were maintained at 37 °C with 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum, penicillin and streptomycin. Transfections for endogenous ChIP assays utilized FuGene6 (Roche) with 1μg of a N-terminal c-myc tagged Rbp-J. Transfections for transient ChIP assays utilized FuGene6 (Roche) with 800 ng of the ECR6-pGL3-Promoter DNA (wildtype or M3), 100 ng of a C-terminal FLAG tagged NICD and 100 ng of a N-terminal c-myc tagged Rbp-J. Both the NICD-FLAG and cmyc-Rbp-J cDNAs were fully sequence verified to exclude any polymerase errors. 48 hours post-transfection cells were washed twice in cold PBS, cross-linked with 1% formaldehyde (Sigma) for 10 min at room temperature and then incubated in 0.14M glycine for 5 min at room temperature to quench the cross-linking reaction. Cells were collected in cold PBS, polluted by centrifugation and resuspended in SDS lysis buffer (10 mm Tris-HCl, pH 8.0, 10mM NaCl, 3mM MgCl2, 1% NP-40, 1% SDS, 0.5% deoxycholic acid and protease inhibitors). Chromatin was sonicated using the BioRuptor (Diagenode) on high for 5 min, cycling 30s on/30s off. Prior to immunoprecipitation, samples were precleared. Chromatin samples were diluted in dilution buffer (16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, 0.01% SDS, 1.1% Triton-X 100), protease inhibitors, protein-G agarose (Invitrogen) and control IgG (rabbit-Santa Cruz, mouse-Calbiochem) were added and then samples were incubated at 4°C for 1 hr while rotating. Following pre-clearance, the lysate was divided and immunoprecipitated with 0.5ug of anti- myc (Millipore), normal-rabbit IgG (Santa Cruz), anti-FLAG (Sigma) or normal-mouse IgG (Calbiochem) for 4 hr at 4°C. Protein-G agarose beads were then added to each reaction for 1 hr at 4°C. The agarose beads were then washed 2X in low salt buffer for 30 min (20 mM Tris-HCl, pH 8.1, 150 mM NaCl, 2mM EDTA, 0.1% SDS, 1% Triton X-100), 2X in high salt buffer for 15 min(20 mM Tris-HCl, pH 8.1, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), 1X LiCl buffer for 10 min (10 mM Tris-HCl, pH 8.1, 0.25M LiCl, 1 mM EDTA, 1% NP-40, 1% deoxycholic acid), and 1X with TE buffer for 5 min (10 mM Tris-HCl, pH 8.1, 1 mM EDTA), all at 4°C. Complexes were eluted in 200 μl elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 15 min. 5M NaCl was added to each sample and incubated overnight at 65°C to reverse chromatin cross-linking. Proteins were degraded following a 1 hr incubation at 45°C in the presence of Proteinase K (Roche) and remaining DNA was purified with the Qiagen PCR Purification kit (Qiagen). Samples were then PCR amplified using JumpStart PCR mix (Sigma). Primers to the endogenous Jagged1 genomic loci:
ECR1-Forward: 5′-TCCCAGCTCATGTATCTTTGCTTGC-3′
ECR1-Reverse: 5′-GGAGGAATGCAGATCAAAGCGAAGTCT-3′
ECR6-Forward: 5′-CTACAACCACTAACAGGAGAGC-3′
ECR6-Reverse: 5′-GCTTCATACTTACAGCAGG-3′
Primers to full length ECR6 vector sequence:
ChIP-Forward: 5′-GGCACCTTCCTTTGCAGTGGT-3′
ChIP-Reverse: 5′-AGGCATGAGAGAAAAGCAGGAGGT-3′
Results
Inhibition of Notch in neural crest impairs Jagged1 expression
In order to determine if Jagged1 expression is regulated by Notch, we examined Jagged1 mRNA and protein expression in animals in which Notch was inhibited by expression of a previously well-characterized dominant negative mastermind-like protein (DNMAML) in neural crest. DNMAML inhibits function of all Notch receptors by binding to NICD and preventing recruitment of coactivators such as p300. 25, 32 R26DNMAML mice allow for Cre-mediated activation of DNMAML expression in a tissue-specific manner, and we crossed R26DNMAML with Pax3Cre 24 mice to inhibit Notch in neural crest.
Inhibition of Notch signaling in neural crest with DNMAML resulted in diminished Jagged1 mRNA surrounding the paired aortic arch arteries that are populated by neural crest derivatives (Figure 1A,E). Jagged1 protein expression was also diminished (Figure 1B,C,F,G) and this correlated with impaired smooth muscle differentiation as assessed by expression of smooth muscle actin (Figure 1D,H). The decrease in Jagged1 expression and smooth muscle differentiation was not a result of absence of neural crest cells from the forming arterial wall. Fate mapping experiments utilizing a Cre-inducible GFP reporter, Z/EG, demonstrated similar quantities of neural crest cells in the region of the forming aortic arch arteries in the presence or absence of the DNMAML allele (Figure S1). Similar decreases in Jagged1 protein expression and smooth muscle differentiation were also observed when DNMAML was activated by Wnt1-Cre (Figure S2), which, like Pax3Cre is expressed by premigratory neural crest. Jagged1 protein expression was quantified in the regions surrounding each of the aortic arch arteries. Notch inhibited animals exhibited a significant decrease in the area of Jagged1 protein expression when compared to control litter-mates, with nearly 3-fold less Jagged1 protein detected overall (Figure S3A). Expression was significantly diminished in each aortic arch artery except the right VI arch, which has largely regressed by E12.5 (Figure S3B). 33 Jagged1 expression was not affected in the descending aorta where vascular smooth muscle is not derived from neural crest precursors and DNMAML was not expressed (Figure S4). 34, 35
Figure 1.

Inhibition of Notch in neural crest impairs Jagged1 expression. (A–D) Frontal sections of E12.5 control embryos. (E–H) Frontal sections of E12.5 Pax3Cre/+ DNMAML embryos. (A, E) In situ hybridization of Jagged1 in control (A) and Notch inhibited mutants (E). Pax3Cre/+ DNMAML embryos exhibit decreased Jagged1 mRNA expression surrounding each of the aortic arch arteries (arrowheads). (B, C, F, G) Immunostaining of Jagged1 in control (B and C) and Notch inhibited mutants (F and G). The left sixth arch arteries (arrowheads B,F) are shown in higher magnification (C, G, respectively). (D, H) Smooth muscle actin (SMA) stained sections adjacent to those shown in C and G respectively. Scale bars: 100μm. (I) Semiquantitative RT-PCR from primary vascular smooth muscle cells stimulated with Fc fragments (cntl) or Fc-Jagged1 (Jag1). Wild type smooth muscle (DNMAML−) cells induce Jagged1 expression following stimulation, while induction is absent in Pax3Cre/+ DNMAML cells (DNMAML+). GAPDH is used as a loading control. E11.5 whole embryo cDNA and H2O were used as positive and negative controls, respectively.
Notch activation in primary vascular smooth muscle induces Jagged1 expression
In order to test whether Notch activation of vascular smooth muscle is sufficient to induce Jagged1 expression, we cultured primary vascular smooth muscle from the aortic arches of control or Pax3Cre;DNMAML E17.5 embryos and cultured these cells on plates coated with control-Fc fragments or with Jagged1-Fc fragments that functionally activate Notch receptors on cultured cells. This method previously demonstrated that control cells exposed to Jagged1-Fc, but not control-Fc, up-regulate the Notch targets Hrt1, Hrt2 and Hrt3 as determined by RT-PCR and that this response is blocked by expression of DNMAML. 4 Interestingly, control cells exposed to Jagged1-Fc also up-regulate Jagged1 message (Figure 1I) and this response is also blocked by DNMAML. These results, consistent with previous studies, suggest that Jagged1 is a downstream target of Notch signaling in vascular smooth muscle. 15, 23
Identification of a Notch-responsive enhancer in the Jagged1 genomic locus
We sought to determine if Jagged1 is a direct Notch target in vascular smooth muscle. Comparative sequence analysis of the Jagged1 genomic locus, including 30kb upstream of the transcription start site, introns, and 10kb downstream of the last exon, identified 15 evolutionarily conserved regions (ECRs) of at least 200 bp bearing a minimum sequence identity of 70% between mouse, human, dog and opposum (Figure 2A, arrow heads). Nine of the 15 ECRs contain putative Rbp-J binding sites (Figure 2A, Rbp-J sites indicated by §). Each of the 15 control regions was cloned upstream of a minimal promoter and a luciferase reporter gene, and these reporter plasmids were transfected with or without NICD to evaluate Notch responsiveness. A Hes1-luciferase reporter was used as a positive control. 36 A single Jagged1 ECR, located within Intron 2, displays Notch responsive activation (Figure 2B). This 617 bp element was denoted “ECR6” and contains a single predicted Rbp-J binding site. This Rbp-J binding site is well conserved across diverse species. Alignments of the Rbp-J binding site and proximal 5′ and 3′ regions exhibit near identical sequence between mouse, human, dog, opposum and chicken (Figure 2C). Further luciferase assays were completed to determine if endogenous Notch signaling could activate ECR6. Cells were plated on either control-Fc fragments or Jagged1-Fc fragments and then transfected with empty vector, Hes1-luciferase reporter or ECR6-luciferase. Parallel experiments examined luciferase activity following no additional treatment, vehicle (DMSO) exposure or incubation with a γ secretase inhibitor (10μM DAPT). In both the non-treated and vehicle treated samples, cells cultured on Jagged1-Fc fragments activated both Hes1 and ECR6 (Figure 2D). This activation was not apparent following culture on control-Fc fragments. The presence of DAPT was sufficient to prevent Hes1 and ECR6 luciferase activity following culture on Jagged1-Fc fragments suggesting that the observed activation was indeed in response to endogenous Notch signaling (Figure 2D). Furthermore, epitope-tagged Rbp-J can occupy the predicted Rbp-J binding site within ECR6 as determined by chromatin immunoprecipitation (ChIP) (Figure 2E). This occupancy is specific for the ECR6 genomic location, as Rbp-J was not bound to a distant ECR, ECR1, which was predicted to contain a Rbp-J binding site, but was not Notch responsive in the luciferase reporter assay (Figure 2A).
Figure 2.

Identification of a Notch-responsive enhancer in the Jagged1 genomic locus. (A) Schematic representation of the Jagged1 genomic locus. White rectangles represent exons; gray rectangles represent 5′ and 3′ untranslated (UTR) regions. Fifteen evolutionary conserved regions (ECRs) are indicated by black arrowheads. The nine predicted Rbp-J binding sites (§) are shown. (B) Results of dual luciferase reporter assays in HeLa cells with each of the 15 ECRs (numbered 1–15) or with negative control luciferase vector (empty) or positive control Hes-1 luciferase reporter are shown. Activity with co-transfected murine NICD1 (mNICD1, white bars) was compared to control expression vector (black bars). Luciferase activity was normalized to the activity of a co-transfected renilla luciferase construct. All experiments were performed in duplicate on at least three separate occasions and data shown are the mean + standard error of the mean (SEM). (C) ECR6 sequence alignment of the conserved Rbp-J site and proximal 5′ and 3′ regions across human, mouse, dog, opposum and chicken. The Rbp-J binding site is in bold, * indicates conservation across species. The genomic locations used in the alignment are human: hg18: chr20: 10593999-10594050, dog: canFam2 chr24: 14663338-14663389, mouse: mm9: chr2:136933564-136933614, opposum: monDom4: chr1:614433896-614433948 and chicken: galGal3 chr3: 14408520-14408570. (D) Dual luciferase reporter assay results from HeLa cells plated on either control-Fc or Jagged1-Fc fragments. HeLa cells were transfected with either empty vector (Empty, negative control), Hes1 reporter (positive control) or ECR6. Cells were either untreated, cultured in the presence of DMSO (vehicle) or DAPT (10μM). Luciferase activity was normalized for transfection efficiency. All experiments were performed in duplicate on at least three separate occasions and data shown are the mean + standard error of the mean (SEM). (E) Chromatin immunoprecipitation (ChIP) with a c-myc tagged Rbp-J protein (+Rbp-J) and endogenous Jagged1 genomic loci. Rbp-J occupancy was compared between the Notch responsive locus, ECR6, and a non-Notch responsive locus, ECR1. Non-transfected (NT) HeLa cells and H2O served as negative controls. Input chromatin served as a positive control.
Successive deletions of up to 305 bp of the 5′ end of ECR6 failed to impair Notch responsiveness in luciferase assays (Figure 3A, B). These deletions left intact the predicted Rbp-J binding site. However, deletion of 308 bp from the 3′ end, which revoved the Rbp-J site, abolished Notch activation. Three independent mutations of the predicted Rbp-J binding site of ECR6 (see methods – each mutation changed 2 or 4 bp) were independently tested, and each abolished Notch-mediated transactivation (Figure 3B). Furthermore, in transfected HEK293T cells, epitope-tagged Rbp-J and NICD can each occupy the putative Rbp-J binding site as determined by ChIP (Figure 3C and 3D). NICD occupancy is not observed when the Rbp-J site is mutated (Figure 3E). These data suggest that Notch activation of Jagged1 transcription is mediated directly via Rbp-J and NICD binding to ECR6.
Figure 3.

Notch activation of ECR6 is Rbp-J dependent. (A) Schematic representation of full length ECR6 with the single Rbp-J binding site (white rectangle). 5′ and 3′ deletions (D1–D6), and three independent Rbp-J point mutants (M1–M3, see methods) were constructed. (B) Dual luciferase reporter assays for the constructs shown in A. Luciferase activity was normalized to the activity of a co-transfected renilla luciferase construct. All experiments were performed in duplicate on at least four separate occasions and data shown are the mean + standard error of the mean (SEM). (C–E) Chromatin immunoprecipitation (ChIP) with a c-myc tagged Rbp-J protein and ECR6 expression plasmid (C), a 3xFLAG tagged mNICD protein and ECR6 expression plasmid (D), or a 3xFLAG tagged mNICD protein and control vs. mutant ECR6 expression plasmid (E) are shown. H2O and diluted (1:10) input chromatin served as negative and positive controls, respectively.
ECR6 directs transgenic expression to vascular smooth muscle in vivo
To determine if ECR6 can mediate expression that recapitulates that of Jagged1 in smooth muscle of the aortic arch arteries, transient transgenic animals were created. Transgenic mice containing ECR6 upstream of a minimal hsp68 promoter and LacZ were examined at E11.5. Whole-mount X-gal staining revealed strong transgenic expression in the distal cardiac outflow tract and great vessels (Figure 4A and 4B). This pattern was observed in 5/7 transient transgenic embryos. Optical projection tomography (OPT) of a genotype positive transgenic animal further validated the robust outflow tract specific expression (Supplemental Movie 1). Upon sectioning, transgene expression was evident in the aortic arch arteries (Figure 4C and 4D) coincident with smooth muscle actin (SMA) (Figure 4E) and Jagged1 (Figure 4J,K) expression. Aortic arch artery expression was eliminated in 10/10 genotype positive transient transgenic embryos derived from a modified construct in which the Rbp-J site within ECR6 was mutated (equivalent to “mutation 3” used for luciferase assays described above) (Figure 4F–I). Thus, ECR6 is sufficient to mediate reporter gene expression in vascular smooth muscle in the aortic arch arteries and this expression pattern requires an intact Rbp-J binding site.
Figure 4.

ECR6 directs transgenic expression to vascular smooth muscle in vivo. (A–E) E11.5 transient transgenic ECR6-lacZ embryos. Whole mount X-gal stained embryos reveal robust expression in the distal outflow tract (OFT, panel B). (C, D) X-gal and eosin stained frontal sections. Expression is visible surrounding the arch arteries including the left sixth arch artery (arrow in C, higher magnification shown in D). Sections were subsequently stained for smooth muscle actin (SMA, panel E). (F–I) E11.5 transient transgenic M3-ECR6-lacZ embryos. Expression in the OFT and surrounding the aortic arch arteries was not evident. The left forth and sixth arteries are indicated with arrowheads in panel H and shown in higher magnification in panel I. (J, K) Frontal sections of wildtype E11.5 embryos stained for Jagged1 protein expression. The left forth and sixth arteries are indicated with arrowheads in panel J and shown in higher magnification in panel K. Scale bars: 100μm. RV-Right Ventricle, LV-Left Ventricle.
To more fully characterize ECR6 expression throughout development, a stable ECR6-hsp68-LacZ mouse line was created (Figure 5). Time course analysis revealed that ECR6 was not active at E9.5 (5A–C), but X-gal activity was readily detectable by E10.5 (5D–F). At E11.5, robust X-gal activity was observed surrounding aortic arch arteries iii, iv and vi (5G–I). Transgenic expression is specific for the three aforementioned arch arteries which are populated by neural crest-derived smooth muscle, as transgenic expression was absent at the second arch arteries (5J). Further, expression was also restricted to neural crest derived vascular smooth muscle, as no transgenic expression was observed surrounding the descending aorta (5K).
Figure 5.
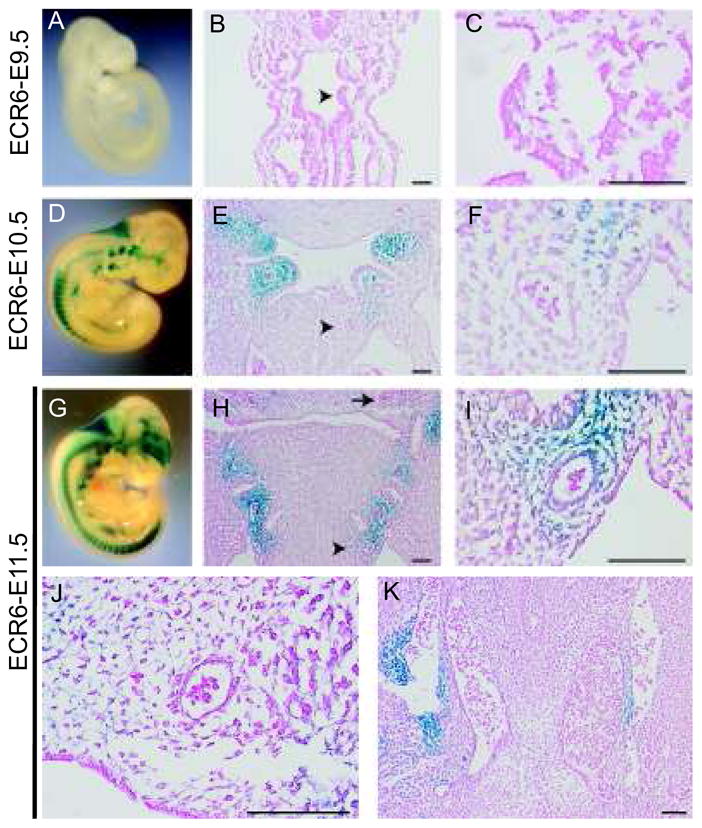
ECR6 is expressed in the developing and mature cardiac outflow tract. (A–C) E9.5 stable transgenic ECR6-lacZ embryos. Whole mount X-gal stained embryos reveal no transgenic expression throughout the embryo (panel A). (B, C) X-gal and eosin stained frontal sections. Expression is absent surrounding the arch arteries (arrowhead in B, higher magnification shown in C). (D–F) E10.5 stable transgenic ECR6-lacZ embryos. Whole mount X-gal stained embryos reveal robust expression at the pharyngeal arches (panel D). (E, F) X-gal and eosin stained frontal sections. Expression is highest surrounding the third and fourth arch arteries, but to a lesser degree the left sixth arch artery (arrowhead in E, higher magnification shown in F). (G–K) E11.5 stable transgenic ECR6-lacZ embryos. Whole mount X-gal stained embryos reveal robust expression at the pharyngeal arches (panel G). (H–J) X-gal and eosin stained frontal sections. Expression is visible surrounding the arch arteries including the left sixth arch artery (arrowhead in H, higher magnification shown in I). (J) No expression was observed at the second arch arteries (arrow in H, higher magnification) or (K) surrounding the descending aorta. Scale bars: 100μm.
Inactivation of Jagged1 in neural crest results in aortic arch remodeling defects
Notch inhibition in neural crest induces aortic arch remodeling defects and congenital heart disease 4 and our present data suggests that Notch activation in neural crest induces Jagged1 expression. Hence, we examined the effects of Jagged1 gene inactivation in neural crest to determine if aortic arch remodeling was affected. We crossed Pax3Cre/+; Jagged1flox/+ mice to Jagged1flox/flox mice and observed a decrease in smooth muscle actin expression in Pax3Cre/+; Jagged1flox/flox embryos, indicative of deficient smooth muscle differentiation (Figure 6A, B). A similar decrease in smooth muscle differentiation was also observed with staining for an earlier marker of differentiation, SM-22 alpha (Figure S5). Further, the hairy and enhancer of split (HES)-related transcription factor 1 (Hrt1), a prototypical Notch target gene 4, 37, 38 was down-regulated in Pax3Cre/+; Jagged1flox/flox embryos when compared to controls (Figure 6C, D). We analyzed aortic arch patterning in 29 Pax3Cre/+; Jagged1flox/flox neonates (see supplemental tables 3 and 4) and identified abnormal aortic arch patterning in 23. Defects included short or absent brachiocephalic artery, common carotid trunk and retroesphageal right subclavian artery (Figure 6E–H).
Figure 6.

Inactivation of Jagged1 in neural crest results in aortic arch remodeling defects. (A–D) Frontal sections of E12.5 mouse embryos. (A, B) Left sixth aortic arch arteries of control (A) and Pax3Cre/+; Jagged1flox/flox (B) embryos stained for smooth muscle actin (SMA). (C, D). In situ hybridization of the Notch target gene Hrt1 in control (C) and Pax3Cre/+; Jagged1flox/flox mutants (D). Arrowheads indicate aortic arch arteries. Scale bars: 100μm. (E–L) Photographs and schematic drawings depicting normal aortic artery neonatal patterning (E, I) and vascular patterning variants that were observed in Pax3Cre/+; Jagged1flox/flox mutants (F–H, J–L). Arrows in panels E and I indicate the normal brachiocephalic (bc) vessel. Arrows in F–H indicate site of abnormal patterning. rsa- right subclavian artery, rca- right carotid artery, lca- left carotid artery, lsa- left subclavian artery.
Discussion
Notch signaling is critical in neural crest-derived smooth muscle differentiation and for proper remodeling of the aortic arch arteries and cardiac outflow tract. Inhibition of MAML-dependent Notch signaling in neural crest impairs the formation of the smooth muscle layer of the aortic arch arteries, though the nascent endothelial tubes form normally. 4 In this study, we identify a Notch-responsive control element within the Jagged1 genomic locus that is capable of directing Jagged1 expression to the smooth muscle layer of the aortic arch arteries. Identification of this Notch responsive ECR in Jagged1 helps to provide a mechanism to explain how a multi-layered smooth muscle wall might form around an endothelial tube to produce a mature artery. We propose that Jagged1 expression by endothelial cells engages Notch receptors on neighboring mesenchyme, including neural crest-derived mesenchyme in the case of the aortic arch arteries. Notch activation in mesenchyme triggers smooth muscle differentiation, resulting in a single layer of smooth muscle around the endothelial tube, and simultaneously directly activates Jagged1 expression via ECR6 (and perhaps additional enhancers in various arterial distributions). This positive feedback, or lateral induction, mechanism results in Notch activation of progressively distant mesenchyme and reiterated smooth muscle differentiation events that produce a multi-layered smooth muscle wall. Eventually, the signal decays, or is terminated by opposing morphogens that have not yet been identified.
This model is supported by significant experimental data. Jagged1 expression by endothelium is required for smooth muscle differentiation of adjacent mesenchyme 3, and Jagged1 is expressed by both endothelium and vascular smooth muscle. Inhibition of Notch by DNMAML in neural crest impairs Jagged1 expression, both in vivo and in explant cultures, as we show in this report. Also, in accord with this model, deletion of Jagged1 in neural crest results in only relative impairment of smooth muscle differentiation, since endothelial Jagged1, and perhaps other redundant Notch receptors, are sufficient to induce some vascular smooth muscle. This results in relatively mild, though penetrant, aortic arch remodeling defects.
The description of Jagged1 as a direct Notch target is in agreement with published data that supports positive feedback between Notch and Jagged1. For example, NIH3T3 cells display Notch-mediated activation of Jagged1 18, and Jagged1 transcript and protein expression decreases following shRNA mediated knock down of either Notch3 or Rbp-J in ovarian cancer cell lines. 39 Notch activation in the basal epidermis causes up-regulation of Notch signaling and Jagged1 expression in both epidermis and dermis 40. In secondary fiber cells of the rat lens, Notch2 is required for Fgf-mediated Jagged1 induction and lens fiber cell differentiation. 22 Finally, Feng et al. have proposed a lateral induction mechanism involving Notch and Jagged1 to explain patent ductus arteriosus in mice lacking Jagged1 in smooth muscle. 15
The identification of a functional role for ECR6 may be relevant for genetic testing in patients with Alagille syndrome, congenital heart disease, or other Notch-related disorders. A recent report predicted that nearly ¼ of human disease-causing mutations may be located within intronic sequences. 41 Given the tissue-specificity of ECR6 in transgenic mice, interesting patient populations to screen for mutations within this conserved region might include those with Alagille syndrome, patent ductus arteriosus, bicuspid aortic valve or aortic arch abnormalities. 10, 11, 15
In summary, we identify a conserved enhancer in the Jagged1 genomic locus that directs expression to the outflow tract and smooth muscle layer of the forming aortic arch arteries. We provide evidence for Notch/Jagged1 lateral induction and for direct regulation of Jagged1 by Notch. Finally, we provide a model to explain how a multilayered smooth muscle wall may form around a primitive endothelial tube.
Supplementary Material
Acknowledgments
We thank Klaus Kaestner and Kathy Loomes for the use of floxed Jagged1 mice, and Warren Pear for R26DNMAML mice. We are grateful to the late Tom Kadesch for Fc-Jagged1.
Funding Sources
This work was supported by NIH U01 HL100405, R01 HL095634, the AHA John Holden DeHaan Myogenesis Center, the WW Smith Professorship and the Spain Cardiovascular Research Fund.
Footnotes
Disclosures
None.
References
- 1.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 2.High FA, Epstein JA. The multifaceted role of notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- 3.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the notch ligand jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth muscle alpha-actin is a direct target of notch/csl. Circ Res. 2006;98:1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- 6.Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Manabe I, Arai M, Nagai R, Kurabayashi M. Jagged1-selective notch signaling induces smooth muscle differentiation via a rbp-jkappa-dependent pathway. J Biol Chem. 2006;281:28555–28564. doi: 10.1074/jbc.M602749200. [DOI] [PubMed] [Google Scholar]
- 7.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in cadasil, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, Baron M, Trump D. An overview of notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol. 2008;96:499–509. doi: 10.1016/j.pbiomolbio.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18:2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human jagged1, which encodes a ligand for notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 11.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human jagged1 gene are responsible for alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 12.McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a jag1 mutation and/or alagille syndrome. Circulation. 2002;106:2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 13.Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T. Embryonic lethality and vascular defects in mice lacking the notch ligand jagged1. Hum Mol Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into alagille syndrome. Development. 2010;137:4061–4072. doi: 10.1242/dev.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific jag1 deletion. Development. 2010;137:4191–4199. doi: 10.1242/dev.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bray SJ. Notch signalling: A simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 17.Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross DA, Kadesch T. Consequences of notch-mediated induction of jagged1. Exp Cell Res. 2004;296:173–182. doi: 10.1016/j.yexcr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Eddison M, Le Roux I, Lewis J. Notch signaling in the development of the inner ear: Lessons from drosophila. Proc Natl Acad Sci U S A. 2000;97:11692–11699. doi: 10.1073/pnas.97.22.11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartman BH, Reh TA, Bermingham-McDonogh O. Notch signaling specifies prosensory domains via lateral induction in the developing mammalian inner ear. Proc Natl Acad Sci U S A. 2010;107:15792–15797. doi: 10.1073/pnas.1002827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foldi J, Chung AY, Xu H, Zhu J, Outtz HH, Kitajewski J, Li Y, Hu X, Ivashkiv LB. Autoamplification of notch signaling in macrophages by tlr-induced and rbp-j-dependent induction of jagged1. J Immunol. 2010;185:5023–5031. doi: 10.4049/jimmunol.1001544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saravanamuthu SS, Gao CY, Zelenka PS. Notch signaling is required for lateral induction of jagged1 during fgf-induced lens fiber differentiation. Dev Biol. 2009;332:166–176. doi: 10.1016/j.ydbio.2009.05.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Kennard S, Lilly B. Notch3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed jagged1. Circ Res. 2009;104:466–475. doi: 10.1161/CIRCRESAHA.108.184846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of cre into the pax3 locus creates a new allele of splotch and identifies unexpected pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, Pear WS. Notch signaling is an important regulator of type 2 immunity. J Exp Med. 2005;202:1037–1042. doi: 10.1084/jem.20050923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loomes KM, Russo P, Ryan M, Nelson A, Underkoffler L, Glover C, Fu H, Gridley T, Kaestner KH, Oakey RJ. Bile duct proliferation in liver-specific jag1 conditional knockout mice: Effects of gene dosage. Hepatology. 2007;45:323–330. doi: 10.1002/hep.21460. [DOI] [PubMed] [Google Scholar]
- 27.Wawersik S, Epstein JA. Gene expression analysis by in situ hybridization. Radioactive probes. Methods Mol Biol. 2000;137:87–96. doi: 10.1385/1-59259-066-7:87. [DOI] [PubMed] [Google Scholar]
- 28.Lepore JJ, Cappola TP, Mericko PA, Morrisey EE, Parmacek MS. Gata-6 regulates genes promoting synthetic functions in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2005;25:309–314. doi: 10.1161/01.ATV.0000152725.76020.3c. [DOI] [PubMed] [Google Scholar]
- 29.Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem. 2005;280:8994–9004. doi: 10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- 30.Degenhardt KR, Milewski RC, Padmanabhan A, Miller M, Singh MK, Lang D, Engleka KA, Wu M, Li J, Zhou D, Antonucci N, Li L, Epstein JA. Distinct enhancers at the pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol. 2010;339:519–527. doi: 10.1016/j.ydbio.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe J, Ahlgren U, Perry P, Hill B, Ross A, Hecksher-Sorensen J, Baldock R, Davidson D. Optical projection tomography as a tool for 3d microscopy and gene expression studies. Science. 2002;296:541–545. doi: 10.1126/science.1068206. [DOI] [PubMed] [Google Scholar]
- 32.Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC, Pear WS. Mastermind critically regulates notch-mediated lymphoid cell fate decisions. Blood. 2004;104:1696–1702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- 33.Hiruma T, Nakajima Y, Nakamura H. Development of pharyngeal arch arteries in early mouse embryo. J Anat. 2002;201:15–29. doi: 10.1046/j.1469-7580.2002.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wasteson P, Johansson BR, Jukkola T, Breuer S, Akyurek LM, Partanen J, Lindahl P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development. 2008;135:1823–1832. doi: 10.1242/dev.020958. [DOI] [PubMed] [Google Scholar]
- 35.Lagha M, Brunelli S, Messina G, Cumano A, Kume T, Relaix F, Buckingham ME. Pax3:Foxc2 reciprocal repression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors. Dev Cell. 2009;17:892–899. doi: 10.1016/j.devcel.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 36.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 37.Fischer A, Gessler M. Hey genes in cardiovascular development. Trends Cardiovasc Med. 2003;13:221–226. doi: 10.1016/s1050-1738(03)00082-3. [DOI] [PubMed] [Google Scholar]
- 38.Iso T, Kedes L, Hamamori Y. Hes and herp families: Multiple effectors of the notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Stoeck A, Lee SJ, Shih Ie M, Wang MM, Wang TL. Jagged1 expression regulated by notch3 and wnt/beta-catenin signaling pathways in ovarian cancer. Oncotarget. 2010;1:210–218. doi: 10.18632/oncotarget.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ambler CA, Watt FM. Adult epidermal notch activity induces dermal accumulation of t cells and neural crest derivatives through upregulation of jagged 1. Development. 2010;137:3569–3579. doi: 10.1242/dev.050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper DN, Chen JM, Ball EV, Howells K, Mort M, Phillips AD, Chuzhanova N, Krawczak M, Kehrer-Sawatzki H, Stenson PD. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.