

Preface
The 'invisible hand' is a term originally coined by Adam Smith in the Theory of Moral Sentiments to describe the forces of self-interest, competition, and supply and demand that regulate the resources in society. This metaphor continues to be used by economists to describe the self-regulating nature of a market economy. The same metaphor can be used to describe the RHO-specific guanine nucleotide dissociation inhibitor (RHOGDI) family, which operates in the background, as an invisible hand, using similar forces to regulate the RHO GTPase cycle.
Introduction
RHO family GTPases control a wide variety of cellular processes, including cell adhesion, migration and proliferation (Box 1). At any given time, only a small fraction of all RHO GTPases present in the cell are in the active state and are associated with membranes. The inactive pool is maintained in the cytosol by associating with RHO-specific guanine nucleotide dissociation inhibitors (RHOGDIs).
Box 1. The RHO switch.
RHO proteins are guanine nucleotide-binding proteins (G proteins also known as GNBPs) that belong to the larger group of GTPases, which include heterotrimeric G proteins, elongation factors, tubulins, septins and large GTPases such as dynamin. Biochemically, GTPases are hydrolase enzymes that bind and hydrolyze GTP. In a similar way to ATP, GTP can act as an energy carrier, but it also has an active role in signal transduction, particularly in the regulation of G protein activity. G proteins, including RHO GTPases, are molecular switches that cycle between an inactive GDP-bound and an active GTP-bound conformation (see the figure). The transition between the two conformational states occurs through two distinct mechanisms: activation by GTP loading and inactivation by GTP hydrolysis. GTP loading is a two-step process that requires the release of the GDP-bound nucleotide and its replacement by a GTP molecule. Nucleotide release is a spontaneous but slow process that has to be catalyzed by RHO guanine-nucleotide exchange factors (RHO GEFs), which associate with RHO GTPases and trigger the release of the nucleotide. The resulting nucleotide-free binary complex has no particular nucleotide specificity. However, the cellular concentration of GTP is markedly higher than that of GDP, which favours GTP loading, resulting in the activation of RHO GTPase. Conversely, to turn off the switch, GTP has to be hydrolyzed. This is facilitated by RHO GTPase-activating proteins (RHOGAPs), which stimulate the intrinsically slow hydrolytic activity of RHO proteins. It is worth noting that, although GEFs and GAPs are the canonical regulators of this cycle, several alternative mechanisms, such as post-translational modifications, may fine-tune the RHO switch. In addition, inactive RHO GTPases are extracted by RHO-specific guanine nucleotide dissociation inhibitors (RHOGDIs) from cell membranes to prevent their inappropriate activation and to protect them from misfolding and degradation.
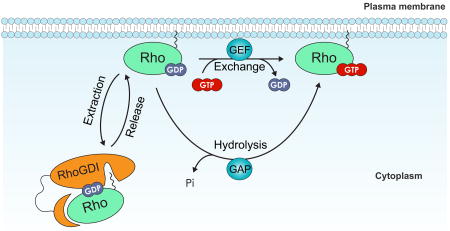
Despite the wide diversity in the RHO GTPase family, there are only three genes encoding RHOGDI in mammals (Box 2) 1. RHOGDI1 (also known as RHOGDIα) is the most abundant and best-characterized member of the family; it is ubiquitously expressed and interacts with several RHO GTPases, including RHOA, RHOC, RAC1, RAC2 and CDC42 2, 3. RHOGDI2 (also known as RHOGDIβ, Ly-GDI and D4-GDI) is expressed in high levels by haematopoietic cells4, 5, but has been also found to be expressed in other tissues, as well as by cancer cells (see below). RHOGDI2 associates with several RHO GTPases in vitro, but with significantly lower affinity than RHOGDI16. However, many of these interactions have not been detected in vivo7. RHOGDI3 (also known as RHOGDIγ) is the most divergent of the three and contains a unique amino-terminal extension that targets it to the Golgi complex and other cellular membranes8. RHOGDI3 is usually expressed at low levels and seems to interact predominantly with RHOB and RHOG8-10. Completing the catalogue of binding specificities of RHOGDIs to the different RHO GTPases, which is far from comprehensive, should shed some light on the differences in the functions of the different isoforms.
Box 2. RHOGDI evolution and orthologues.
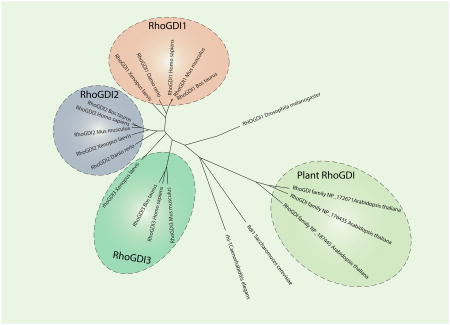
Guanine nucleotide dissociation inhibitors (GDIs) can be categorized according to their guanine nucleotide-binding protein (G protein) specificity, but not all G proteins have GDIs (for example Ras does not, but the RHO and Rab families do). The different GDI families share some common functional and structural features, but they usually have unrelated amino acid sequences, suggesting that the families emerged independently during evolution. The RHOGDI family (see the figure) is defined by a conserved RHOGDI backbone. In humans, there are three RHOGDI proteins: RHOGDI1 (also known as RHOGDIα and encoded by ARHGDIA), which is expressed ubiquitously; RHOGDI2 (also known as RHOGDIβ, Ly-GDI, D4-GDI and encoded by ARHGDIB), which is expressed by haematopoietic cells among others and is commonly upregulated in certain tumours; and RHOGDI3 (also known as RHOGDIγ and encoded by ARHGDIG), which is expressed mostly in the brain. Because RHOGDIs are evolutionarily conserved proteins, they have several orthologues in other eukaryotes (see the figure). There is a single RHOGDI in many unicellular and organisms such as Saccharomyces cerevisiae (Rdi1), and also in protostomes such as Caenorhabditis elegans (RHI-1) and Drosophila melanogaster (RHOGDI). Other metazoans have two or three RHOGDIs: for example, Danio rerio has two and Xenopus laevis has three. Tetrapods, including avians and mammals, generally have three RHOGDIs. Interestingly, plants such as Arabidopsis thaliana have their own particular set of Rho proteins (ROPs) and their own specific RHOGDI proteins, which highlights both their differences with the animal kingdom and the universal necessity to have RHOGDIs despite these differences. From the study of RHOGDIs in different organisms, it emerges that RHOGDIs have fundamentally conserved functions in eukaryotes: the regulation of RHO protein cycling between lipid membranes and the cytosol, the stabilization of RHO proteins and the inhibition of nucleotide release. The figure was generated by aligning the sequences of the RHOGDIs using ClustalW and by compiling node distances using the neighbour-joining method.
Although originally considered to be passive regulators, recent studies have shown that the RHOGDI family have a key role in the regulation of RHO GTPases. Like an 'invisible hand'—a term originally coined by Adam Smith in the Theory of Moral Sentiments to describe the forces of self-interest, competition and supply and demand that regulate the resources in society11—RHOGDIs operate in the background, controlling key aspects of the RHO GTPase cycle. In this Review, we discuss how RHOGDIs regulate RHO GTPases, by modulating the extent of their expression, their membrane localization and their activation state. We also describe the mechanisms by which the interaction between RHOGDIs and RHO GTPases is regulated, and how, in many cases, this regulation can be selective for each type of RHO GTPase. Changes in the levels of expression of the different RHOGDIs have marked effects on the overall levels and activities of RHO GTPases and have been correlated with several types of cancer.
RHOGDI functions
RHOGDIs were initially characterized as simply RHO GTPase inhibitors; however, recent work indicates that their function is more complex 12.
RHOGDIs as negative regulators of RHO GTPases
The first RHOGDI (RHOGDI1) was originally discovered in the pre-genomics era, when new proteins were still identified on the basis of their biochemical properties and function. RHOGDI (or RHOB p20 GDI, as it was named then) was initially purified from rabbit intestine and later cloned from bovine brain tissue2, 13. It was characterized as a protein capable of inhibiting some of the basic features of RHO proteins, (and not of RAS, RAP and RAB), such as the release of GDP and the loading of GTP to RHO-GDP. RHOGDIs do not prevent loading of GDP or GTP to a nucleotide-free RHO; this means that they inhibit the release of the nucleotide and not its binding to the RHO GTPase 13. It was therefore clear that its biochemical activity was different from that of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). RHOGDIs were also found to inhibit the GTPase activity of RHO proteins, preventing both intrinsic and GAP-stimulated hydrolysis of GTP 14. This dual inhibitory function would later be understood from the structure of RHOGDI complexed with a RHO GTPase. The N-terminal domain of RHOGDI interacts with the switch I and switch II domains of the RHO protein (see below) and restricts the spatial flexibility that is required for the transition between different nucleotide-bound forms of the RHO GTPase, essentially 'locking up' the GTPase. Conceptually, RHOGDI seemed to function in the RHO switch (Box1) simply as an inhibitory molecule that bound mostly to inactive RHO GTPases and preventing nucleotide exchange. This view of RHOGDI as an inhibitory regulator was extended by the observation that RHOGDI can extract RHO proteins from membranes and hold them in a complex in the cytosol away from their sites of action at membranes3.
RHOGDIs as chaperones
Given that RHO GTPases are prenylated and act at membranes, it is somewhat surprising that the largest fraction of each family member is found in the cytosol 12, 15. One of the main functions of RHOGDIs is to maintain a stable soluble pool of inactive RHO GTPases, which at any given time may account for most (90–95%) of the RHO proteins in the cell 12, 15. A generally accepted explanation for this cytosolic pool is that it acts as a reservoir, allowing inactive RHO GTPases to be rapidly translocated to any membrane in the cell for activation in response to specific signals. This 'instant deployment' strategy would allow cells to respond quickly to any stimulus or challenge that requires the activation of a particular RHO GTPase. It is important to note here that most of the studies have examined the effects on the cytosolic pool of the major RHO GTPases — RAC1, RHOA and CDC42 — and findings may not apply to all family members.
One of the problems associated with maintaining a soluble RHO GTPase pool is the hydrophobic isoprenoid moiety at the carboxyl terminus of all RHO proteins. Isoprenylation of RHO proteins is essential for their proper subcellular localization and signalling16. However, in the absence of membranes, the isoprenoid moiety, which is highly hydrophobic, impairs the ability of RHO GTPases to fold properly12. Binding to RHOGDI stabilizes the cytosolic RHO pool and shields the isoprenyl group from exposure to water by inserting it into a hydrophobic pocket in the C-terminal half of the molecule. Consistent with this, phosphorylation of RHOA on Ser188 increases its affinity for RHOGDI and protects it from degradation (see below)17.
This function of RHOGDIs has always been considered to be more of a 'housekeeping' role and has not been characterized in detail. However, it was recently found that, in the absence of RHOGDI1, the cytosolic pool of RHO GTPases is unstable and rapidly degraded in a proteasome-dependent manner (Fig. 1)12. In the absence of the isoprenyl group, RHO GTPases are much more stable, although their signalling is impaired because they fail to localize to their sites of action at specific membranes. In this way, RHOGDIs act as chaperones, not as much in assisting the folding of newly synthesized proteins (although it is possible that this occurs), but by stabilizing the mature RHO GTPases when they are not associated with membranes. Even though a chaperone function for RHOGDIs has been long recognized18, their role in protecting against degradation was generally overlooked.
Figure 1. The RHOGDI cycle.

(a) Newly synthesized RHO family GTPases are geranylgeranylated and then post-translationally modified by the protease Ras-converting enzyme 1 (RCE1) and by isoprenylcysteine carboxyl methyltransferase (ICMT) at the cytoplasmic face of the endoplasmic reticulum (ER). (b) After geranylgeranylation, RHO proteins associate with RHO specific guanine nucleotide dissociation inhibitors (RHOGDIs), which sequester them in the cytosol and protect them from degradation. (c) Free prenylated cytosolic RHO GTPases are unstable and are rapidly degraded by the proteasome. (d) Several RHO GTPases can associate with RHOGDI and compete for its binding. Overexpression of a GTPase can displace the endogenous RHO proteins from RHOGDI targeting them for degradation. (e) The rate of cycling of the RHOGDI -RHO GTPase complex between the cytosol and the membrane can be regulated by post-translational modifications on both the RHO GTPases and the RHOGDI, which modulate the affinity of the interaction. A slower pathway for recycling RHO proteins through vesicle trafficking has also been postulated. (f) Once at the membrane, the RHO GTPases can be activated by guanine nucleotide exchange factors (GEFs) and bind to downstream effectors. Following inactivation by GTPase-activating proteins (GAPs), RHO GTPases are extracted from the membrane by RHOGDI. (g) Active RHOA can also be targeted for degradation by the ubiquitin ligase SMAD ubiquitylation regulatory factor 1 (Smurf1). GGTase, geranylgeranyl transferase.
RHOGDI shuttle RHO GTPases between membranes
Since the discovery that RHOGDIs can extract RHO proteins from membranes, the idea that RHOGDIs may shuttle RHO GTPases between different membrane compartments has been prevalent. However, evidence has been presented both for and against this, and the debate continues. In a series of papers, Wilson-Delfosse and colleagues19-21 designed RHO GTPase mutants (Arg66Glu in RAC1 and CDC42, Arg68Glu in RHOA) that cannot bind to RHOGDIs and hypothesized that, if RHOGDI is required to shuttle RHO proteins between membranes, the mutated proteins should be mislocalized and not functional. By expressing constitutively active RHO GTPases, they found no significant difference in the signalling between the forms that were able or unable to bind to RHOGDIs. Similarly, RHOGDI-null cells responded the same way as their wild-type counterparts when constitutively active mutants of RAC1 and CDC42 were overexpressed19, 20. This led them to conclude that RHOGDIs are not required for the translocation of RHO GTPases to their membrane destinations 19-21. Consistent with this, yeast cells lacking Rdi1 (the RHOGDI orthologue) and RHOGDI1-knockout mice had mild phenotypes 22-24. The RDI1 deletion yeast strain was indistinguishable from wild-type cells in terms of growth, cell morphology and mating, but exhibited defects in pseudohyphal growth and mitosis exit22. Similarly, RHOGDI1-knockout mice reached adulthood normally, suggesting there are other mechanisms that can control the cycling of RHO GTPases 23, 24.
By contrast, Cerione's group found that, although a fast-cycling mutant of CDC42 (Phe28Leu) induced cell transformation, this property was lost when a second mutation (Arg66Ala) that impaired binding to RHOGDI was introduced 25. CDC42 Phe28Leu can undergo spontaneous GTP–GDP exchange while maintaining full GTPase activity26, and, unlike GTPase-defective CDC42 mutants (which are toxic to cells), can be stably expressed and exhibit properties of oncogenic transformation. Unable to associate with RHOGDI, the CDC42 double mutant, accumulated in membranes in the perinuclear region rather than reaching the plasma membrane25. Similarly, another group found that the Arg66Glu RAC1 mutant, which cannot bind RHOGDI, failed to translocate to the plasma membrane in response to hepatocyte growth factor (HGF) stimulation (which normally activates RAC1)27. Fractionation of cells has revealed a biphasic distribution of membrane-associated RHOA, RAC1 and CDC42, with most found in the endoplasmic reticulum (ER) and a smaller amount of each one in the plasma membrane fraction. Depletion of RHOGDI1, however, causes a marked reduction in the levels of each GTPases that are associated with the plasma membrane 12, suggesting that RHOGDIs are involved in the transport of RHO GTPases to the plasma membrane.
How can this contrasting evidence be reconciled? One possibility is that there are other RHOGDI-like molecules compensating for the loss of RHOGDIs. For example, recent work shows that caveolin 1 binds to CDC42 and sequesters it in its GDP-bound form at secretory granules 28. CDC42 is required for insulin granule exocytosis, during which it is proposed to function in targeting the secretory granules to specific sites at the plasma membrane29. Upon stimulation by glucose, CDC42 is dissociated from caveolin 1 and subsequently activated, allowing the secretory granules to fuse with the plasma membrane. Mechanistically, caveolin 1 assumes some of the properties of RHOGDIs by sequestering inactive CDC42 and restricting fusion of secretory granules. It will be interesting to discover whether other membrane-bound RHOGDI-like molecules contribute to the spatial and temporal regulation of RHO GTPases.
However, there is evidence against this theory: in the absence of RhoGDIs, essentially no RHO GTPases are detected in the cytosol 12. An alternative explanation, which we favour, comes from studies in yeast, in which there is evidence supporting the coexistence of two mechanistically distinct recycling systems for CDC42, a fast-cycling mechanism involving Rdi1 and a slower one involving vesicle trafficking (Figure 1)30. At the early stages of bud formation, CDC42 is targeted to a small region in the plasma membrane that later becomes the bud site, where it functions to orchestrate the polarization of the actin cytoskeleton (which is required to direct secretory traffic to the budding site) 31. Deletion of the Rdi1 significantly decreases the rate of exchange of CDC42 between the plasma membrane and the cytosol30 but has no obvious growth phenotype, owing to the presence of the endocytic recycling pathway. However, when both pathways are inhibited, the polarization of CDC42 at the bud site is rapidly lost.
We favour the idea of vesicle trafficking accounting for the small amount of RHO proteins that can reach the plasma membrane in mammalian cells lacking RHOGDI1. The existence of two pathways by which RHO GTPases reach their membrane destinations provides an explanation for how constitutively active RHO proteins that cannot interact with RHOGDI can still exert their effects19, 20. Without binding to RHOGDI, a fraction of RHO GTPases continues to be transported to the plasma membrane through vesicle trafficking; this occurs at a reduced rate, but one sufficient to account for their phenotype. We suspect that the mild phenotype of the RHOGDI1-knockout mice also results from sufficient transport of RHO proteins occurring through vesicle trafficking.
Structural analysis of RHOGDI–RHO GTPase
RHOGDI1 has been crystallized in complex with CDC42, RHOA and RAC132-34 (Figure 2), whereas the structure of RHOGDI2 has been solved in complex with RAC235. These structures provide snapshots of the key residues involved in regulating the interactions and have provided the information necessary for mutagenesis studies that characterized the importance of these residues.
Figure 2. Structure of the RHOGDI–RHO GTPase complex.
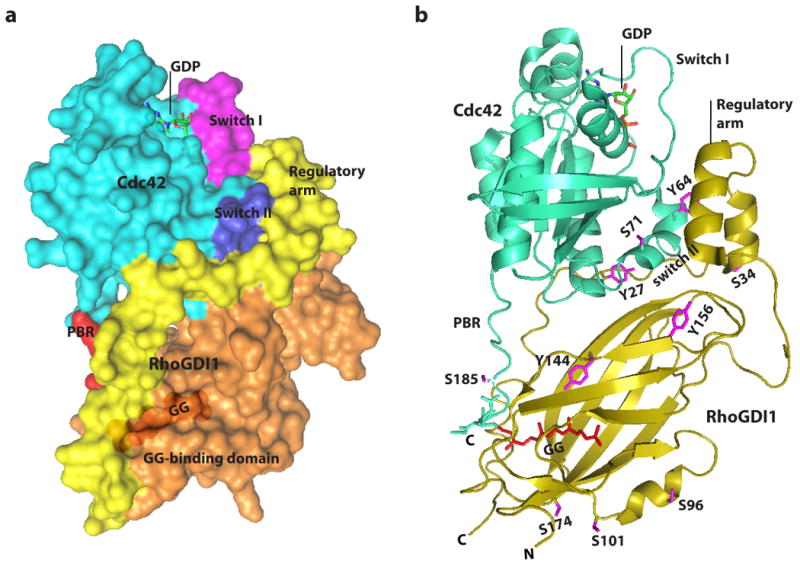
(A) Space filling model showing CDC42 in complex with RHO-specific guanine nucleotide dissociation inhibitor 1 (RHOGDI1)32. The domains that participate in the interaction are highlighted and labelled on the struucture. (B) Cartoon representation of the crystal structure of prenylated CDC42 in complex with RHOGDI 32. The phosphorylated residues are shown in magenta, CDC42 is shown in green, and RHOGDI1 is shown in gold. These structures are reproduced from the NCBI Molecular Modeling Database(ID: 1275). C, carboxyl terminus; GG, geranylgeranyl; N, amino terminus; PBR, Poly Basic Region.
The structure of RHOGDIs comprises two main domains: a C-terminal domain (amino acids74–204) — which includes the geranylgeranyl-binding pocket and is required to extract RHO GTPases from the membrane — and a 'regulatory arm' at the N-terminus (amino acids 5–55) — which inhibits exchange and hydrolysis through interactions with the switch I and switch II domains in the RHO GTPases. The isoprenyl-binding domain adopts an immunoglobulin-like fold, and the surface of the geranylgeranyl-binding pocket is lined with hydrophobic residues 32, 34. Insertion of the isoprenyl moiety perturbs the structure of the RHOGDI, displacing the β-strands βH and βI by 2.0Å, as well as several individual amino acids, to overcome steric hindrances imposed by the isoprenyl moiety. The N-terminal region of RHOGDIs, which is flexible and disordered when in solution, folds into two antiparallel helices upon formation of the complex and interacts with the switch I and switch II domains of the RHO GTPases (Figure 2)32, 34, 35.
It is interesting to note that RHOGDIs can accommodate both GTP- and GDP-bound forms of RHO GTPases 18, 36. Structural studies have shown that the main interaction sites between RHOGDIs and RHO GTPases are virtually unaffected by the nucleotide state 37.
Numerous key residues have been identified in the interface between RHOGDIs and RHO GTPases, including Thr35, Tyr64, Arg66, His103 and His104 (amino acid numbering refers to RAC1 and CDC42), which form hydrogen bonds with RHOGDIs. Thr35, located in the switch I domain, is particularly important for the interaction, as it is conserved in all GTPases and is required for the coordination of Mg2+ (which is essential for stabilizing the nucleotide). The regulatory arm in RHOGDIs interacts with Thr35 in CDC42 by forming a hydrogen bond with Asp45. Arg66, which is located in the switch II region and forms hydrogen bonds with Asp185, Pro30 and Ala31, is another key residue in the RHOGDI -RHO GTPase interaction; substitution of the RHO GTPase Arg66 for Glu (or of Arg68 in RHOA) abolishes the interaction with RHOGDIs 19-21. Interestingly, the RHOGDI- RHO GTPase interaction can be rescued with a charge-reversing substitution in Asp185 in RHOGDI (Asp185Arg) 20.
Based on structural and experimental data, a two-step mechanism has been proposed to explain how RHOGDIs extract RHO GTPases from the membrane 36. First, the N terminus of the RHOGDIs associates with the switch I and switch II domains of the RHOGTPase. This initial binding step is followed by a slower isomerization event that results in the isoprenyl group swapping from the membrane to the hydrophobic pocket 32, 38. The acidic patch in the RHOGDI geranylgeranyl-binding pocket may contribute to the extraction by competing with acidic phospholipids for binding to the polybasic tail of the RHO GTPase. It was recently shown that, in the presence of membranes, RHOGDIs have a much higher affinity for CDC42-GDP (inactive) than for CDC42-GTP (active) 38. By selectively extracting the GDP-bound (inactive) forms of RHO GTPases from membranes, RHOGDI could contribute to the concentration of active GTPases at the membrane.
Regulation of RHOGDI–RHO GTPase interactions
Selective activation of a single RHO GTPase by a signalling pathway requires the release of that RHO family protein from the RHOGDI. But here there is a conundrum because each of the three mammalian RHOGDIs interacts with multiple RHO GTPases. How can a single RHO GTPase be released from the RHOGDI selectively? In addition, our recent findings argue that displacement of a RHO GTPase from RHOGDI has to be coupled to its association with cell membranes (or some other entity) to prevent its degradation 12. Based on similar G proteins, such as members of the RAB family, the existence of a GDI dissociation factor (GDF) has been postulated. By definition, a GDF interacts with RHOGDIs and promotes the dissociation of the RHOGDI-RHO GTPase complex, rendering the RHO GTPase available for activation by GEFs. However, in contrast to the RAB system, a conserved GDF for RHO proteins has not been found. Evidence for a range of mechanisms has been provided, which are discussed below. Some of them regulate the specific displacement of a single RHO GTPase from RHOGDI, whereas others displace all the RHO GTPases equally.
Release by lipids
Early work revealed that RHOGDI–RHO GTPase complexes could be disrupted by acidic lipids 39, and subsequent work showed that phosphoinositide lipids could open up RHOGDIs, thereby facilitating GEF-mediated exchange on bound RHOA 40. A role for lipids in the release of RHO GTPases from RHOGDI was shown during integrin-mediated adhesion 41, 42. In suspended cells RAC1-GTP was found to be retained in the cytosol complexed with RHOGDI; however, integrin-mediated adhesion could mediate the dissociation of RAC1 from the RHOGDI. A lipid raft seemed to be responsible for the recruitment of RAC1 to sites of integrin engagement, and liposomes that were similar in lipid composition to lipid rafts were found to dissociate Rac1-GTP from RHOGDI 41.
Another study using liposomes found that prenylated RAC1 bound to RHOGDI was inaccessible for nucleotide exchange mediated by the DBL homology domain (DH domain) and pleckstrin homology domain (PH domain) of the GEF T lymphoma invasion and metastasis-inducing 1 (TIAM1). However, nucleotide exchange could be observed when liposomes were added, and this corresponded specifically to a fraction of RAC1 dissociating from RHOGDI and binding to the liposomes 43. On the basis of these results, the authors proposed a two-step model for the activation of RHO GTPases: their release from RHOGDIs is promoted by lipids (FIG. 3a), which then leads to their activation by GEFs.
Figure 3. Mechanisms of regulation of the RHOGDI-RHOGTPase interaction.

a | Release by lipids: The presence of acidic phospholipids can promote the release of RHOGTPases from RHO-specific guanine nucleotide dissociation inhibitors (RHOGDIs). Phospholipids mediate a partial opening of the complex that exposes the GTPases to RHO-specific guanine nucleotide exchange factors (RHOGEFs) or other dissociation factors such as the ones described in b. b | Release by protein–protein interactions: p75 neurotrophin receptor (p75NTR) and ezrin, radixin and moesin (ERM) proteins interact with RHOGDIs and facilitate the release of RHOA, which can then be activated by specific RHOGEFs. The interaction of p75NTR with RHOGDIs is enhanced by myelin derived proteins such as myelin-associated glycoprotein (MAG) and neurite outgrowth inhibitor (NOGO). ERM proteins also need to be activated to interact with RHOGDI. c | Release by phosphorylation. Phosphorylation of RHOGDI Ser, Thr and Tyr residues promote the release of RHO GTPases. Depending on the residues phosphorylated, this release can be specific for a single RHO GTPase, or affect multiple RHO GTPases simultaneously (Table 1). For example p21-activated kinase (PAK)- or FER-mediated phosphorylation promote the specific release of RAC1, but not RHOA or CDC42. Some kinases act in concert to target the RAC1–RHODI complex to the membrane and regulate the local release of RAC1 at specific site on the membrane, where it is subsequently activated by RHOGEFs. Diacylglycerol kinase-ζ (DGKζ) forms a complex with PAK, RAC1 and RHOGDI. In response to platelet derived growth factor (PDGF), DGKζ stimulates the production of phosphatidic acid (PA), which induces PAK activity. Active PAK then phosphorylates RHOGDI and releases RAC1 for activation. Positively charged phospholipids are indicated in blue, negatively charged phopholipids in red. DAG, diacylglycerol; PDGFR, PDGF receptor.
To examine this model further, another group measured exchange using liposomes of defined composition with RHOGDI bound to prenylated RAC1 and active DH and PH domains of the GEFs TIAM1 or TRIO. They found that liposomes containing phosphoinositides co-operated with active GEFs to promote the release of RAC1 from the RHOGDI 44. Like the previous group, these investigators favoured a two-step model, although the nature of their experimental conditions could not rule out a one-step model in which the GEF activity promoted dissociation of the RHOGDI–RAC1 complex.
Interestingly, in these experiments it was observed that if GTP was replaced with GDP, RAC1 did not dissociate from the RHOGDI and did not become associated with the liposomes44. This is consistent with the finding that, in the presence of membranes, RHOGDI has a lower affinity for GTP-bound RHO GTPases than GDP-bound ones 38. Nevertheless, exchange did occur in the RHOGDI complex, indicating somewhat unexpectedly that the GEFs used in this experiment (TIAM and TRIO) could act on RAC1 complexed with RHOGDI. This observation suggests a one-step mechanism of RHO GTPase activation, but one in which the bound nucleotide (GTP versus GDP) is crucial for RHO GTPase release.
Release by GEFs
RHOGEFs have always been considered natural candidates for the role of GDFs in the RHO GTPase system. They act directly downstream of the dissociation event, and because they discriminate between individual RHO proteins and are targets of the signalling pathways that activate RHO GTPases, they could provide specificity to the reaction. However, according to structural analysis, both RHOGEFs and RHOGDIs interact with the switch I and switch II domains of RHO GTPases, which suggests that the two interactions may be mutually exclusive32, 45.
Although findings indicate that GEFs can function as GDFs44, in particular in the RAB GTPase subfamily 46, 47, there is little evidence suggesting that GEFs catalyze displacement directly. Furthermore, even though a role for GEFs has been proposed in the two-step mechanism of RHO GTPase activation (see above), the two steps are mechanistically distinct 43; more work is needed to establish whether RHOGEFs have direct roles as GDFs or whether their actions are confined to the second step, activating the RHO protein that has been released from RHOGDI into the membrane.
Release by specific protein interactions
Another possibility is that specificity in the dissociation of the RHOGDI–RHO GTPase complex is afforded by distinct protein–protein interactions. For example, ezrin, radixin, and moesin (ERM) proteins, p75 neurotrophin receptor (p75NTR; also known as NGFR) and the Tyr kinase ETK have all been reported to promote the displacement of RHO GTPases from RHOGDI 48-51(FIG.3b). The N terminus of ERM proteins associates with RHOGDIs and can compete for binding to all RHO GTPases tested, thereby inhibiting RHOGDI function and promoting the activation of RHO proteins 50, 52. However, in vivo data suggest that ERM proteins may regulate the preferential release of RHOA50, 53. Interestingly, radixin also interacts with the GEF DBL, suggesting it may couple release of the GTPase from RHOGDI with activation52.
p75 NTR is a neurotrophin receptor that functions in the regulation of axonal elongation by neurotrophins and by several myelin-derived proteins. Neurotrophins stimulate neurite outgrowth by inhibiting RHOA activity, whereas myelin-derived proteins activate RHOA and inhibit axonal elongation. The ability of p75NTR to modulate RHOA activity depends on its ability to interact with RHOGDI to promote RHOA release and subsequent activation 51. Myelin-derived proteins, such as neurite outgrowth inhibitor (NOGO) or myelin-associated glycoprotein (MAG), enhance the association of p75 NTR with RHOGDIs, which suggests that they activate RHOA by inducing its release from the RHOGDI (Figure 3)51.
ETK is a non-receptor Tyr kinase that belongs to the Bruton Tyr kinase (BTK) family and has been shown to have a role in the regulation of various cellular processes, including cytoskeletal reorganization and cell motility 54. ETK promotes the specific displacement of RHOA (but not RAC1 and CDC42) from RHOGDI1 in a kinase-independent manner 48. ETK interacts with RHOA through an N-terminal PH domain and competes for its binding to RHOGDIs, promoting its release and subsequent activation 48.
Release by phosphorylation
In recent years, phosphorylation has emerged as one of the key post-translational modifications that can selectively alter the affinities of the different RHO GTPases for RHOGDIs for one another. In general terms, phosphorylation of RHOGDIs decreases their affinity for RHO GTPases, promoting RHO GTPase release and making them available for activation. By contrast, phosphorylation of RHO GTPases increases their affinity for RHOGDIs, promoting their sequestration and inactivation in the cytosol (Table 1).
Table 1. Post-translational modifications that modulate the RHOGDI–RHO GTPase interaction.
| Phosphorylation site | Kinase | GDI or GTPase | Effect | Refs |
|---|---|---|---|---|
| RhoGDIs*‡ | ||||
| Ser 34 | PKCα | GDI1 | Promotes dissociation of RhoA | 56,89§ |
| Ser 96 | PKCα | GDI1 | Promotes dissociation of RhoA and RhoG | 58,90 |
| Ser 101 | PAK1 | GDI1 | Promotes dissociation of Rac1 (and to a lesser extent Cdc42) | 55 |
| Ser 174 | PAK1 | GDI1 | Promotes dissociation of Rac1 (and to a lesser extent Cdc42) | 55 |
| Ser 174 | PKA | GDI1 | Inhibits interaction with RhoA | 91 |
| Ser 148 | ND | GDI1 | ND | 92 |
| Thr ? | PKCζ | GDI1 | Promotes dissociation of RhoA, Rac1 and Cdc42 | 93 |
| Tyr 27‖ | Src | GDI1/2 | Promotes dissociation of RhoA, Rac1 and Cdc42 | 94,95 |
| Tyr 156‖ | Src | GDI1/2 | Promotes dissociation of RhoA, Rac1 and Cdc42 | 94,95 |
| Tyr 156?¶ | Fer | GDI1 | Promotes dissociation of Rac1 | 96 |
| RhoGTPases | ||||
| Ser188 | PKA/A/PKG/SLK | RhoA | Increased association with GDI | 97-102 |
| Ser187 | PKA | RhoG | Increased association with GDI | 101 |
| Ser185 | PKA | Cdc42 | Increased association with GDI | 103 |
| Tyr64 | Src | Cdc42 | Increased association with GDI | 104 |
| Ser71 | Akt | Rac1, Cdc42 | Increased association with GDI | 105,106 |
ND, not determined; PAK1, p21-activated kinase 1; PK, protein kinase; RHOGDI, RHO-specific guanine nucleotide dissociation inhibitor; SLK, STE20-like kinase.
Amino acid numbering corresponds to the sequence of human RHOGDI1.
Indicates RHOGDI1 residues; Tyr27, Ser34, Ser148 and Tyr156 are conserved in RHOGDI2 and RHOGDI3, whereas RHOGDI1 Ser101 and Ser174 are Thr in RHOGDI2, and Ser96 is present only in RHOGDI1).
This article gave the first description of RHOGDI phosphorylation by PKCα. However, the phosphorylated residue was not identified.
The equivalent Tyr residues in RHOGDI2 (Tyr24, Tyr153) are also phosphorylated by SRC95. Mainly Tyr156 is phosphorylated, with Tyr27 being only weakly phosphorylated.
The phosphorylated residue is in the carboxy terminal half of RHOGDI1, where Tyr156 is located.
This suggests that, at least to a certain extent, some kinases can function as bona fide GDFs. For example, phosphorylation of RHOGDI1 on Ser101 and Ser174 by p-21-activated kinase (PAK1) reduces its affinity for RAC1 but not RHOA, promoting the release of RAC1 and its subsequent activation 55. Similarly, phosphorylation of RHOGDI1 on Ser34 by protein kinase Cα (PKCα) selectively releases RHOA (but not RAC1or CDC42) and promotes its activation 56. Ser101 and Ser174 are located in the hydrophobic binding cleft of RHOGDI on its solvent-exposed surface, suggesting their phosphorylation could perturb binding of the isoprenyl moiety of the RHO GTPase32 (FIG. 2). However, as the phosphorylation-mediated release is specific for RAC1, it is possible that interactions with the polybasic region, the most divergent sequence between RHO GTPases, may also be affected by the negative charge afforded by the phosphorylation and contribute to release. Ser34 is located in the N-terminal regulatory arm of RHOGDIs, which interacts primarily with the switch II domain of RHO GTPases 32. This region folds into a helix-loop-helix motif that is maintained mainly by hydrophobic interactions, so it is possible that the negative charge from the phosphate group interferes with the RHO GTPase interaction (FIG. 2). In addition, Ser34 is surrounded by many key residues in the N terminus of the RHOGDI, which interact with Arg66 (in CDC42 and RAC1), a residue in RHO GTPases that is essential for the interaction with RHOGDIs (and correlates with Arg68 in RHOA, RHOB and RHOC) 19-21. It is therefore possible that phosphorylation of Ser34 disrupts or interferes with the interactions surrounding Arg66, triggering displacement. However, Arg66 is conserved in most RHO GTPases, so it is not clear how Ser34 affects specifically RHOA but not RAC1 and CDC42 release.
It is tempting to speculate that a RHOGDI phosphorylation code (similar to the histone modification code) controls which RHO GTPase is to be released from the complex in response to a stimulus. This attractive idea, which has been previously postulated 1, would solve the requirement for a GDF in an elegant way. However, this hypothesis is hard to justify based on the sequence and structural data. The residues that are phosphorylated in RHOGDIs are either concentrated around the geranylgeranyl-binding pocket or in the regulatory arm region that interacts primarily with the switch II region in the RHO GTPase (FIG. 2 and Table 1), so interfering with binding to the prenylated region of RHO GTPases should affect all RHO proteins the same way. The same can be argued for the switch II region, which is identical in the major RHO GTPases (RHOA, RAC1 and CDC42).
An alternative hypothesis is that selective release of a particular RHO GTPase from the RHOGDI is achieved by the recruitment of the complex to a specific cellular location, where the RHOGDI is phosphorylated. This could be mediated by scaffolding proteins that, in response to a signal, can recruit a particular RHOGDI– RHO GTPase complex to a site of stimulation. A specific kinase would then phosphorylate the RHOGDI, triggering the release of the RHO GTPase and allowing its subsequent activation by a RHOGEF. Consistent with this, a recent report showed that, upon platelet-derived growth factor treatment, diacylglycerol kinase-ζ (DGKζ) forms a complex with RAC1 and RHOGDI and promotes the release of RAC1. DGKζ stimulates the production of phosphatidic acid, which induces PAK activity. PAK subsequently phosphorylates RHOGDI, releasing RAC1 for activation57 (FIG. 3c). In DGKζ-deficient fibroblasts PAK1 phosphorylation and RHOGDI–RAC1 dissociation were attenuated and PAK failed to target properly to focal adhesions.
Similarly, following stimulation by HGF, DGKα was found to promote the formation of phosphatidic acid, which in this case recruits atypical PKCζ (aPKCζ)– aPKCι, in complex with RHOGDI and RAC1. The activation of aPKCζ– aPKCι mediates the release of RAC1 from the RHOGDI, allowing its activation 27.
Another example involves the transmembrane receptor syndecan4, a heparan sulfate proteoglycan. Syndecan4 and its adaptor, synectin (which binds to the intracellular tail of syndecan 4), form a complex with RHOGDI58, an interaction that increases RHOGDI's affinity for RHOG. Binding of syndecan4 to its ligand, fibroblast growth factor 2 (FGF2), promotes the activation of PKCα, which in turn phosphorylates RHOGDI. This mediates the release of RHOG (but not of RAC1), leading to its activation and further downstream effects59, 60.
RHOGTPase crosstalk through RHOGDIs
Most RHO GTPases bind to at least one of the three RHOGDIs that are expressed in mammals 1. In addition, it has been previously shown that the total amount of the most abundant RHOGDI is roughly equivalent to the sum of the levels of the three major RHOGTPases: RAC1, CDC42 and RHOA 61. We have recently shown that RHO proteins can only exist in complex with RHOGDI or associated with cellular membranes 12. These results imply that the total levels of the major RHO proteins are determined by the amount of RHOGDIs in the cell. The limited amount of RHOGDIs generates competitive pressure between the different GTPases to bind to RHOGDI to avoid degradation (FIG. 1). At steady state, the RHO GTPases must reach an equilibrium between their ability to bind RHOGDI, the competition from the other RHO GTPases and their turnover by degradation. This previously unnoticed crosstalk between RHO GTPases through RHOGDI predicts that any change in the level of a particular RHO GTPase, or in the affinity of a particular RHO GTPase for RHOGDI, whether physiologically or experimentally induced, should interfere with this equilibrium.
We have recently shown that the exogenous overexpression of a single RHO GTPase competes with endogenous RHO proteins and displaces them from RHOGDI in a dose-dependent manner, triggering their subsequent degradation 12. In addition, the remaining membrane-bound RHO proteins are eventually extracted by RHOGDIs, displaced by the overexpressed protein and degraded. The result is the virtual silencing of both the protein levels and activity of all endogenous RHO proteins associated with RHOGDIs.
In spite of this result, there is evidence that in some situations the released RHO proteins may become activated. For example, a recent study observed that increased PKG-mediated phosphorylation of Ser188 in RHOA in smooth muscle cells increased the binding of RHOA to RHOGDI and displaced RAC162. However, the released RAC1 became activated, leading to cell migration. A similar situation was observed in PtK1 epithelial cells, in which PKA activity was observed to be synchronized with protrusion at the leading edge63. PKA-mediated phosphorylation of RHOA on Ser188 increased its affinity for RHOGDI and reduced its activity. It will be interesting to learn whether a similar displacement and activation of RAC1 is involved in this situation too.
This could have a significant role when the levels of a particular RHO GTPase or RHOGDI are affected, as occurs in many cancer types (see below), by indirectly affecting the homeostasis of the RHO GTPase system as a whole, magnifying the effects of altered expression levels.
Biological roles of RHOGDIs
The only known functions of RHOGDIs are in the context of their interactions with and regulation of RHO GTPases (for reviews on RHO GTPase functions, see Refs 64, 65). Briefly, some of the best-characterized effects of RHO GTPases are on the cytoskeleton. For example, RHOA, RAC1 and CDC42 can all stimulate actin polymerization by interacting with different effectors. The resulting phenotypes are typically, although not always, distinct: RAC1 promotes cell spreading, inducing the formation of lamellae at the cell periphery; CDC42 induces the formation of filopodia; and RHOA induces the formation of filamentous actin bundles, such as stress fibres, although RHOA-induced lamellae have also been described in some cell types66. These three ubiquitously expressed RHO family members exert other effects on the cytoskeleton and coordinately regulate many types of cell migration. RHO GTPases also play important parts in cell adhesion (both to other cells and to the matrix), cell polarity, vesicle trafficking, cell cycle progression, gene expression and differentiation. Indeed, there seem few cellular activities in which RHO proteins do not have some regulatory role64, 65.
When RHOGDI1 has been investigated by knockdown of protein expression or gene knockout, the overall levels of all RHO GTPases with which it interacts decrease. There was a loss of detectable cytosolic RHO proteins, but the remaining membrane-bound fraction of each RHO protein is highly activated 12. A similar situation was seen in mammalian cells, when RHOGDI1 was knocked down, and in yeast when the single RHOGDI was deleted12, 22, 24. Given these results, a prominent phenotype might be expected; however, surprisingly, there is only a mild phenotype in yeast and mice22, 24. In mice, the loss of RHOGDI1 does not affect embryonic development, but the adult mice develop progressive renal defects, that ultimately lead to death and are attributed to increased RAC1 activity23. In addition, the male mice are infertile, with impaired spermatogenesis, and there are problems with implantation of RHOGDI1-null embryos in female mice 24. One possible explanation for the mild phenotype is that RHOGDI2 compensated for the loss of RHOGDI1. However, the double knockout of RHOGDI1 and RHOGDI2 produces a phenotype that is only slightly more severe than the single RHOGDI1 knockout, with additional immunological defects superimposed on those induced by the loss of RHOGDI1 67.
Although mice lacking RHOGDIs are viable and develop normally, some effects have been observed when the behaviour of individual cells has been examined. For example, knockdown of RHOGDI1 in bladder cancer cells increased their migration, but knockdown of RHOGDI2 had no effect68. By contrast, silencing RHOGDI1 in a rapidly migrating melanoma cell line inhibited cell migration 12. At first sight, these contradictory results seem difficult to reconcile. One potential explanation relates to the two modes of cycling RHO proteins to the plasma membrane (see above). In the absence of RHOGDIs, the slow delivery of RHO proteins to the plasma membrane by vesicle trafficking may be sufficient to satisfy the requirements of slowly migrating cells but inadequate for those migrating rapidly. The fact that slow cells actually increase their speed of migration is consistent with the remaining fraction of RHO proteins (particularly RAC1) being highly activated when RHOGDIs are depleted. So, although the amount reaching the plasma membrane is greatly decreased, those that do get to this site are activated and stay at the membrane for longer, thus stimulating migration. By contrast, fast-migrating cells may need to recycle RAC1 or other RHO proteins to newly formed protrusions at a speed that exceeds that of vesicle trafficking.
Other phenotypic changes have also been observed in cells depleted of RHOGDI1. For example, mesangial cells from the knockout mice revealed decreased rates of cell spreading 69. When vascular permeability was studied in RHOGDI1-knockout mice, an increase in the basal permeability of the pulmonary vascular endothelium was observed70. This correlated with increased activity of RHOA in these cells and opening of the interendothelial junctions.
RHOGDIs and cancer
Changes in RHOGDI1 and RHOGDI2 expression levels have been associated with many cancers71. However, the changes vary depending on the tumour type. For instance, RHOGDI1 expression is upregulated in colorectal and ovarian cancer, and in this case high expression levels correlates with increased invasion and resistance to chemotherapy72-74. By contrast, RHOGDI1 expression is reduced in brain cancers and correlates with reduced expression of RHOA and RHOB but not RAC1. The expression levels of the three proteins are inversely correlated with the degree of malignancy75. In breast cancers, conflicting results have been reported, with RHOGDI1 expression being increased or decreased in different studies76, 77. Finally, RHOGDI1 is downregulated in hepatocellular carcinoma, and its expression was found to be controlled in part by a recently characterized microRNA, miR-151, which is localized in an intron of the gene encoding focal adhesion kinase (FAK) and is expressed with it78. During hepatocellular tumour progression, FAK expression increases together with miR-151, promoting the downregulation of RHOGDI1 and facilitating tumour invasion and metastasis78.
The expression levels of RHOGDI2 are also severely altered in a range of cancers79, 80. RHOGDI2 expression is increased in pancreatic cancers and this correlates with increased invasiveness 80, 81. The opposite occurs in bladder cancers, in which RHOGDI2 is downregulated in invasive tumour cells 68, 82, and this decrease correlates with decreased patient survival 83. Similarly, RHOGDI2 is lost in Hodgkin's lymphoma cell lines, although here there is no clear correlation with growth or survival 84. In breast tumours the pattern of expression of RHOGDI2 is complex. There is evidence that RHOGDI2 expression is biphasic, increasing at the early stages of progression but decreasing sharply during metastasis85. RHOGDI has also been linked to oestrogen receptor expression and was identified as part of a predictive gene signature for poor prognosis for tumours that express oestrogen receptor 86.
The effects of the RHOGDIs on cancers are clearly complex and do not fit a simple explanatory model. It has to be remembered that the consequences of changes in their expression are manifested through their actions on multiple RHO GTPases, and that the levels and activities of these vary significantly in different cell types and different cancers. Even a single RHO family member can have opposite effects in different tumour types. This is well illustrated by work on the RAC-specific GEF TIAM1: although it was originally discovered in a search for genes that promoted T cell invasion and metastasis 87, in epithelial cells it has the opposite effect, delaying migration but enhancing stable cell–cell junctions88. In both cases, the effects were due to the TIAM1-mediated activation of RAC1, which, actually, has different effects in the two cell types. As the interaction of RHOGDI with RHO GTPases is also governed by phosphorylation on multiple sites, the activities of specific kinases will likely affect how RHOGDI affects the phenotype of particular cancer cells.
Conclusion
In this Review we have discussed some of the emerging ideas about RHOGDIs, how they act as chaperones for RHO GTPases, regulating RHO protein stability, and how RHOGDIs shuttle the GTPases to membranes for activation and interaction with their downstream targets.
Understanding the factors that control the release of RHO proteins from RHOGDIs is one of the unresolved but exciting areas in the field. We anticipate that answers to the questions in this area will come from continued biochemical analysis of this process, using reconstituted in vitro model systems, as well as from imaging techniques that allow the visualization of RHOGDI complexes in living cells.
Another topic of considerable interest relates to the crosstalk between RHO GTPases that occurs through their competitive binding to RHOGDIs. The recent work showing that phosphorylation of RHOA by PKA increases its binding affinity to RHOGDI1 and results in displacement and activation of RAC162, illustrates a level of interaction between RHO proteins that had not been anticipated. The discovery of other examples of crosstalk mediated by RHOGDIs will strengthen the idea that they contribute actively to RHO protein dynamics and signalling and are not simply passive inhibitors of the RHO GTPase cycle.
Finally, little is known about how the expression of RHOGDI1 or RHOGDI2 is regulated. The fact that the level of RHOGDI1 seems to be related to the sum of the expression levels of the major RHO proteins suggests that there are feedback mechanisms coordinating the expression of both the RHO GTPases and RHOGDIs. The discovery that miR-151 regulates RHOGDI1 expression opens a new window on this problem78. We suspect that other factors influencing the expression of the RHOGDIs remain to be discovered and that many of these may be altered in cancer cells. RHOGDIs have been suggested as potential targets for cancer therapy71, but if this direction is pursued it will be crucial to learn more about the factors regulating RHOGDI expression particularly in tumour cells.
Acknowledgments
RGM gratefully acknowledges support from the Simmons Scholars Program, EB thanks Fellowship from the city of Nice (to E.B.) and an Allocation INSERM InCa/AVENIR (#R08227AS.). KB gratefully acknowledges the Kenan Foundation for support and NIH grants GM029860 and HL080166.
Glossary
- Prenylated
Modified by the attachment of an isoprenoid to a C terminal cysteine residue.
- Isoprenoid
Compounds that are derived from isoprene (2-methyl-1,3-butadiene) linked together in head-to-tail or tail-to-tail conformations and that comprise farnesyl and geranylgeranyl molecules that are used in the covalent modification of proteins on cysteine residues.
- Isoprenylation
A post-translational modification of proteins by the attachment of an isoprenoid to a C-terminal Cys. The isoprenoids used, farnesyl diphosphate or geranylgeranyl diphosphate, are derived from the same biochemical pathway that produces cholesterol.
- geranyl-geranyl
A 20-carbon isoprenoid precursor to geranyl-geranylated proteins. The source of the geranyl-geranyl group is geranyl-geranyl pyrophosphate, an intermediate in the HMG-CoA reductase pathway used by organisms in the biosynthesis of terpenes and terpenoids.
- switch I and II
Two regions in RHOGTPases that undergo a conformational change in the active GTP-bound form and provide a platform for the selective interaction with downstream effectors.
- immunoglobulin-like fold
A type of protein domain that consists of a 2-layer sandwich of between 7 and 9 antiparallel β-strands arranged in two β-sheets.
- steric hindrance
occurs when the size of groups within a molecule prevents chemical reactions that are observed in related smaller molecules.
- Isomerization
The process by which one molecule is transformed into another molecule that has exactly the same molecular formula, but with a different structure.
- DH domain (Dbl homology)
A domain of about 200 amino acids that has been shown to encode GEF activity specific many RHO family members.
- PH domains (Pleckstrin homology)
A protein domain of approximately 120 amino acids that occurs in a wide range of proteins involved in intracellular signaling. PH domains frequently bind to cell membranes by interacting with acidic phospholipids, such as phosphoinositides.
- Biphasic
Having two distinct phases.
- Lipid raft
Membrane microdomains that are enriched in cholesterol, sphingolipids and lipid-modified proteins such as GPI-linked proteins and palmitoylated proteins. These microdomains often function as platforms for signalling events.
Biographies
RGM Biography: Rafael Garcia-Mata obtained his PhD from the University of Alabama at Birmigham in 2002, and after postdoctoral research at the University of North Carolina at Chapel Hill joined the faculty at the University of North Carolina where he is now Assistant Professor and Simmons Scholar. The research in his lab is centered in understanding the mechanisms of regulation of RHO family of small GTPases.
RGM Lab webpage: http://rgmlab.webnode.com/
EB Biography: Etienne Boulter obtained his Ph.D. in Cell Biology from the University of Nice Sophia-Antipolis, Nice, France, and followed with a postdoctoral training at The University of North Carolina at Chapel Hill. He is currently is a Postdoctoral Fellow at the University of Nice Sophia-Antipolis, Nice, France, where he studies the regulation of Rho proteins by CD98hc in the epidermis.
KB's biography: Keith Burridge obtained his Ph.D. from Cambridge University and after postdoctoral research at Cold Spring Harbor Laboratory joined in 1981the faculty at the University of North Carolina, Chapel Hill, where he is currently Kenan Distinguished Professor of Cell and Developmental Biology. He has a longstanding interest in cell adhesion and the cytoskeleton, and has worked for many years on various aspects of RHO GTPase signaling.
KB Lab webpage: http://www.med.unc.edu/cellbio/faculty-research/burridge
References
- 1.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–63. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Fukumoto Y, et al. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene. 1990;5:1321–8. [PubMed] [Google Scholar]
- 3.Leonard D, et al. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J Biol Chem. 1992;267:22860–8. [PubMed] [Google Scholar]
- 4.Lelias JM, et al. cDNA cloning of a human mRNA preferentially expressed in hematopoietic cells and with homology to a GDP-dissociation inhibitor for the rho GTP-binding proteins. Proc Natl Acad Sci U S A. 1993;90:1479–83. doi: 10.1073/pnas.90.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherle P, Behrens T, Staudt LM. Ly-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proc Natl Acad Sci U S A. 1993;90:7568–72. doi: 10.1073/pnas.90.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platko JV, et al. A single residue can modify target-binding affinity and activity of the functional domain of the Rho-subfamily GDP dissociation inhibitors. Proc Natl Acad Sci U S A. 1995;92:2974–8. doi: 10.1073/pnas.92.7.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorvel JP, Chang TC, Boretto J, Azuma T, Chavrier P. Differential properties of D4/LyGDI versus RhoGDI: phosphorylation and rho GTPase selectivity. FEBS Lett. 1998;422:269–73. doi: 10.1016/s0014-5793(98)00020-9. [DOI] [PubMed] [Google Scholar]
- 8.Brunet N, Morin A, Olofsson B. RhoGDI-3 regulates RhoG and targets this protein to the Golgi complex through its unique N-terminal domain. Traffic. 2002;3:342–57. doi: 10.1034/j.1600-0854.2002.30504.x. [DOI] [PubMed] [Google Scholar]
- 9.Zalcman G, et al. RhoGDI-3 Is a New GDP Dissociation Inhibitor (GDI). IDENTIFICATION OF A NON-CYTOSOLIC GDI PROTEIN INTERACTING WITH THE SMALL GTP-BINDING PROTEINS RhoB AND RhoG. J Biol Chem. 1996;271:30366–30374. doi: 10.1074/jbc.271.48.30366. [DOI] [PubMed] [Google Scholar]
- 10.Adra CN, et al. RhoGDIgamma: a GDP-dissociation inhibitor for Rho proteins with preferential expression in brain and pancreas. Proc Natl Acad Sci U S A. 1997;94:4279–84. doi: 10.1073/pnas.94.9.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith A. The theory of moral sentiments. A. Millar; London: 1759. [Google Scholar]
- 12.Boulter E, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–83. doi: 10.1038/ncb2049. This paper shows that not only does RHOGDI1 act as a chaperone, protecting multiple Rho proteins from proteolytic degradation, but that RHO proteins compete for binding to RHOGDI1, such that over-expression of one leads to the displacement and degradation of the others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ueda T, Kikuchi A, Ohga N, Yamamoto J, Takai Y. Purification and characterization from bovine brain cytosol of a novel regulatory protein inhibiting the dissociation of GDP from and the subsequent binding of GTP to rhoB p20, a ras p21-like GTP-binding protein. J Biol Chem. 1990;265:9373–80. [PubMed] [Google Scholar]
- 14.Hart MJ, et al. A GDP dissociation inhibitor that serves as a GTPase inhibitor for the Ras-like protein CDC42Hs. Science. 1992;258:812–5. doi: 10.1126/science.1439791. [DOI] [PubMed] [Google Scholar]
- 15.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–85. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox AD, Der CJ. Protein prenylation: more than just glue? ; Curr Opin Cell Biol. 1992;4:1008–16. doi: 10.1016/0955-0674(92)90133-w. [DOI] [PubMed] [Google Scholar]
- 17.Rolli-Derkinderen M, et al. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–60. doi: 10.1161/01.RES.0000170084.88780.ea. [DOI] [PubMed] [Google Scholar]
- 18.Hancock JF, Hall A. A novel role for RhoGDI as an inhibitor of GAP proteins. Embo J. 1993;12:1915–21. doi: 10.1002/j.1460-2075.1993.tb05840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gandhi PN, et al. An activating mutant of Rac1 that fails to interact with Rho GDP-dissociation inhibitor stimulates membrane ruffling in mammalian cells. Biochem J. 2004;378:409–19. doi: 10.1042/BJ20030979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson RM, et al. An activating mutant of Cdc42 that fails to interact with Rho GDP-dissociation inhibitor localizes to the plasma membrane and mediates actin reorganization. Experimental Cell Research. 2004;301:211–22. doi: 10.1016/j.yexcr.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 21.Gibson RM, Wilson-Delfosse AL. RhoGDI-binding-defective mutant of Cdc42Hs targets to membranes and activates filopodia formation but does not cycle with the cytosol of mammalian cells. Biochem J. 2001;359:285–94. doi: 10.1042/0264-6021:3590285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiedje C, Sakwa I, Just U, Hofken T. The Rho GDI Rdi1 regulates Rho GTPases by distinct mechanisms. Mol Biol Cell. 2008;19:2885–96. doi: 10.1091/mbc.E07-11-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shibata S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–6. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 24.Togawa A, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–80. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 25.Lin Q, Fuji RN, Yang W, Cerione RA. RhoGDI is required for Cdc42-mediated cellular transformation. Curr Biol. 2003;13:1469–79. doi: 10.1016/s0960-9822(03)00613-4. [DOI] [PubMed] [Google Scholar]
- 26.Lin R, Bagrodia S, Cerione R, Manor D. A novel Cdc42Hs mutant induces cellular transformation. Curr Biol. 1997;7:794–7. doi: 10.1016/s0960-9822(06)00338-1. [DOI] [PubMed] [Google Scholar]
- 27.Chianale F, et al. Diacylglycerol kinase alpha mediates HGF-induced Rac activation and membrane ruffling by regulating atypical PKC and RhoGDI. Proc Natl Acad Sci U S A. 2010;107:4182–7. doi: 10.1073/pnas.0908326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nevins AK, Thurmond DC. Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J Biol Chem. 2006;281:18961–72. doi: 10.1074/jbc.M603604200. [DOI] [PubMed] [Google Scholar]
- 29.Kowluru A, et al. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J Clin Invest. 1996;98:540–55. doi: 10.1172/JCI118822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slaughter BD, Das A, Schwartz JW, Rubinstein B, Li R. Dual modes of cdc42 recycling fine-tune polarized morphogenesis. Dev Cell. 2009;17:823–35. doi: 10.1016/j.devcel.2009.10.022. Proposes a dual recycling mechanism for Cdc42 in yeast. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruyne D, Bretscher A. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. Journal of cell science. 2000;113(Pt 3):365–75. doi: 10.1242/jcs.113.3.365. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman GR, Nassar N, Cerione RA. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–56. doi: 10.1016/s0092-8674(00)80670-4. Solved the crystal structure of prenylated Cdc42 in complex with RHOGDI1. [DOI] [PubMed] [Google Scholar]
- 33.Longenecker K, et al. How RhoGDI binds Rho. Acta Crystallogr D Biol Crystallogr. 1999;55:1503–15. doi: 10.1107/s090744499900801x. [DOI] [PubMed] [Google Scholar]
- 34.Grizot S, et al. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry. 2001;40:10007–13. doi: 10.1021/bi010288k. [DOI] [PubMed] [Google Scholar]
- 35.Scheffzek K, Stephan I, Jensen ON, Illenberger D, Gierschik P. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat Struct Biol. 2000;7:122–6. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- 36.Nomanbhoy TK, Cerione R. Characterization of the interaction between RhoGDI and Cdc42Hs using fluorescence spectroscopy. J Biol Chem. 1996;271:10004–9. doi: 10.1074/jbc.271.17.10004. [DOI] [PubMed] [Google Scholar]
- 37.Phillips MJ, Calero G, Chan B, Ramachandran S, Cerione RA. Effector proteins exert an important influence on the signaling-active state of the small GTPase Cdc42. J Biol Chem. 2008;283:14153–64. doi: 10.1074/jbc.M706271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson JL, Erickson JW, Cerione RA. New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J Biol Chem. 2009;284:23860–71. doi: 10.1074/jbc.M109.031815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chuang TH, Bohl BP, Bokoch GM. Biologically active lipids are regulators of Rac.GDI complexation. J Biol Chem. 1993;268:26206–11. [PubMed] [Google Scholar]
- 40.Faure J, Vignais PV, Dagher MC. Phosphoinositide-dependent activation of Rho A involves partial opening of the RhoA/Rho-GDI complex. Eur J Biochem. 1999;262:879–89. doi: 10.1046/j.1432-1327.1999.00458.x. [DOI] [PubMed] [Google Scholar]
- 41.del Pozo MA, et al. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–42. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 42.Del Pozo MA, et al. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 2002;4:232–9. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- 43.Robbe K, Otto-Bruc A, Chardin P, Antonny B. Dissociation of GDP dissociation inhibitor and membrane translocation are required for efficient activation of Rac by the Dbl homology-pleckstrin homology region of Tiam. J Biol Chem. 2003;278:4756–62. doi: 10.1074/jbc.M210412200. Evidence is provided for a two-step model of RHO protein activation, in which release from RHOGDI is promoted by lipids, leading to membrane translocation and subsequent GEF-mediated nucleotide exchange. [DOI] [PubMed] [Google Scholar]
- 44.Ugolev Y, Berdichevsky Y, Weinbaum C, Pick E. Dissociation of Rac1(GDP).RhoGDI complexes by the cooperative action of anionic liposomes containing phosphatidylinositol 3,4,5-trisphosphate, Rac guanine nucleotide exchange factor, and GTP. J Biol Chem. 2008;283:22257–71. doi: 10.1074/jbc.M800734200. Liposomes containing phosphoinositides were found to co-operate with active GEFs to release and activate RAC1 from RHOGDI1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408:682–8. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]
- 46.Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science. 2007;318:974–7. doi: 10.1126/science.1149121. [DOI] [PubMed] [Google Scholar]
- 47.Schoebel S, Oesterlin LK, Blankenfeldt W, Goody RS, Itzen A. RabGDI displacement by DrrA from Legionella is a consequence of its guanine nucleotide exchange activity. Molecular cell. 2009;36:1060–72. doi: 10.1016/j.molcel.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 48.Kim O, Yang J, Qiu Y. Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J Biol Chem. 2002;277:30066–71. doi: 10.1074/jbc.M201713200. [DOI] [PubMed] [Google Scholar]
- 49.Maeda M, Matsui T, Imamura M, Tsukita S. Expression level, subcellular distribution and rho-GDI binding affinity of merlin in comparison with Ezrin/Radixin/Moesin proteins. Oncogene. 1999;18:4788–97. doi: 10.1038/sj.onc.1202871. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi K, et al. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J Biol Chem. 1997;272:23371–5. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nature neuroscience. 2003;6:461–7. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K, et al. Interaction of radixin with Rho small G protein GDP/GTP exchange protein Dbl. Oncogene. 1998;16:3279–84. doi: 10.1038/sj.onc.1201874. [DOI] [PubMed] [Google Scholar]
- 53.Hirao M, et al. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19:5651–61. doi: 10.1038/sj.onc.1203958. [DOI] [PubMed] [Google Scholar]
- 55.DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell. 2004;15:117–27. doi: 10.1016/j.molcel.2004.05.019. RHOGDI1 was shown to be a substrate for PAK1 and phosphorylation by PAK1 resulted in the selective release of RAC1 from RHOGDI1. [DOI] [PubMed] [Google Scholar]
- 56.Dovas A, et al. Serine34 phosphorylation of RHO guanine dissociation inhibitor (RHOGDI{alpha}) links signaling from conventional protein kinase C to RHO GTPase in cell adhesion. J Biol Chem. 2010;285:23296–23308. doi: 10.1074/jbc.M109.098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abramovici H, et al. Diacylglycerol kinase zeta regulates actin cytoskeleton reorganization through dissociation of Rac1 from RhoGDI. Mol Biol Cell. 2009;20:2049–59. doi: 10.1091/mbc.E07-12-1248. Shows that the PA produced by DGKzeta initiates RHOGDI release and RAC1 activation through PAK1-mediated phosphorylation of RHOGDI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elfenbein A, et al. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J Cell Biol. 2009;186:75–83. doi: 10.1083/jcb.200810179. The authors show that not only is RHOG affinity for RHOGDI1 increased when RHOGDI forms a complex with Syndecan4 and its binding partner synectin, but also that in response to FGF2 binding to Syndecan4, RHOG is released and activated as a result of phosphorylation by PKCα. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brugnera E, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–82. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 60.Gumienny TL, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 61.Michaelson D, et al. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–26. doi: 10.1083/jcb.152.1.111. A seminal paper analyzing the factors that affect the subcellular distribution of RHO GTPases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rolli-Derkinderen M, Toumaniantz G, Pacaud P, Loirand G. RhoA phosphorylation induces Rac1 release from guanine dissociation inhibitor alpha and stimulation of vascular smooth muscle cell migration. Mol Cell Biol. 2010;30:4786–96. doi: 10.1128/MCB.00381-10. The authors show that RHOA phosphorylation by PKG in vascular smooth muscle cells increases RHOA binding to RhoGDI1, thereby displacing and activating bound RAC1, resulting in increased cell migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tkachenko E, et al. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat Cell Biol. 2011;13:661–8. doi: 10.1038/ncb2231. Activation of PKA at the leading edge of cells coordinates protrusion and retraction, through a mechanism involving PKA-mediated phosphorylation of RHOA on Ser188, which promotes RHOA binding to RhoGDI and inhibition of RHOA activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 65.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 66.O'Connor KL, Nguyen BK, Mercurio AM. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J Cell Biol. 2000;148:253–8. doi: 10.1083/jcb.148.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishizaki H, et al. Defective chemokine-directed lymphocyte migration and development in the absence of Rho guanosine diphosphate-dissociation inhibitors alpha and beta. J Immunol. 2006;177:8512–8521. doi: 10.4049/jimmunol.177.12.8512. [DOI] [PubMed] [Google Scholar]
- 68.Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res. 2009;69:2838–44. doi: 10.1158/0008-5472.CAN-08-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bielek H, Anselmo A, Dermardirossian C. Morphological and proliferative abnormalities in renal mesangial cells lacking RhoGDI. Cell Signal. 2009;21:1974–83. doi: 10.1016/j.cellsig.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorovoy M, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007;101:50–8. doi: 10.1161/CIRCRESAHA.106.145847. [DOI] [PubMed] [Google Scholar]
- 71.Harding MA, Theodorescu D. RhoGDI signaling provides targets for cancer therapy. Eur J Cancer. 2010;46:1252–9. doi: 10.1016/j.ejca.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones MB, et al. Proteomic analysis and identification of new biomarkers and therapeutic targets for invasive ovarian cancer. Proteomics. 2002;2:76–84. [PubMed] [Google Scholar]
- 73.Zhao L, Wang H, Li J, Liu Y, Ding Y. Overexpression of Rho GDP-dissociation inhibitor alpha is associated with tumor progression and poor prognosis of colorectal cancer. J Proteome Res. 2008;7:3994–4003. doi: 10.1021/pr800271b. [DOI] [PubMed] [Google Scholar]
- 74.Zhao L, Wang H, Sun X, Ding Y. Comparative proteomic analysis identifies proteins associated with the development and progression of colorectal carcinoma. The FEBS journal. 2010;277:4195–4204. doi: 10.1111/j.1742-4658.2010.07808.x. [DOI] [PubMed] [Google Scholar]
- 75.Forget MA, et al. The expression of rho proteins decreases with human brain tumor progression: potential tumor markers. Clin exp metastasis. 2002;19:9–15. doi: 10.1023/a:1013884426692. [DOI] [PubMed] [Google Scholar]
- 76.Fritz G, Lang P, Just I. Tissue-specific variations in the expression and regulation of the small GTP-binding protein Rho. Biochimica et biophysica acta. 1994;1222:331–338. doi: 10.1016/0167-4889(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 77.Jiang WG, et al. Prognostic value of rho GTPases and rho guanine nucleotide dissociation inhibitors in human breast cancers. Clin Cancer Res. 2003;9:6432–40. [PubMed] [Google Scholar]
- 78.Ding J, et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12:390–9. doi: 10.1038/ncb2039. During hepatocellular carcinoma progression and tumor cell invasion, RHOGDI1 expression is downregulated by microRNA miR-151, which is encoded within an intron of the FAK gene. [DOI] [PubMed] [Google Scholar]
- 79.Harding MA, Theodorescu D. RhoGDI2: a new metastasis suppressor gene: discovery and clinical translation. Urologic oncology. 2007;25:401–6. doi: 10.1016/j.urolonc.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 80.Abiatari I, et al. Consensus transcriptome signature of perineural invasion in pancreatic carcinoma. Molecular cancer therapeutics. 2009;8:1494–1504. doi: 10.1158/1535-7163.MCT-08-0755. [DOI] [PubMed] [Google Scholar]
- 81.Koide N, et al. Establishment of perineural invasion models and analysis of gene expression revealed an invariant chain (CD74) as a possible molecule involved in perineural invasion in pancreatic cancer. Clin Cancer Res. 2006;12:2419–2426. doi: 10.1158/1078-0432.CCR-05-1852. [DOI] [PubMed] [Google Scholar]
- 82.Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin Exp Metastasis. 2000;18:519–525. doi: 10.1023/a:1011819621859. [DOI] [PubMed] [Google Scholar]
- 83.Theodorescu D, et al. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:3800–3806. doi: 10.1158/1078-0432.CCR-03-0653. [DOI] [PubMed] [Google Scholar]
- 84.Ma L, et al. Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. British journal of haematology. 2007;139:217–223. doi: 10.1111/j.1365-2141.2007.06782.x. [DOI] [PubMed] [Google Scholar]
- 85.Hu LD, Zou HF, Zhan SX, Cao KM. Biphasic expression of RhoGDI2 in the progression of breast cancer and its negative relation with lymph node metastasis. Oncology reports. 2007;17:1383–1389. [PubMed] [Google Scholar]
- 86.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 87.Habets GG, et al. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–49. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 88.Hordijk PL, et al. Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science. 1997;278:1464–6. doi: 10.1126/science.278.5342.1464. [DOI] [PubMed] [Google Scholar]
- 89.Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem. 2001;276:22614–20. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 90.Knezevic N, et al. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol. 2007;27:6323–33. doi: 10.1128/MCB.00523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiao J, et al. Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am J Physiol Cell Physiol. 2008;295:C1161–8. doi: 10.1152/ajpcell.00139.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guerrera IC, Keep NH, Godovac-Zimmermann J. Proteomics study reveals cross-talk between Rho guanidine nucleotide dissociation inhibitor 1 post-translational modifications in epidermal growth factor stimulated fibroblasts. J Proteome Res. 2007;6:2623–30. doi: 10.1021/pr070078f. [DOI] [PubMed] [Google Scholar]
- 93.Kuribayashi K, et al. Essential role of protein kinase C zeta in transducing a motility signal induced by superoxide and a chemotactic peptide, fMLP. J Cell Biol. 2007;176:1049–60. doi: 10.1083/jcb.200607019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DerMardirossian C, Rocklin G, Seo JY, Bokoch GM. Phosphorylation of RhoGDI by Src Regulates Rho GTPase Binding and Cytosol-Membrane Cycling. Mol Biol Cell. 2006;17:4760–4768. doi: 10.1091/mbc.E06-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu Y, et al. Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proc Natl Acad Sci U S A. 2009;106:5807–12. doi: 10.1073/pnas.0810094106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fei F, et al. The Fer tyrosine kinase regulates interactions of Rho GDP-Dissociation Inhibitor alpha with the small GTPase Rac. BMC Biochem. 11:48. doi: 10.1186/1471-2091-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sauzeau V, Rolli-Derkinderen M, Marionneau C, Loirand G, Pacaud P. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem. 2003;278:9472–80. doi: 10.1074/jbc.M212776200. [DOI] [PubMed] [Google Scholar]
- 98.Lang P, et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. Embo J. 1996;15:510–9. [PMC free article] [PubMed] [Google Scholar]
- 99.Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 1998;273:22554–62. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- 100.Tamma G, et al. cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI. J Cell Sci. 2003;116:1519–25. doi: 10.1242/jcs.00355. [DOI] [PubMed] [Google Scholar]
- 101.Ellerbroek SM, Wennerberg K, Burridge K. Serine Phosphorylation Negatively Regulates RhoA in Vivo. J Biol Chem. 2003;278:19023–19031. doi: 10.1074/jbc.M213066200. [DOI] [PubMed] [Google Scholar]
- 102.Guilluy C, et al. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ Res. 2008;102:1265–74. doi: 10.1161/CIRCRESAHA.107.164764. [DOI] [PubMed] [Google Scholar]
- 103.Forget MA, Desrosiers RR, Gingras D, Beliveau R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–54. doi: 10.1042/0264-6021:3610243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tu S, Wu WJ, Wang J, Cerione RA. Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J Biol Chem. 2003;278:49293–300. doi: 10.1074/jbc.M307021200. [DOI] [PubMed] [Google Scholar]
- 105.Schoentaube J, Olling A, Tatge H, Just I, Gerhard R. Serine-71 phosphorylation of Rac1/Cdc42 diminishes the pathogenic effect of Clostridium difficile toxin A. Cell Microbiol. 2009;11:1816–26. doi: 10.1111/j.1462-5822.2009.01373.x. [DOI] [PubMed] [Google Scholar]
- 106.Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–8. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]