

Abstract
Cells use protein quality control (PQC) systems to protect themselves from potentially harmful misfolded proteins. Many misfolded proteins are repaired by molecular chaperones, but irreparably damaged proteins must be destroyed. Eukaryotes predominantly destroy these abnormally folded proteins through the ubiquitin-proteasome pathway, which requires compartment-specific ubiquitin ligase complexes that mark substrates with ubiquitin for proteasome degradation. In the yeast nucleus, misfolded proteins are targeted for degradation by the ubiquitin ligase San1, which binds misfolded nuclear proteins directly and does not appear to require chaperones for substrate binding. San1 is also remarkably adaptable, as it is capable of ubiquitinating a structurally diverse assortment of abnormally folded substrates. We attribute this adaptability to San1's high degree of structural disorder, which provides flexibility and allows San1 to conform to differently shaped substrates. Here we review our recent work characterizing San1's distinctive mode of substrate recognition and the associated implications for PQC in the nucleus.
Keywords: ubiquitin ligase, intrinsic disorder, unstructured, protein quality control, ubiquitin, San1, proteasome
Introduction
To function properly, proteins must adopt specific conformations formed through a series of folding and assembly processes. Although essential to function, protein folding is error-prone and vulnerable to disruption by metabolic and environmental stresses. The failure of proteins to fold properly has a profound impact on cellular physiology as abnormally folded proteins often acquire harmful aggregation-prone states and underlie many human pathologies, including many prominent neurodegenerative diseases.1 Cells must therefore manage misfolded proteins appropriately or face deleterious consequences from their accumulation.
Cells protect themselves from the toxic effects caused by misfolded proteins by employing various protein quality control (PQC) systems that either repair or destroy abnormally folded proteins. Molecular chaperones facilitate the restoration of misfolded proteins to their natively folded states. However, some misfolded proteins cannot be repaired and must ultimately be destroyed by the cell's PQC degradation machinery. In eukaryotes, PQC degradation primarily proceeds through compartment-specific ubiquitin ligase complexes that ubiquitinate abnormally folded proteins for subsequent destruction by the proteasome. The best characterized of these PQC degradation systems exist in the endoplasmic reticulum (ER) and cytoplasm.2,3 These compartments have been extensively studied for PQC degradation in part because of their participation in the protein biosynthetic pathway, where errors in translation and folding yield misfolded proteins during synthesis. However, the nuclear compartment also requires robust PQC, as evidenced by the large number of human neurodegenerative disorders characterized by protein inclusions in the nucleus.4
Compared to the cytoplasm and ER, PQC degradation in the nucleus is poorly characterized. Recent studies have identified ubiquitin ligases involved in nuclear PQC degradation. In yeast, the nuclear-localized ubiquitin ligase San1 specifically targets abnormal proteins for proteasome-mediated degradation.5 The mammalian ubiquitin ligases PML-IV and UHRF-2 may also serve similar functions.6–8 Until recently, how nuclear ubiquitin ligases target their substrates remained unknown. To resolve this question and improve our understanding about how nuclear PQC degradation systems recognize their substrates, we focused our efforts on the characterizing how the yeast ubiquitin ligase San1 binds its substrates.9 The results of those efforts are the focus of this Extra View.
San1 Directly Interacts with Substrates
Confronted with the task of recognizing a diverse assortment of potential substrates, PQC ubiquitin ligases can use one of two approaches. The first approach and simplest, involves the ubiquitin ligase binding substrates directly. This approach requires that the ubiquitin ligase possesses some means of distinguishing abnormally folded from normal proteins. Alternatively, ubiquitin ligases can appropriate the substrate recognition features of chaperones or other adaptor proteins.
For most known ubiquitin ligases involved in PQC degradation, the degradation of their substrates requires the action of protein chaperones. In the mammalian cytoplasm, the ubiquitin ligase CHIP interacts with Hsp70 chaperones and ubiquitinates Hsp70 client proteins that cannot fold properly.10 In the yeast cytoplasm, the ubiquitin ligase Ubr1 requires Hsp70 and Hsp110 chaperones to ubiquitinate substrates.11,12 In the yeast ER, the ubiquitin ligases Hrd1 and Doa10 require several associated proteins to ubiquitinate substrates. Hrd1 requires its partner Hrd3, the Hsp70 chaperone Kar2, and the lectin Yos9 to ubiquitinate ER lumenal substrates, while Doa10 requires cytoplasmic Hsp70 and Hsp40 chaperones to ubiquitinate ER membrane proteins with cytosolic lesions.13–17
The active role of chaperones in so many PQC degradation pathways implies that they are generally required for substrate recognition. While this appears to be the case for cytoplasmic and ER-localized ubiquitin ligases involved in PQC degradation, the nuclear-localized ubiquitin ligase San1 lacks chaperone-binding motifs. This does not preclude an adaptor requirement for San1, so we conducted genetic and biochemical assays to determine if San1 interacts with substrates directly.
We first assayed San1-substrate interactions in yeast, beginning with a two-hybrid genetic selection to figure out how San1 interacts with known substrates. Our genetic analysis revealed that San1 selectively interacts with abnormally folded, missense substrates, but not their normally folded, wild-type counterparts. In addition to the two-hybrid selection, we also used a formaldehyde-crosslinking San1 immunoprecipitation approach coupled with mass spectrometry (IP-MS) to ascertain whether San1 binds substrates directly or, if not, to identify the adaptor proteins San1 uses for substrate recognition. From the results of the IP-MS study, we observed enrichment for a misfolded, missense San1 substrate, but not its normally folded, wild-type variant, confirming San1's selectivity for abnormal proteins. We also failed to observe enrichment for chaperones or any other adaptor protein in the San1 IP-MS analysis, suggesting a direct interaction between San1 and its substrates.
Although our yeast-based assays implied a direct interaction between San1 and its substrates, we sought more definitive evidence of this interaction. We first used an in vitro ubiquitination assay, reconstituting the ubiquitin cascade with San1 as the ligase component and substrates purified from E. coli. However, isolating abnormally folded San1 substrates from the E. coli expression system proved difficult due to their aggregation, so we used the model substrate luciferase instead. We observed denaturation-dependent and San1-dependent ubiquitination of luciferase, similar to results observed in a previously published assay using the ubiquitin ligase CHIP.18 Unlike the CHIP ubiquitination assay, San1-dependent ubiquitination did not require chaperones. To approximate this assay using the difficult-to-isolate San1 substrates, we reconstituted the ubiquitination cascade in E. coli cells, avoiding the difficult purification procedure. Using this assay, we were able to demonstrate San1-dependent ubiquitination of substrates derived from yeast without the confounding presence of potential yeast adaptor proteins.
Potential Role for Chaperones in San1-Mediated PQC Degradation
Our results described a nuclear PQC degradation pathway where San1 recognizes substrates by direct interaction. We did not observe a requirement for chaperones in San1 substrate recognition, but this does not preclude chaperone involvement in San1-mediated degradation. From our in vitro ubiquitination assay, we observed that San1 is unable to recognize aggregated forms of luciferase. Chaperones participate in the kinetic partitioning of proteins between various folding states and prevent client proteins from aggregating.19 Therefore, nuclear-localized chaperones could contribute to San1 substrate recognition by maintaining substrates in their soluble states (Fig. 1).
Figure 1.

Chaperones contribute to San1-mediated degradation without directly interacting with San1. Chaperones promote San1-dependent degradation by maintaining the solubility of misfolded proteins, but antagonize this process by repairing misfolded proteins. Processes that enhance San1 degradation, such as protein misfolding and substrate solubilization, are indicated by solid arrows. Processes that decrease San1 degradation, like protein repair and substrate aggregation, are indicated by dotted arrows.
Conversely, chaperones might act to suppress San1-mediated degradation by competing for substrates (Fig. 1). Consistent with this, in a recent study examining San1's involvement the degradation of misfolded cytoplasmic substrates, the authors found that the Hsp110 chaperone Sse1 negatively impacted San1's ability to ubiquitinate a misfolded substrate in vitro.11 In considering the triage model of PQC, the decision to repair or destroy misfolded proteins is thought to be determined at the level of protein chaperones, such that repairing misfolded proteins when possible is preferable to degrading them.20 One way the cell might favor chaperone-mediated repair in the nucleus in a competitive mode is by expressing nuclear chaperones more abundantly than nuclear PQC ubiquitin ligases like San1, thus allowing the chaperones to outcompete San1 for folding-competent proteins. Indeed, the steady-state levels of nuclear-localized chaperones normally exceed those of San1 by a hundred-fold or more.21 Even more compelling is that during heat shock SAN1 gene transcription is decreased whereas chaperone transcription is increased,22 thus biasing nuclear PQC towards chaperone function more so under stress conditions.
Finally, it is possible that San1 does not interact with chaperones because chaperone-mediated folding and San1-dependent PQC degradation are conducted in separate subregions of the nucleus. This seems unlikely in yeast as San1 appears to be uniformly distributed throughout the nucleus as do nuclear chaperones.5,23,24 However, subcompartmentalization for refolding and degradation could exist in the mammalian nucleus where a number of different subnuclear bodies have been identified.
Determining the relative contributions of chaperones to San1-mediated degradation in the nucleus is complicated, and how chaperone-mediated processes affect San1-dependent degradation remains unclear. Understanding these processes in the context of PQC degradation is necessary for a more complete understanding of PQC in the nucleus. Further study is needed to determine the extent to which chaperones act in competition with, collaboration with, or parallel to San1-mediated degradation.
San1 is Intrinsically Disordered
Having established that San1 binds substrates directly, we examined San1's sequence for conserved features that indicate how substrate binding occurs. However, San1 lacks domain structure outside of its RING domain. Because the RING domain mediates the activity-conferring interaction with ubiquitin conjugating enzymes, it is unlikely to be involved in substrate recognition.5,25
Without domains that indicate how San1 recognizes substrates, we turned our attention to San1's sequence characteristics for clues about San1's substrate-binding capacity. Noting the relatively low frequency of core-forming hydrophobic residues outside of its RING domain, we considered the possibility that San1 is intrinsically disordered. To explore this possibility, we applied several disorder prediction methods to San1's sequence. We first used the Predictor of Naturally Disordered Regions (PONDR) VL-XT, which uses a neural network algorithm trained on sequence attributes typically found in regions absent in X-ray and nuclear magnetic resonance structures.26 Confirming our expectations, PONDR identified San1 as highly disordered. We found similar results using IUPred, which identifies disordered regions based on their inability to form stabilizing interresidue connections and FoldIndex, which predicts disordered residues based on the ratio of charged to hydrophobic surrounding residues.27,28
The consensus prediction on San1's disorder prompted us to use several biochemical methods to confirm this feature experimentally. Using size exclusion chromatography, we observed that San1 migrates much more quickly than bovine serum albumin (BSA), a globular protein of similar size. This is a strong indicator of structural disorder, as disordered proteins have much larger hydrodynamic radii than globular proteins.29 We confirmed this result using limited proteolysis, which takes advantage of the fact that protease cleavage sites are usually inaccessible in structured regions but readily available in regions of disorder.30 Consistent with the expectation for a disordered protein, San1 was more rapidly digested by the proteases trypsin and thermolysin than BSA. Lastly, we evaluated San1's disorder using circular dichroism spectroscopy (CD), which provides a general estimate of a protein's secondary structure composition based on its absorbance of left-hand and right-hand polarized light.31 San1 possesses the characteristic spectra of a protein with a large amount of random coil structure, which is typical for disordered proteins. These data confirmed the consensus prediction that San1 is intrinsically disordered, but the significance of disorder remained a mystery. How could intrinsic disorder contribute to San1's role in PQC degradation?
San1 Contains Potential Binding Sites in its Disordered Regions
Intrinsic disorder is a common property in eukaryotic proteins and is thought to provide these proteins with the ability to adopt multiple conformational states.32 This conformational flexibility is especially useful for proteins that interact with multiple partners, such as hubs in protein-protein interaction networks.33 Such flexibility might also be useful for PQC components that engage differently shaped misfolded substrates. Indeed, structural disorder is associated with the function of several protein chaperones, most notably the disordered N-terminal region of small heat shock proteins (sHSPs), which is required for substrate recognition.34–36
Considering the role of disorder in sHSP substrate recognition, we hypothesized that San1 uses its disordered regions to bind substrates. If this were the case, we would anticipate the presence of one or more binding sites in San1's disordered regions. We can identify binding sites in disordered regions based on distinct sequence characteristics. Notably, we observed the presence of alternating ordered and disordered sequences in San1's PONDR profile. Such small ordered regions are thought to be motifs used in the binding of other proteins. We examined these potential interaction sites using ANCHOR, an algorithm that predicts binding sites in disordered regions based on their capacity to form favorable interactions.37 The binding sites identified by ANCHOR conformed perfectly to those predicted by PONDR, giving us greater confidence in the results found using these independently devised algorithms.
Given our expectation that San1 uses its disordered regions to bind substrates, the predicted binding sites in those regions should be indispensable and therefore conserved in San1 homologs. We evaluated San1 homologs from several Saccharomyces species, finding that they reproducibly complemented the san1Δ allele for substrate degradation when expressed in Saccharomyces cerevisiae and share the same predicted disorder topology. We then aligned the sequences of San1 and its homologs using ClustalW.38 Interestingly, we found that the overall San1 sequence was only partially conserved, with the highest divergence of conservation in its regions of disorder. By contrast, each predicted binding site in San1's sequence was strongly conserved, with most predicted binding sites sharing complete identity among San1 homologs.
San1 Distinctly Binds Each Substrate Using Its Disordered Regions
We hypothesized that if San1 uses its disordered N- and C-terminal domains for substrate interactions, then each of the predicted binding sites within those domains should help provide distinct specificity for the differently shaped misfolded substrates that San1 encounters. We also considered the possibility that some of these sites might mediate the interaction between San1 and cofactors. Notably, both prediction methods accurately identified the ordered segments of the RING domain as potential binding sites; these segments contain the zinc-coordinating cysteine and histidine residues required for interaction with ubiquitin conjugating enzymes. Aside from identifying substrate and cofactor interaction sites, there remained the possibility that some or all of the binding sites predicted by the PONDR and ANCHOR algorithms do not exist, or that these sites mediate interactions completely unrelated to San1's function in PQC degradation.
To resolve these ambiguities, we needed to evaluate experimentally the importance of these predicted sites for San1-substrate interactions. We decided to conduct a systematic deletion analysis of San1 and test the resulting deletions for their ability to bind substrates using the two-hybrid assay. If San1 contains multiple substrate interaction sites, then no single deletion should eliminate San1-substrate interaction. Moreover, if these sites provide distinct specificities for different abnormally folded substrates, then each deletion would have varying effects on San1's interaction with different substrates. However, if San1 recognizes substrates through a single binding site, each deletion would have identical effects on San1's interaction with each substrate.
To rigorously test our hypothesis that San1 binds each of its substrates distinctly, we needed a larger collection of substrates. Because the two-hybrid assay faithfully reported San1's interaction with known substrates, we used another two-hybrid selection to identify more substrates. In this selection, we tested the interaction between San1 and a yeast-derived cDNA library and identified 28 unique interactors. Notably, no interacting chaperones or other adaptors came out of the library used in our genetic selection. To verify that the San1 interactors were in fact substrates, we tested the identified interactors for San1-dependent degradation using cycloheximide-chase assays. Most of the identified interactors (25 of 28) underwent San1-mediated degradation to some degree, confirming that we had identified San1 substrates and expanding our pool of substrates to 31 distinct, abnormally folded proteins.
Having developed a sufficiently sized catalog of San1 substrates, we were able to conduct an exhaustive San1 deletion and interaction analysis. We made 20 small deletions in San1, covering all of the conserved predicted binding sites and non-conserved disordered segments in San1's sequence. We then tested the San1 deletions for interaction with each substrate in our collection. We conducted our two-hybrid assays on three types of media, each with varying stringency for two-hybrid interaction, to distinguish more clearly the effects of each San1 deletion on each substrate interaction. In the results of our extensive two-hybrid analysis, we observed that each of the San1 deletions had distinct effects on San1's interaction with each substrate, supporting the hypothesis that San1 uses its multiple binding sites to specifically interact with each of its differently shaped substrates. We also observed that many of the San1 deletions only had observable effects on substrate interaction when tested on higher stringency media for two-hybrid interaction, indicating variable affinities for each substrate. We also tested a deletion of San1's RING domain in the two-hybrid and observed no effect on San1-substrate interaction, demonstrating that ubiquitin conjugating enzymes do not contribute to San1's substrate recognition.
We then tested the effects of San1 deletions on its ability to degrade its substrates in vivo. Because of the difficulty involved in testing each San1 deletion against our collection of substrates using conventional cycloheximide-chase assays, we opted to measure the effects of San1 deletions on the steady-state levels of green fluorescent protein-tagged (GFP) variants of representative substrates using flow cytometry. Using this assay, we observed distinct effects of San1 deletions on the steady-state levels of each substrate tested. Our results were analogous, but not identical to the results obtained using the two-hybrid, with the discrepancy largely attributable to changes in the constructs used and the differences in what the assays report. Taken together, the results of the two-hybrid and steady-state experiments confirmed the hypothesis that San1 uses multiple binding sites embedded in its disordered regions to distinctly interact with each of its differently shaped substrates.
By using multiple sites embedded in disordered regions to bind substrates, San1 is unusual for PQC-involved ubiquitin ligases. For example, the ubiquitin ligases CHIP, Hrd1, Doa10, PML-IV and UHRF-2 do not possess the extent of disorder seen in San1 (our unpublished observations). Intriguingly, San1's disordered binding regions do bear remarkable similarities to the disordered N-terminal regions of sHSPs. Notably, the disordered N-terminal domain of the Pisum sativum (garden pea) sHsp 18.1 contains at least six residues involved in substrate interactions, as demonstrated by p-benzoyl-L-phenylalanine (Bpa) crosslinking.36 These residues are dispersed throughout the disordered domain, indicating the presence of multiple substrate-binding sites in the PsHsp18.1 N-terminal region.
How Does San1 Engage Substrates?
Although we were able to glean new insights into San1's substrate binding mechanism, many questions remain about how San1 specifically engages substrates. For example, does San1 undergo disorder-order transitions when binding substrates? One possibility is that San1 overall adopts a single ordered conformation when bound by a substrate. Another possibility is that San1 adopts a local ordered conformation in part of its sequence upon binding a substrate. A final possibility is that San1 remains completely disordered upon binding substrates.
Another question about San1's mode of substrate interaction is how its many binding sites in an individual San1 molecule work together to bind a single substrate molecule. We determined that for every substrate, at least two of San1's binding sites possess some degree of affinity for that substrate. Therefore, San1 can engage each substrate in multiple ways, which increases its avidity for substrates. However, we do not yet know whether San1 uses these multiple binding sites independently or in conjunction with each other. Therefore, we envision two models that describe how San1 uses its multiple binding sites to interact with substrates.
In our first model, San1 uses its binding sites as a group, engaging substrates with multiple contacts. One way of envisioning this model is that San1 binds each substrate through specific interactions, gradually adopting conformations where other binding sites engage the substrate. Another possibility is that San1 interacts with substrates using all of the required binding sites simultaneously. In either interpretation of this model, San1 “grasps” substrates using multiple binding sites to maintain the San1-substrate interaction long enough for ubiquitination to occur (Fig. 2A).
Figure 2.
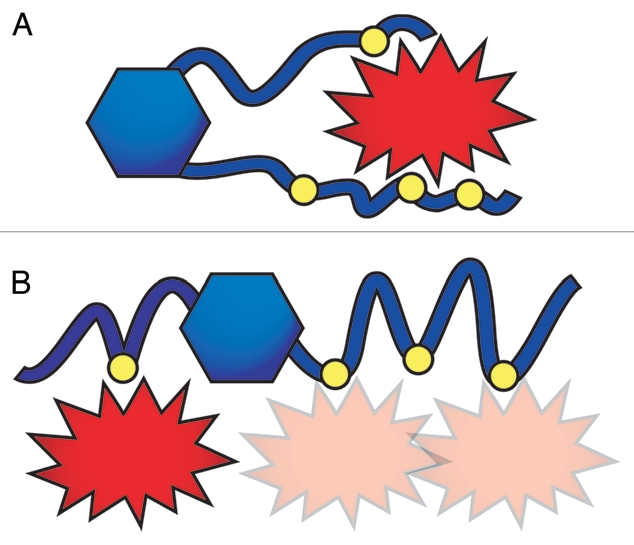
Two models describe how San1 possibly interacts with substrates. (A) San1 “grasps” substrates using several of its binding sites simultaneously and at several contact points. (B) Each San1 binding site independently interacts with substrates. In this model, San1 increases its avidity for substrates by possessing multiple binding sites for each substrate.
The second model describes San1 using its binding sites independently. In this model, substrate-binding sites lie along San1's disordered sequence like “beads on a string”, each possessing its own affinity for a given substrate (Fig. 2B). Multiple contacts are possible but not required for substrate interaction. With each binding event occurring independently, San1-substrate interactions in this model are weaker than those described by the “grasping” model. However, we favor this model for two reasons. First, we observed that San1-substrate interactions require crosslinking to survive standard immunoprecipitation conditions. This indicates that the San1-substrate interaction is either relatively weak or transient. Our second reason for favoring the “beads on a string” model is that several of our identified San1 substrates are short peptides fused to a two-hybrid or fluorescent reporter protein. San1 does not recognize the isolated reporter proteins, and the peptides are probably too small for more than one San1 binding site to engage them at the same time. This would mean that a single binding event could be sufficient for San1-mediated ubiquitination in these cases.
To resolve the two models, we will require some structural information as to how San1 interacts with its substrates. While a 3D crystal structure of San1 bound to substrates is unlikely at this time due to San1's high intrinsic disorder, it might be possible to use NMR, SAXS or hydrogen-deuterium exchange mass spectrometry to probe San1's substrate interactions. Alternatively, if the “beads on a string” model is correct, we anticipate that the substrate-binding sites embedded within the disordered regions would not be dependent upon their location within San1 and could be switched among themselves. Furthermore, the intervening disordered segments would be not be subject to strict size limits and could vary in length. Both seem to be the case when we examined the San1 homologs from more distantly related yeast species (S. pombe and C. albicans). While this is suggestive of the second model, we will ultimately need some additional structural insight into how San1 engages substrates in the future to improve our understanding of San1's substrate binding mode.
Nuclear PQC Degradation via Disordered Ligases
While PML-IV and URHF-2 have been implicated in mammalian nuclear PQC degradation,5–7 neither possesses the disordered topology of San1. Thus, a true San1 functional homolog has yet to be discovered in mammalian systems. One explanation for this is that highly disordered proteins like San1 evolve rapidly in their disordered regions because linear sequence in these regions does not have to be conserved, only their flexibility needs to be maintained. This would confound any conventional homology analyses.39 We have now taken a different approach to identify functional San1 analogs based on San1's definitional characteristics: a high degree of predicted disorder containing multiple embedded binding sites. Using these criteria, we've identified many putative human ubiquitin ligases that match San1's characteristics, and we will test them for a role in nuclear PQC degradation in the future.
It could also be the case that PQC degradation in higher multicellular eukaryotes evolved differently than in simpler single-cell eukaryotes. For example, higher eukaryotes have multiple different subnuclear regions (or intranuclear bodies) where PQC degradation could be compartmentalized, whereas single-cell eukaryotes tend to lack these subnuclear regions. In the case of the single-cell eukaryotes, they may have evolved a generalist PQC degradation system like San1 that “cruises” the nucleoplasm looking for misfolded proteins that could present a problem if they persist. In multicellular eukaryotes that have larger nuclei with multiple subregions, a more formal process of subcompartment delivery could have evolved, wherein chaperones recognize misfolded proteins in the nucleoplasm and deliver them to intranuclear bodies specializing in PQC degradation. Interestingly, the ubiquitin ligase PML-IV has been implicated in the PQC degradation of polyQ-expansion proteins in mammalian cells and localizes specifically to intranuclear PML clastosomes,6,8 suggesting this might be a possibility. Whether or not a generalist PQC degradation system like San1 exists in high eukaryotes is still an open question.
Understanding the complexities of nuclear PQC degradation in higher eukaryotes remains relatively uncharted territory. However, given that >25 human diseases are characterized by protein inclusions in the nucleus,4 we anticipate that nuclear PQC will garner more attention in the future. We hope our studies in yeast will be able to provide continued insight from which to build a base for nuclear PQC degradation studies in higher eukaryotes.
Acknowledgments
This work was supported by NIH/NIGMS training grant T32GM007750, NIH/NIA grant R01AG031136, NIH/NCRR grant R21RR025787, an Ellison Medical Foundation New Scholar Award in Aging and a Marian E. Smith Junior Faculty Award.
Extra View to: Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004.
References
- 1.Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 2.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Goeckeler JL, Brodsky JL. Molecular chaperones and substrate ubiquitination control the efficiency of endoplasmic reticulum-associated degradation. Diabetes Obes Metab. 2010;12:32–38. doi: 10.1111/j.1463-1326.2010.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol. 2007;33:2–42. doi: 10.1111/j.1365-2990.2006.00819.x. [DOI] [PubMed] [Google Scholar]
- 5.Gardner RG, Nelson ZW, Gottschling DE. Degradation-mediated protein quality control in the nucleus. Cell. 2005;120:803–815. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Fu L, Gao YS, Tousson A, Shah A, Chen TL, Vertel BM, et al. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol Biol Cell. 2005;16:4905–4917. doi: 10.1091/mbc.E05-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata A, Nagashima Y, Matsumoto L, Suzuki T, Yamanaka T, Date H, et al. Intra-nuclear degradation of polyglutamine aggregates by the ubiquitin proteasome system. J Biol Chem. 2009;284:9796–9803. doi: 10.1074/jbc.M809739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janer A, Martin E, Muriel MP, Latouche M, Fujigasaki H, Ruberg M, et al. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J Cell Biol. 2006;174:65–76. doi: 10.1083/jcb.200511045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nillegoda NB, Theodoraki MA, Mandal AK, Mayo KJ, Ren HY, Sultana R, et al. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol Biol Cell. 2010;21:2102–2116. doi: 10.1091/mbc.E10-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 14.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 15.Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- 16.Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283:32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 20.Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- 21.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 22.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chughtai ZS, Rassadi R, Matusiewicz N, Stochaj U. Starvation promotes nuclear accumulation of the hsp70 Ssa4p in yeast cells. J Biol Chem. 2001;276:20261–20266. doi: 10.1074/jbc.M100364200. [DOI] [PubMed] [Google Scholar]
- 24.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta A, Ramsey KL, Smith JS, Auble DT. Sir antagonist 1 (San1) is a ubiquitin ligase. J Biol Chem. 2004;279:26830–26838. doi: 10.1074/jbc.M400894200. [DOI] [PubMed] [Google Scholar]
- 26.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 28.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, et al. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 29.Receveur-Brechot V, Bourhis JM, Uversky VN, Canard B, Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 30.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 31.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, et al. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9:1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, et al. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haslbeck M, Ignatiou A, Saibil H, Helmich S, Frenzl E, Stromer T, et al. A domain in the N-terminal part of Hsp26 is essential for chaperone function and oligomerization. J Mol Biol. 2004;343:445–455. doi: 10.1016/j.jmb.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 35.Jaya N, Garcia V, Vierling E. Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc Natl Acad Sci USA. 2009;106:15604–15609. doi: 10.1073/pnas.0902177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stromer T, Fischer E, Richter K, Haslbeck M, Buchner J. Analysis of the regulation of the molecular chaperone Hsp26 by temperature-induced dissociation: the N-terminal domail is important for oligomer assembly and the binding of unfolding proteins. J Biol Chem. 2004;279:11222–11228. doi: 10.1074/jbc.M310149200. [DOI] [PubMed] [Google Scholar]
- 37.Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]