

Abstract
Supramolecular structures based on organized assemblies of macrocyclic chromophores, particularly porphyrin-based dyes, have attracted widespread interest as components of molecular devices with potential applications in molecular electronics, artificial light harvesting, and pharmacology. We report the formation of J-aggregates of two porphyrin-based dyes, 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin (TSPP, 4) and an amino tris-sulfonate analog (5) in water using a functionalized norbornene-based homopolymer, synthesized by ring opening metathesis polymerization (ROMP). Ionic interactions of the cationic side chains (ammonium groups) of the polymer under acidic conditions with the negatively charged sulfonate groups of the porphyrins facilitated polymer template enhanced J-aggregation of the porphyrin dyes. J-aggregation behavior was investigated photophysically by UV-vis absorption along with steady-state and time-resolved fluorescence studies. Two-photon absorption (2PA) was enhanced by about an order of magnitude for the J-aggregated TSPP relative to its free base. Significantly, the 2PA cross section of the polymer-templated TSPP J-aggregate was up to three times higher than the J-aggregated TSPP in the absence of the polymer template while the 2PA cross section for polymer-templated J-aggregates of 5 increased substantially, up to ca. 10,000 GM, suggesting a prominent role of polymer-templating to facilitate porphyrin aggregation and greatly enhance nonlinear absorption.
Introduction
Supramolecular structures, based on organized assemblies/aggregation of macrocyclic chromophores, have attracted widespread interest as molecular devices with potential applications in molecular electronics, artificial light harvesting, and pharmacology.1–5 In the late 1930s, Gunter Scheibe6 and Edwin E. Jelly7 independently discovered the phenomenon of J- and H-aggregation for cyanine-based dyes. Aggregation, driven by non-covalent interactions, causes remarkable changes in the optical and electronic properties of the molecular aggregates. For example, J-aggregation (side-by-side) and H-aggregation (face-to-face) is characterized by strong, narrow, red-shifted absorption bands (J) and blue-shifted absorption bands (H) with respect to its monomeric form. Strong intermolecular interactions between the dyes and delocalized excitonic energy over the whole assembly of the aggregate are characteristic of this phenomenon. Kasha et al.8 proposed the following equation to explain the dipole–dipole coupling interaction energy (V) in aggregates:
where M is the transition dipole moment, r is the distance from center-center between dipoles, and θ represents the geometrical factor related to mutual inclination of the aligned monomers.
Among the known molecular building blocks, porphyrins constitute a highly attractive class of “synthons” for functional nanomaterials due to their unique photonic and electronic properties, specifically for their potential applications in photodynamic therapy, nonlinear optics, and for investigation of artificial light harvesting systems that mimic natural photosynthetic receptors.9,10 5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrin (TSPP) is, perhaps, one of the most studied synthetic porphyrins. Under acidic aqueous conditions (usually pH <1), the diacid species of the TSPP porphyrin forms J-aggregates due to hydrophobic π-π stacking and electrostatic interaction between the anionic sulfonated phenyl group and the cationic core.11 The tuning factors of porphyrin aggregation in aqueous solution vary depending on the porphyrin structure and concentration, as well as the pH, ionic strength,12 and counter ions of inorganic salts in the media. Also, the medium, such as copolymer micelles,13,14 ionic liquids,15 nucleic acids, polypeptides, proteins, and carbon nanotubes, is capable of influencing porphyrin aggregation behavior.
Investigation of polymer-based self-assembled J-aggregation of dyes is intriguing and of current interest. Santoro et al. demonstrated that the tetraanionic meso-tetrakis(4-sulfonatophenyl)porphine (H2TSPP) in the pH range 5–12 exists in a monomeric form, and its fluorescence is not pH-dependent.16 However, in the presence of polylysine, absorption, circular dichroism, and resonant light scattering data indicate extensive polymer-induced self-aggregation of the porphyrins. In particular, at low pH (<7), the protonated polylysine promotes porphyrin binding and self-aggregation with consequent strong quenching of fluorescence.16 Periasamy et al. observed that poly-(L, D, or DL)-lysine, depending on optical chirality, induced J-aggregation of TSPP more efficiently than monomeric lysine.17a Only micromolar concentrations of polylysine were required for complete conversion of the porphyrin monomer to its J-aggregate.17 Quite impressively, Whitten et al. demonstrated `superquenching' of polyelectrolytes containing cyanine pendant polylysines (repeat unit: 1–900) both in solution and after adsorption onto silica nanoparticles.18 The self-assembled polymer-initiated surface activated quenching and led to formation of J-aggregates due to enhanced binding with an increasing number of repeat units of the polymer.18 Laponite clay exerted a similar effect with cyanine dyes to induce J-aggregation.19 Zhao et al. recently reported the micellization of poly(ethylene glycol)-block-poly(4-vinylpyridine) (PEG114-b-P4VP61) induced by TPPS in acidic solution, where the core contained TSPP/PV4P and the shell was structured with PEG. TSPP formed J-aggregates and H-aggregates in the micellar core at pH 1.5–2.5 and 3.0–4.0, respectively.14 Kano et al. found that the TSPP-acid form was stabilized to induce J-aggregation by binding with ferric myoglobin (metMb) in water at neutral pH due to encapsulation and fixation by the relatively rigid protein molecules. The hydrophobic core of the J-aggregate caused the deformation of the secondary structure of the metMb, and, thus, denaturation of the protein.20 Chmelka et al. reported that mesostructured silica-block copolymer thin films provided orientationally-ordered host matrixes for stable alignment of co-assembled porphyrin J-aggregates with anisotropic optical properties.21 Smith et al. reported the induction of J- and H-aggregation of TSPP by the cationic polyelectrolyte poly(diallyldimethylammonium chloride) (PDDA) on films deposited on Si. The films were made by dipping in alternating aqueous solutions containing film components (layer-by-layer deposition).22 From these and other reports, it is evident that there is substantial interest in inducing and controlling porphyrin aggregation.
Herein, our main focus is to determine whether a functional polymer can serve as the foundation to build a supramolecular structure containing a porphyrin-based dye and facilitate J-aggregation. Of particular interest is the potential modulation and enhancement of two-photon absorption (2PA) by J-aggregates relative to the corresponding unaggregated monomers. We report the synthesis and characterization of a norbornene-based monomer, containing dimethylamino pendant groups, and the corresponding homopolymer by ring opening metathesis polymerization (ROMP). ROMP was selected for its tolerance to a variety of functional groups and the ability to control molecular weight. J-aggregation of two porphyrin-based dyes, commercial TSPP (4) and an analog (5) in acidic aqueous solution was investigated without and with presence of the homopolymer template. The aggregation properties were evaluated by UV-vis absorption, fluorescence, and fluorescence lifetime decay studies, clearly demonstrating J-aggregation was more pronounced in dyes in the presence of the polymer, possibly due to enhanced stabilization of the anionic dye's periphery by the cationic nature of the pendant ammonium moieties (−NHMe2+) of the polymers at low pH. Finally, 2PA was determined for free (unaggregated) and aggregated (without and with the polymer template) porphyrins 4 and 5.
Experimental
Materials
5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrin (TSPP) and tetraphenylporphyrin (TPP) were purchased from Strem and Aldrich, respectively. 6-N, N-Dimethylaminohexanol was obtained from TCI America. Grubbs' first generation catalyst and norbornene carboxylic acid were purchased from Aldrich. The norbornenyl acid chloride 1 was prepared as previously reported.23 All solvents were purified and dried according to standard procedures.
Synthesis
Synthesis of 6-(N, N-dimethylamino)hexyl bicyclo[2.2.1]hept-5-ene-2-carboxylate (2)
Acid chloride (1) (10.8 g, 0.069 M) was dissolved in freshly dried THF. Then, a mixture of 6-N, N-dimethylaminohexanol (14.22 mL, 0.086 M) and NaHCO3 (11.6 g, 0.14 M) was added to the solution at room temperature under N2 and refluxed overnight. After the reaction was complete, the mixture was filtered to remove the salt, and THF was removed under reduced pressure. This was followed by washing with water and extraction with CH2Cl2, then drying over anhydrous Na2SO4. Colorless oil was obtained after column chromatography with 3:1 CH2Cl2:MeOH, solvent removal, and vacuum drying. (13.73g, 75%). 1H NMR (500 MHz, CDCl3) δ: 8.98 (NH, 1H), 6.18–5.79 (m, 2H, HC=CH), 4.09 – 3.82 (m, 2H), 3.13 (s, 0.5H), 3.04–2.68 (m, 2H), 2.42–2.06 (m, 8H), 1.92 – 1.73 (s, 1H), 1.64 –1.06 (m, 11H). 13C NMR (125 MHz, CDCl3) δ: 176.24, 174.69, (C=O exo and endo) 138.02, 137.55, 135.74, 132. 36, 64.29, 63.38, 59.40, 46.74, 45.76, 43.07, 43.31, 30.40, 30.22, 29.27, 28.88, 28.34, 26.90, 25.79. HR-MS-ESI theoretical m/z [M+H]+ = 266.21, found 266.21.
Synthesis of polymer 3
ROMP of monomer 2 with Grubbs' first generation catalyst was performed as shown in Scheme 1. The glassware was dried, evacuated under vacuum, and purged with N2 in a Schlenk line several times prior to conducting the polymerization reaction. A solution (0.2 M) of monomer 2 (265 mg, 1 × 10−3 M, 175 eq) was prepared in dry CH2Cl2 under N2. The catalyst solution was prepared by dissolving the catalyst in anhydrous CH2Cl2 under N2 in a glove box. The catalyst solution (8.5 mg, 1 × 10−6 M in 0.5 mL CH2Cl2, 1 eq) was added to the reaction mixture and stirred for 1 h at 30 °C. The polymerization reaction mixture was terminated with excess ethyl vinyl ether (300 eq relative to catalyst) and stirred for another 1 h. The reaction mixture was then poured into cold methanol, stirred, collected by filtration, and dried under vacuum, yielding flaky white solid in 82% yield. 1H NMR (500 MHz, CDCl3) δ: 5.54 –5.09 (b, -HC=CH-), 4.22– 3.84, 3.26– 2.61, 2.58–2.22, 2.15–1.85, 1.80–1.05(b). Mw =3400 (by NMR, see Supporting Information)24, n ~ 13.
Scheme 1.

Monomer and polymer synthesis.
Characterization
1H NMR and 13C NMR spectra were acquired on a Varian NMR spectrometer at 500 and 125 MHz, respectively, using CDCl3 as the solvent for all monomers and polymers. High resolution mass spectrometry (HR-MS) analysis was performed in the Department of Chemistry, University of Florida, Gainesville, FL. Samples for the absorption spectroscopy measurements were prepared by dissolving the dyes in ultrapure water and acidified with 0.2 M HCl solution. Different buffer solutions (pH: 2.2–1.0) were prepared according to the literature.25
Linear photophysical properties were investigated in spectroscopic-grade solvents (DMSO and ultrapure water) at room temperature. Absorption spectra were obtained with an Agilent 8453 UV-visible spectrophotometer using 10 mm path length quartz cuvettes with dye concentrations of 1 ×−5 M. Fluorescence spectra were obtained with a Photon Technologies, Inc. (PTI) QuantaMaster spectrofluorimeter, using 10 mm spectrofluorometric quartz cuvettes and low concentration C ≤ 10−6 M. All fluorescence spectra were corrected for the spectral sensitivity of the PTI emission monochromator and photomultiplier tube (PMT) detector. Fluorescence lifetimes were measured with a time-correlated single photon counting system (PicoQuant PicoHarp 300) under linear polarized femtosecond excitation.
Two-photon absorption (2PA) cross sections (δ2PA) were measured by an open aperture Z-scan method using an amplified Ti:sapphire laser system (Coherent, Legend Elite seeded with a Verdi-pumped Mira 900) with an optical parametric amplifier (Coherent OPerA Solo) providing laser pulses of ~100 fs full-width at half maximum (FWHM) duration at 1 kHz repetition rate.
Results and Discussion
Synthesis
The synthesis of monomer 2 is shown in Scheme 1. Acid chloride 1 was reacted with 6-N, N-dimethylaminohexanol under basic conditions, affording ester 2. The NMR spectra and MS results confirmed product formation. The homopolymer of 2 was prepared via ROMP, using Grubbs' first generation catalyst, and characterized by NMR. The polymer was soluble in organic solvents such as CHCl3 and CH2Cl2. However, the solubility of the homopolymer in THF was poor, which restricted measurement of the molecular weight via GPC. Therefore, the molecular weight of the polymer was estimated by comparison of its diffusion coefficient with the diffusion coefficients of three polyethylene glycol standards via an NMR technique (see the Supporting Information),24 resulting an estimated molecular weight of ca. 3400. The polymer became water soluble after lowering the pH with HCl due to protonation of the pendant amino groups. As porphyrin derivatives generally undergo aggregation at low pH (<1) in aqueous medium, polymer 3 should serve as a suitable cationic template.
An analog (5) of TSPP (4) was synthesized by a three-step process.26 Briefly, tetraphenylporphyrin was selectively mono-nitrated with fuming nitric acid. The resulting nitro group was reduced with SnCl2/conc. HCl, transforming it into an amine, followed by exhaustive sulfonation to produce water soluble porphyrin derivative 5.
Solutions of two porphyrin dyes (4 and 5) were prepared in aqueous solution at low pH by acidification with various buffers (HCl/KCl, pH 2.2, 2.0, 1.5, and 1.0). Solutions of the dyes were prepared under neutral conditions by dissolution in ultra-pure water (pH ~ 7.0) while other solutions were prepared under acidic conditions by adding 0.2 M HCl (without adding any salt, pH 2.0). All the solutions were prepared at the same concentration of porphyrin dye ([C] = 4 × 10−6 M). To investigate the use of a polymer template for porphyrin aggregation, homopolymer 3 was dissolved in the same pH buffers as mentioned above and a certain amount (0.4g/L) of the porphyrin dye was gradually added (Scheme 2). All solution preparation and photophysical studies were done in the dark. The aggregation properties were studied by time-dependent UV-vis absorption, steady-state fluorescence, and fluorescence lifetime decay for both the dye solutions themselves as well as polymer-templated dye solutions.
Scheme 2.

Porphyrin complexation with the polymeric template.
UV-vis Absorption Spectroscopy
The absorption spectra of TSPP (4) and its analog (5) in acidified water without or with the polymer template at 10−5 M concentration of the dyes were measured (Figure 1). TSPP (4) is known to form J-aggregates under strong acidic conditions. In neutral aqueous solution, TSPP remains as a monomeric free base form due, in part, to electrostatic anionic repulsion of the sulfonate groups. The absorption bands correspond to its non-protonated form (Figure 1 (a)); exhibiting an intense Soret band at 414 nm and weak Q bands at 516, 550, 581, and 635 nm.
Figure 1.
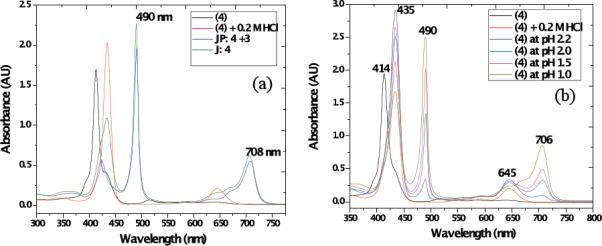
(a) UV-vis absorption spectra of 4 in solution (black: TSPP, red: TSPP with 0.2 M HCl acidic conditions, green: TSPP in pH 1 buffer formed J-aggregates, and blue: TSPP in pH 1 buffer with polymer 3 template), and (b) formation of J-aggregates of (4) at different pH (HCl/KCl aqueous buffer, (black: TSPP, red: TSPP with 0.2 M HCl, blue: TSPP in pH 2.2, green: TSPP in pH 2.0, pink: TSPP in pH 1.5, and tan: TSPP in pH 1.0 buffer).
In the presence of acid, protonation takes place at the two nitrogens of the porphyrin core. Consequently, the absorption maxima of the Soret band shifted to 435 nm and the two Q bands shifted to 590 and 645 nm. The presence of J-type aggregation is usually signaled by an intense and narrow absorbance at 490 nm (J-band), which is bathochromically shifted relative to the monomer absorption band at about 435 nm (the Soret band), accompanied by a weaker, broad band at 705 nm.27,28 The 490 nm J-band of the aggregates has been assigned to a Frenkel exciton transition.27,28,29,30 Porphyrins tend to self-assemble through balancing π-π oblique stacking interactions of their hydrophobic porphyrin rings, and charged substituent groups, present at the inner (cationic N) core and outer surface (anionic sulfonate groups) by electrostatic forces.11
The induction of J-aggregation of 4 was studied using HCl/KCl buffers (pH range 2.2 –1.0) at room temperature in the dark (Figure 2). Porphyrin 4 formed intense J-bands with decreasing pH (Figure 2 (a)). At pH 1.0, with increasing time, a substantial percentage of molecules of 4 participated in J-aggregation, as shown in Figure 2 (b), behavior that is consistent with other reports.31
Figure 2.
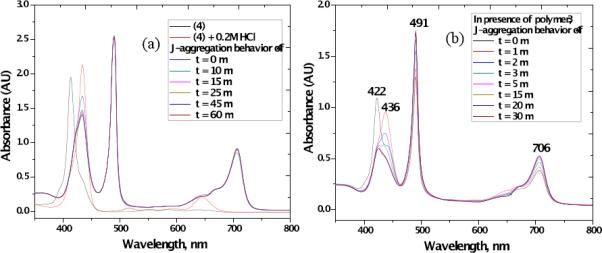
(a) Kinetic study of J-band of 4 at pH 1.0 at different time intervals (a 1.0 cm path length cuvette cell was used with ([4]= 40 μM) at room temperature), (b) Formation of stable J-aggregates of 4 in the presence of polymer 3 in aqueous buffer (pH = 1.0) at different time intervals.
In the presence of polymer 3, porphyrin 4 produced a well-defined strong and sharp J-band at 491 nm along with a broad band at 706 nm, while an H band at 422 nm appeared at slightly higher pH (>1.0). Formation of the H-aggregates is generally attributed to π interactions of the porphyrin rings with the combined effect of attractive σ-π and repulsive π-π interactions.32 Also, with increasing time periods (0–30 min), more molecules were transformed from H-aggregates to J-aggregates, producing a very sharp and narrow J-band at 490 nm, possibly due to a stabilization effect of cationic polymer 3 as shown in Figure 2 (b). Also, the spectral width (FWHM) of the absorption peak of the J-aggregate varied with the coherence length as N−1/2, where N is the spectroscopic aggregation number,33,34 and the effect of external ammonium ions were reported.17b
To the best of our knowledge the J-aggregation of porphyrin 5 has not been reported. Porphyrin 5, in its non-protonated form, exhibited a Soret band at 416 nm along with a shoulder at 444 nm and a weak band at 654 nm. In presence of 0.2 M HCl, the Soret band of 5 bathochromically shifted to 433 nm and the Q band underwent a hypsochromic shift to 647 nm. Porphyrin 5 behaved differently with respect to 4 in the pH range 1.0–2.2 due to the presence of the amine group instead of the fourth sulfonate group at the para-position of one of the porphyrin phenyl substituents. Porphyrin 5 showed an increasing J-band with increasing pH (Figure 3 (a, b)) with a sharp J-band absorption at 484 nm and another red shifted band at 696 nm. With addition of polymer 3, stronger J-aggregation of 5 occurred at the same concentration of the dye at pH 2.2 relative to without the polymer, which is clear from the higher ratio and sharper nature of the peak corresponding to the J band at 484 and 697 nm relative to the peak at 432 nm (Figure 3 (e, f)).
Figure 3.

UV-vis absorption spectra of 5 (a) at different pH in HCl/KCl aqueous buffer and formation of J-aggregates of 5, (b) in presence of polymer 3 at pH 1.0 (HCl/KCl buffer), (c, d) J-aggregation behavior as a function of varying concentration of 5 without polymer (c) and in presence of polymer 3 (d) [exact concentration of the added 5 is shown] All experiments were conducted with [5] = 10−5 M in 1 cm cuvette at room temperature, *denotes measurement after storing overnight (orange line in (d)). (e, f) Ratiometric UV-vis absorbance study of the J-bands to the monomer peak ratio of 5 with its increasing concentration (e) without polymer and (f) in presence of polymer 3.
The effect of concentration of dye 5, on the formation of J-aggregates by itself as well as in presence of particular concentrations of polymer 3 was also investigated, as shown in Figure 3 (c, d). From these studies, it is evident that J-aggregation of the dye was facilitated with increasing dye concentration in both cases. The results are consistent with the following interpretation. The polymeric template led to more efficient aggregation of 5, which is reflected in the higher ratio of the J-band population at nearly similar concentrations. In addition to π-π hydrophobic interactions and the electrostatic attraction between the imidazolium ring and phenyl sulfonate groups, the interaction of the cationic dimethylammonium groups in the J-aggregated species likely reduced repulsive forces among porphyrin rings, thus making J-aggregation more stable in aqueous solution.
Fluorescence Spectroscopy
Steady state fluorescence emission spectra were recorded for TPPS (4) under neutral conditions, acidic conditions (10−6 M), and after formation of J-aggregates with or without the polymer (10−5 M) at room temperature in 1 cm cuvettes using a PTI Quantamaster spectrofluorimeter (Figure 4). The emission spectra did not change whether excited at the B or Q bands, as reported in literature.11, 35 Figure 4 shows the fluorescence spectra of 4 resulting from the excitation of the Soret band (B-band) of the species at 413, 434, and 490 nm for the free base, dianion monomer, and J-aggregated species, respectively. The emission spectra did not change whether excited at the B or Q bands, consistent with that reported previously.11 Emission of the aggregated species (with or without polymer) was considerably weaker, with almost negligible Stokes shifts, compared to the monomer and dianion species, again likely due to the predominant effect of radiative quenching pathways.11
Figure 4.
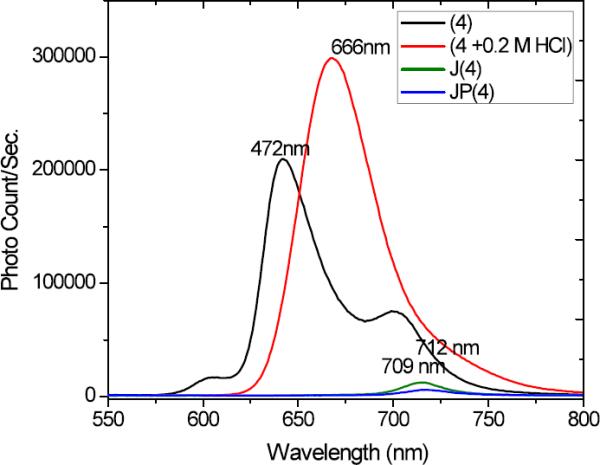
Steady state fluorescence spectra of dye 4 (black line is [4] = 10−6 M in water (λexc = 413 nm (free base)), red line is [4] = 10−5 M (λexc (dianion) = 434 nm), and green and blue lines are [4] = 10−5 M for J-aggregated species without and with polymer (λexc = 490 nm), respectively, at room temperature.
Lifetime studies
To shed light on the photophysics of porphyrin J-aggregation, fluorescence lifetime decay was studied using a time-correlated single photon counting system (PicoHarp 300) under linear polarized femtosecond excitation (Table 1). Fluorescence lifetime decays of TPPS, 4, are shown in Figure S2. The free base and dianion monomer had a single exponential decay with excited state lifetimes of 6.87 and 3.74 ns for nonlinear (two-photon) excitation at 826 and 868 nm, respectively. As anticipated, the lifetime of the J-aggregated species was very short when excited at the Q band (710 nm). Specifically, with the polymer template a shorter (250 ps) lifetime was observed, as shown in Table 1. Fluorescence quenching and shortening of the excited state lifetime is characteristic of J-aggregation.36, 37
Table 1.
Photophysical properties of the TPPS (4 free base and dianion) and corresponding J-aggregation with or without polymer template.
| λabs (nm) | λem (nm) | λexc (nm) | Lifetime (ns) | |||
|---|---|---|---|---|---|---|
| t1 | t2 | R2 | ||||
| 4 | 413, 515, 635 | 472 | 413(826) | 6.87 | 0.99246 | |
| 4 (0.2 M HCl) | 434, 644 | 466 | 434 (868) | 3.74 | 0.99788 | |
| J (4) * | 434, 490, 708 | 709 | 710 | 0.32 | 0.080 | 0.99711 |
| JP (4 + 3) * | 421, 491, 711 | 712 | 710 | 0.25 | 0.99728 | |
J-aggregates were measured at pH 2.2
Fluorescence lifetime decays of porphyrin 5 are shown in Figure S3. For 5 the lifetimes for free monomer and its anioinic form were 6.47 and 2.01 ns, respectively, as shown in Table 2. While at a higher concentration of the dye, with or without polymer, its forms partially J-aggregated species with short fluorescence lifetimes along with the contribution from its anionic form.
Table 2.
Photophysical properties of the 5 (free base and anion) and corresponding J-aggregation with or without polymer template.
| λabs (nm) | λem (nm) | λexc (nm) | Lifetime (ns) | |||
|---|---|---|---|---|---|---|
| t1 | t2 | R2 | ||||
| 5 | 415, 659 | 606, 653, 706 | 413 (826) | 6.47 | 0.980 | |
| 5 (0.2 M HCl) | 433,647 | 671 | 430 (860) | 2.01 | 0.9964 | |
| J (5) | 433, 485, 698 | 671 | 698 | 2.22 (64%) | 0.35(36%) | 0.9982 |
| JP (5+3) | 433, 485, 698 | 671 | 698 | 1.87(80%) | 0.24 (20%) | 0.9984 |
Nonlinear absorption studies
In order to broaden the potential applications of porphyrin aggregates and probe the effect of aggregation on nonlinear absorption, two-photon absorption (2PA) cross sections (δ2PA) were measured by an open aperture Z-scan method using an amplified Ti:sapphire laser system with an optical parametric amplifier providing laser pulses of ~100 fs (FWHM) duration at 1 kHz repetition rate (Figure 5) in a 1 mm quartz cuvette at room temperature. The concentration of TSPP (4) and TSPP analog (5) solutions for Z scan measurements was 10−2 M. J-aggregated TSPP solutions at pH 1 and polymer-templated J-aggregated TSPP were used at 10−3 M due to precipitation at higher concentrations.
Figure 5.

Z-scan experiment setup, (1) 100% reflection mirrors, (2) focusing lens, (3) pinhole, (4) neutral density filter, and (5) beam splitter.
As shown in Figure 6, the 2PA cross section of TSPP was 30–95 GM (GM, Goppert-Mayer units, 1 × 10−50 cm4 s photon−1 molecule–1), depending on excitation wavelength. Significantly, after aggregation at pH 1, the 2PA cross section values increased by a factor of four. In addition, after polymer-templated aggregation, the 2PA cross section values increased an additional 2.5 times higher than aggregation at pH 1. Therefore, the polymer template not only increased J-aggregation but the increased J-aggregation resulted in a substantial increase in two-photon absorption.
Figure 6.
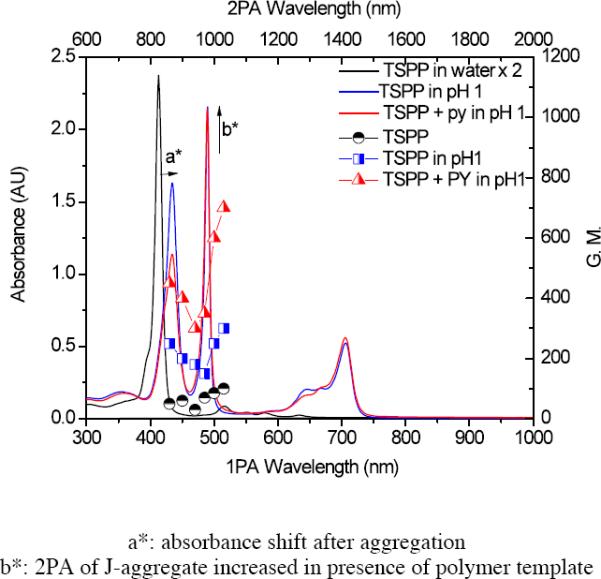
Absorption spectra (bottom x-axis and left y-axis) of TSPP (4) in 10−5 M water (black line), 4 at pH 1 (blue line), and 4 with polymer 3 in pH 1 solution (red line), and corresponding 2PA cross sections (top x-axis and right y-axis, each half filled symbols, respectively).
In the case of the TSPP analog (5), the trend is same as 4 in that 2PA cross section value was enhanced by aggregation and the polymer template enhanced 2PA further shown as in Figure 7. The aggregation enhancement effect of 5 is significant, with data revealing 2PA cross sections of 160–300 GM (three time higher than 4). When 5 underwent aggregation at pH 1, the increase in 2PA cross section values were up to 6000 GM at 1000 nm. Moreover, after polymer-templated aggregation, the 2PA cross section values increased by a factor of 2, resulting in 2PA cross sections up to ca. 10,000 GM. Though the origin of this enhancement is not fully understood and will be the subject of future experimental and theoretical studies, polymer-templated formation of porphyrin J-aggregates provides a means to modulate and greatly enhance nonlinear absorption, an aspect that may be useful in applications ranging from bioimaging and photodynamic therapy (singlet oxygen quantum yield for 5 was determined to be 0.15 in H2O, significant for a photosensitizer in aqueous solution) to optical power limiting.
Figure 7.
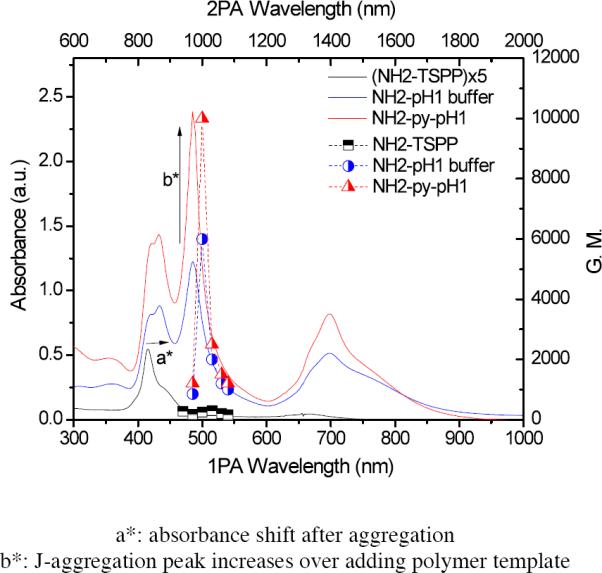
Absorption spectra (bottom x-axis and left y-axis) of TSPP analog 5, in water (black line), 5 at pH 1 (blue line), and 5 with polymer in pH 1 solution (red line), and corresponding 2PA cross sections (top x-axis and right y-axis, each half filled symbols, respectively).
Conclusion
The effects exerted by a ROMP-based water soluble polymeric template, containing pendant amino groups, on the J-aggregation properties of two porphyrin-based dyes (TSPP, 4, and its amine-containing analog 5) were investigated. J-aggregation of the two dyes (4 and 5) was confirmed with UV-vis absorption and time-resolved fluorescence spectroscopy, revealing characteristic long wavelength absorption bands and markedly shorter lifetimes of J-aggregates relative to monomer alone while steady state fluorescence emission decreased significantly with aggregation. The photophysical properties were consistent with J-aggregation that was facilitated by the amphiphilic polymer, with the polymer inducing J-aggregation faster than the dye alone under similar conditions. Not surprisingly, substitution of one sulfonic acid group by an amine group may have affected the ionic interaction of the dyes, thereby influencing aggregation properties and structure. These results support the use of cationic polymer templates to enhance the J-aggregation in solution through possible attractive interaction between anionic porphyrin periphery and pendant cationic moieties on the polymer template, facilitating the design of polymer-templated supramolecular aggregates. Significantly, two-photon absorption of the J-aggregates was higher than the corresponding unaggregated porphyrins. In addition, aggregation was firmly enhanced by the polymer template, resulting in a significant increase in 2PA, up to 10,000 GM for polymer-templated 5. Both pH and the template provides a means to modulate the nonlinear absorptivity of these porphyrin derivatives and achieve relatively high 2PA cross sections, an aspect that is important for a number of applications such as optical data storage, optical power limiting, bioimaging, and photodynamic therapy.
Supplementary Material
Acknowledgements
We wish to acknowledge the National Science Foundation (CHE-0840431, CHE-0832622, and ECCS-0925712), the National Institutes of Health (1 R15 EB008858-01), the U. S. Civilian Research and Development Foundation (UKB2-2923-KV-07), the Ministry of Education and Science of Ukraine (grant M/49-2008), and Dr. David Richardson for assistance with NMR experiments (in Supporting Information).
Footnotes
Supporting Information Available. Molecular weight determination via NMR experiments, time-resolved fluorescence data, and data for ratiometric UV-vis absorbance studies on J-aggregates of 4 as a function of concentration by itself and in presence of polymer 3 are included in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Nam YS, Shin T, Park H, Magyar AP, Choi K, Fantner G, Nelson KA, Belcher AM. J. Am. Chem. Soc. 2010;132:1462. doi: 10.1021/ja908812b. [DOI] [PubMed] [Google Scholar]
- (2).Hunter C. Nature. 2011;469:39. doi: 10.1038/469039a. [DOI] [PubMed] [Google Scholar]
- (3).Ochiai T, Nagata M, Shimoyama K, Amano M, Kondo M, Dewa T, Hashimoto H, Nango M. Langmuir. 2010;26:14419. doi: 10.1021/la102869w. [DOI] [PubMed] [Google Scholar]
- (4).Astruc D, Boisselier E, Ornelas C. Chem. Rev. 2010;110:1857. doi: 10.1021/cr900327d. [DOI] [PubMed] [Google Scholar]
- (5).(a) Heim D, Écija D, Seufert K, Auwärter W, Aurisicchio C, Fabbro C, Bonifazi D, Barth JV. J. Am. Chem. Soc. 2010;132:6783. doi: 10.1021/ja1010527. [DOI] [PubMed] [Google Scholar]; (b) Würthner F, Kaiser TE, Saha-Möller CR. Angew.Chem. Int. Ed. 2011;50:3376. doi: 10.1002/anie.201002307. [DOI] [PubMed] [Google Scholar]
- (6).(a) Scheibe G. Angew. Chem. 1936;49:563. [Google Scholar]; (b) Scheibe G. Ber. dtsch. chem. Ges. 1921;54:786. [Google Scholar]; (c) Scheibe G, Kandler L, Ecker H. Naturwissenschaften. 1937;29:75. [Google Scholar]; d) Scheibe G. Angew. Chem. 1937;50:210. [Google Scholar]
- (7).Jelly EE. Nature. 1936;138:1009. [Google Scholar]
- (8).Kasha M, Rawls HR, El-Bayoumi MA. Pure Appl. Chem. 1965;11:371. [Google Scholar]
- (9).Mirsky VM, Yatsimirsky A. Artificial Receptors for Chemical Sensors. Wiley-VCH; 2010. [Google Scholar]
- (10).Guldi DM. Chem. Soc. Rev. 2001;31:22. doi: 10.1039/b106962b. [DOI] [PubMed] [Google Scholar]
- (11).Kobayashi T. J-aggregates. World Scientific; Singapore: 1996. [Google Scholar]
- (12).Ohno O, Kaizu Y, Kobayashi H. J. Chem. Phys. 1993;99:4128. [Google Scholar]
- (13).Lin J, Ding W, Hong K, Mays JW, Xu Z, Yuan Y. Soft Mater. 2008;4:1605. doi: 10.1039/b804838j. [DOI] [PubMed] [Google Scholar]
- (14).Zhao L, Ma R, Li J, Li Y, An Y, Shi L. Biomacromolecules. 2008;9:2601. doi: 10.1021/bm8004808. [DOI] [PubMed] [Google Scholar]
- (15).Wu JJ, Li N, Li KA, Liu F. J. Phys. Chem. B. 2008;112:8134. doi: 10.1021/jp802482f. [DOI] [PubMed] [Google Scholar]
- (16).Purrello R, Bellacchio E, Gurrieri S, Lauceri R, Raudino A, Scolaro LM, Santoro AM. J. Phys. Chem. B. 1998;102:8852. [Google Scholar]
- (17).(a) Koti ASR, Periasamy N. Chem. Mater. Comm. 2003;15:369. [Google Scholar]; (b) Koti ASR, Taneja J, Periasamy N. Chem. Phys. Lett. 2003;375:171. [Google Scholar]
- (18).Jones RM, Lu L, Helgeson R, Bergstedt TS, McBranch DW, Whitten DG. Proc. Natl. Acad. Sci. 2001;98:14769. doi: 10.1073/pnas.251555298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lu L, Jones RM, McBranch D, Whitten D. Langmuir. 2002;18:7706. [Google Scholar]
- (20).Kano K, Watanabe K, Ishida Y. J. Phys. Chem.B. 2008;112:14402. doi: 10.1021/jp802567b. [DOI] [PubMed] [Google Scholar]
- (21).Steinbeck CA, Ernst M, Meier BH, Chmelka BF. J. Phys. Chem. C. 2008;112:2565. [Google Scholar]
- (22).Smith ARG, Ruggles JL, Yu A, Gentle IR. Langmuir. 2009;25:9873. doi: 10.1021/la900953a. [DOI] [PubMed] [Google Scholar]
- (23).Biswas S, Belfield KD, Das RK, Ghosh S, Hebard AF. Chem. Mater. 2009;21:5644. [Google Scholar]
- (24).Walderhaug H. J. Phys. Chem. B. 1999;103:3352. [Google Scholar]
- (25).Lide DR. Handbook of chemistry and physics. CRC Pr I Llc; 2003. [Google Scholar]
- (26).Kruper WJ, Jr, Chamberlin TA, Kochanny M. J. Org. Chem. 1989;54:2753. [Google Scholar]
- (27).Fleischer RF, Palmer JM, Srivastava TS, Chatterjee AJ. J. Am. Chem. Soc. 1971;93:3162. doi: 10.1021/ja00742a012. [DOI] [PubMed] [Google Scholar]
- (28).Pasternack RF, Huber PR, Boyd P, Engasser G, Francesconi L, Gibbs E, Fasella P, Venturo GC, Hinds LD. J. Am. Chem. Soc. 1972;94:4511. doi: 10.1021/ja00768a016. [DOI] [PubMed] [Google Scholar]
- (29).Akins DL, Ozcelik S, Zhu HR, Guo C. J. Phys. Chem. A. 1996;100:14390. [Google Scholar]
- (30).Kano H, Saito T, Kobayashi T. J. Phys. Chem. B. 2001;105:413. [Google Scholar]
- (31).Smith ARG, Ruggles JL, Yu A, Gentle IR. Langmuir. 2009;25:9873. doi: 10.1021/la900953a. [DOI] [PubMed] [Google Scholar]
- (32).(a) Hunter CA, Sanders JKM. J. Am. Chem. Soc. 1990;112:5525. [Google Scholar]; (b) Ribo JM, Crusats J, Farrera J-A, Valero ML. J. Chem. Soc. Chem. Commun. 1994:681–682. [Google Scholar]; (c) Barber DC, Freitag-Beeston RA, Whitten DG. J. Phys. Chem. 1991;95:4075. [Google Scholar]
- (33).Knapp EW. Chem. Phys. Lett. 1984;85:73. [Google Scholar]
- (34).Knapp EW. Chem. Phys. Lett. 1984;111:481. [Google Scholar]
- (35).Akins DL, Ozcelik S, Zhu H-R, Guo C. J. Phys. Chem. 1996;100:14390. [Google Scholar]
- (36).Misawa K, Kobayashi T. J. Chem. Phys. 1999;110:5844. [Google Scholar]
- (37).Maiti NC, Ravikanth M, Mazumdar S, Periasamy N. J. Phys. Chem. 1995;99:17192. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.