

Abstract
The pluripotent genome is characterized by unique epigenetic features and a decondensed chromatin conformation. However, the relationship between epigenetic regulation and pluripotency is not altogether clear. Here, using an enhanced MEF/ESC fusion protocol, we compared the reprogramming potency and histone modifications of different embryonic stem cell (ESC) lines (R1, J1, E14, C57BL/6) and found that E14 ESCs are significantly less potent, with significantly reduced H3K9ac levels. Treatment of E14 ESCs with histone deacetylase (HDAC) inhibitors (HDACi) increased H3K9ac levels and restored their reprogramming capacity. Microarray and H3K9ac ChIP-seq analyses, suggested increased extracellular matrix (ECM) activity following HDACi treatment in E14 ESCs. These data suggest that H3K9ac may predict pluripotency and that increasing pluripotency by HDAC inhibition acts through H3K9ac to enhance the activity of target genes involved in ECM production to support pluripotency.
Keywords: chromatin plasticity, embryonic stem cells, histone acetylation, histone modifications
Introduction
Nuclear reprogramming resets the transcriptional state of the somatic genome to a configuration that is compatible with developmental pluripotency.1,2 Reprogramming of somatic cells to a pluripotent state, giving rise to all cell types, was first demonstrated by somatic cell nuclear transfer (SCNT) experiments resulting in the production of clonally derived frogs.3 It was later shown that oocytes can also be employed for the cloning of mammals.4,5 However, the efficiency of mammalian cloning, as measured by the production of live-birth clones, is low and it has been suggested that this is due to the incomplete reprogramming of the somatic nucleus.6
Embryonic stem cells (ESCs), like oocytes, contain reprogramming activity, and can be employed to reprogram somatic genomes through fusion.7 Developmental pluripotency has been demonstrated in hybrids derived from the fusion of murine ES and somatic cells,8–12 as well as thymocytes.7 In addition, embryonal carcinoma (EC) and embryonic germ (EG) cells have also been shown to contain reprogramming activities in fusion experiments.13–15 Human fibroblasts have been successfully reprogrammed as well upon fusion with human ESC8 or myeloid precursor cells.16 The reprogramming activity of ESCs likely resides within the nuclear compartment because karyoplasts, but not cytoplasts, derived from these cells, can bring about reprogramming of somatic cells upon fusion.17 The hybrid cells can give rise to a variety of cell types7,8,15 and can be continuously propagated under appropriate culture conditions. They maintain a stable tetraploid DNA content and their morphology, growth rate and antigen expression patterns are characteristic of ESCs.8,11
Interestingly, unlike reprogramming by transduction with the transcription factors Oct4, Sox2, Klf4 and c-Myc,18 reprogramming by fusion was shown to be rapid and occurring without cell division,19 making it an extremely convenient method for studying the mechanisms of reprogramming and for conducting comparisons between different ESC lines. It is known that ESCs derived from different strains of mice vary considerably in their potency to generate germline chimeras20 and it was shown that changes in copy number variants of the ESC genome correlates with the proliferation and differentiation potentials of these lines.21
The molecular mechanism(s) and factors responsible for reprogramming the epigenome are largely unknown. DNA methylation and histone modifications play important roles in regulating gene activity via alterations of chromatin structure.22,23 Histone tail modifications24 include acetylated, methylated or ubiquitinated lysine residues, methylated arginines and phosphorylated serine residues.25 These epigenetic modifications, often referred to as the ‘histone code’26,27 can determine the state of a cell's expression pattern.
In this study we investigated the reprogramming potency of different lines of ESCs to reprogram MEFs, with relation to chromatin state. We optimized virus-mediated fusion conditions and compared various established ESC lines in this setting. We found that the reprogramming potency of the E14 ESC line was relatively low compared with others, but could be raised to the level of R1 cells upon incubation with histone deacetylase (HDAC) inhibitors. Concomitantly, we found that the levels of H3K9ac were low in E14 cells but increased significantly after incubation with a low dose of HDAC inhibitors. Microarray analyses of E14 before and after 16 hrs of low level treatment with HDAC inhibitors revealed enrichment for extracellular matrix (ECM) activity suggesting that the ECM niche is secreted by ESCs to maintain pluripotency. Using chromatin immunoprecipitation (ChIP) of H3K9ac before and after HDAC inhibition followed by SOLiD sequencing (ChIP-seq), we identify the genomic regions that are associated with increased pluripotency. Taken together, these results indicate that H3K9 acetylation levels of pluripotent cells during the fusion events are highly correlated with the reprogramming efficiency of the somatic cells, and that HDAC inhibition can significantly increase the reprogramming potential by promoting ECM production.
Results
Baculovirus significantly enhances MEF/ESC fusion.
To investigate the capacity of ESCs to reprogram somatic cells, we fused R1 or E14 mouse ESCs with Oct4/GFP/neoR/ROSA26-β-geo MEFs (henceforth, MEFs) (Fig. S1A and B). In order to increase the fusion efficiency we fused the cells by introducing baculovirus under low pH conditions.34 ESCs stained with PKH26-red were plated on MEFs and transfected with baculovirus. Fusion efficiency (∼70–85%) was measured by the number of stained MEFs after fusion with stained ESCs (Fig. S1C–E). ESCs were subjected to dual G418/HAT drug selection. After 6–8 d, GFP-positive and drug-resistant colonies emerged, and picked at 10–14 d (Fig. S1B, bottom left). R1 ESCs fused with MEFs produced ∼40 hybrid colonies per 107 MEFs. Such colonies (Fig. S1B, bottom right) behaved similarly to ESCs. As expected, the hybrids stained blue with X-gal due to the β-gal inside the ROSA26 locus (Fig. S1F), and PCR showed that hybrids contained neo, GFP and LacZ (Fig. S1G). G418 and HAT failed to produce colonies if MEFs and ESCs were separately subjected to fusion conditions. In all fusion experiments we included the R1 ESCs as a baseline and positive control. We next analyzed the chromosomal content of the hybrids. As expected, all hybrids were near-tetraploid (Fig. S1H and I). RT-PCR demonstrated that hybrids do not express MEF markers e.g., Coll1α, Igf2 and wisp1, but express Oct4, Nanog, Rex1 and Sox2 (Fig. S1J). Further, the hybrid lines formed embryoid bodies (EBs) with growth and differentiation properties similar to ESCs. Pooled EBs at different time points demonstrated that the hybrids exhibit a remarkable degree of developmental potential to form all three germ layers (Fig. S1K), similar to ESCs. These data suggest that via fusion with ESCs, the MEFs acquired ESC features, replacing their own.
One of the unique features of ESCs is their dynamic association of chromatin proteins with chromatin and their paucity in condensed heterochromatin foci.31,35 We therefore analyzed chromatin structure by light microscopy and measured the dynamic association of HP1-GFP with chromatin using fluorescence recovery after photobleaching (FRAP). DAPI staining demonstrated that the chromatin structure in the fusion colonies appeared morphologically similar to that of ESCs with fewer, less pronounced, heterochromatin foci (Fig. S2A and B), as previously demonstrated,31,36 and in stark contrast to the heterochromatin organization in MEFs (Fig. S2C). In addition, hybrid cells displayed HP1-GFP dynamic similar to ESCs, whereas HP1-GFP recovery in MEFs was significantly slower (Fig. S2D, p < 0.05).31 Our observations demonstrate that the MEF/ESC hybrids, although tetraploid, adopt transcriptional, morphological, developmental and nuclear/chromatin characteristics of ESCs.
Different ESC lines have varying reprogramming capacities.
In an effort to determine the relative potency of different ESC lines to reprogram MEFs, we compared fusion efficiency of four established ESC lines, J1, C57BL/6, R1 and E14. Fusion of MEFs with R1, J1 or C57BL/6 ESC lines resulted in a range of 17–41 reprogrammed GFP-positive ESC-like colonies per 2 × 107 MEFs, whereas this yield dropped significantly for E14 ESCs (p < 0.05, Mann-Whitney U-test, p < 0.005, 2-tailed t-test; Table 1A).
Table 1A.
Summary of MEF/ESC fusion experiments
| Comparison of MEF/ESC hybrids after fusion of MEF with different ESC lines | ||
| ESC line | Exp. 1 | Exp. 2 |
| C57BL/6 | 18 | 19 |
| J1 | 29 | 25 |
| R1 | 41 | 37 |
| E14 | 4 | 6 |
Numbers of GFP hybrid colonies/20 million MEFs.
Reprogramming capacity is correlated with H3K9 acetylation.
To investigate whether the differences in ESC potency are correlated with chromatin structure and the epigenetic state, we analyzed a variety of chromatin marks. All 4 lines exhibited similar levels of the majority of the chromatin marks that we analyzed, including acetylated histones (H3ac, H4ac), H3K4me3, H3K9m3, H3K27me3, phospho-serine5 RNA polymerase 2 (Pol2pS5) and HP1α (Fig. 1A and B). In contrast, H3K9ac was significantly lower in E14 compared to all other ESC lines tested (Fig. 1A, bottom). This prompt the idea that the low reprogramming capacity of E14 ESCs might be related to the low levels of H3K9 acetylation observed in these cells.
Figure 1.

Decreased H3K9ac in E14 ESCs. (A) Immunofluorescence (IF) images of pan-acetylated H4 (H4ac), tri-methylated H3 on lysine 4 and 9 (H3K4me3, H3K9me3), RNA polymerase II phosphorylated on serine 5 (Pol2pS5), HP1α and H3 acetylated on lysine 9 (H3K9ac). (B) Quantitative assessment of (A). Y axis depicts arbitrary fluorescent units. Values represent at least 300 cells from three independent experiments.
HDAC inhibitors can restore pluripotency of inferior ESC lines.
To test this, we added the HDACi trichostatin A (TSA) to our ESC colonies. As expected, a 4 h period of incubation of E14 ESCs with 5 or 25 nM TSA resulted in a dose-dependent increase in H3ac (Fig. 2A, top) and H3K9ac (Fig. 2A, bottom). Interestingly, H3K9ac displayed a more significant increase than H3ac (Fig. 2B). This was also true for a 14–16 h incubation of E14 cells with 5 nM of TSA (Fig. 2A–B). H3K9ac in R1 cells reached similar levels to that of E14 cells following TSA treatment, but since the initial levels were significantly higher (Fig. 1), the observed change was markedly lower. To rule out pleitropic effects, we further tested the levels of H3K9ac in E14 cells by western blots following 16 h of the HDACi valproic acid (VPA) and sodium butyrate (NaBu) (Fig. 2C). An increase was observed for VPA and almost no change for NaBu. We thus tested the reprogramming potency of E14 versus R1 ESCs before and after 5 nM of TSA or 0.5 mM of VPA. Whereas in R1 ESCs no significant change in the reprogramming capacity was observed, TSA treatment in E14 cells increased formation of reprogrammed colonies by about 5–6-fold (p < 0.05, Mann-Whitney U-test, p < 0.005, 2-tailed t-test; Table 1B), while 0.5 mM of VPA resulted in an increase from 1 to 13 colonies per 107 MEFs. In R1 ESCs, VPA increased the number of colonies from 10–15. These results demonstrate that increasing histone acetylation levels using HDACi increases the reprogramming capacity of less potent ESC lines.
Figure 2.
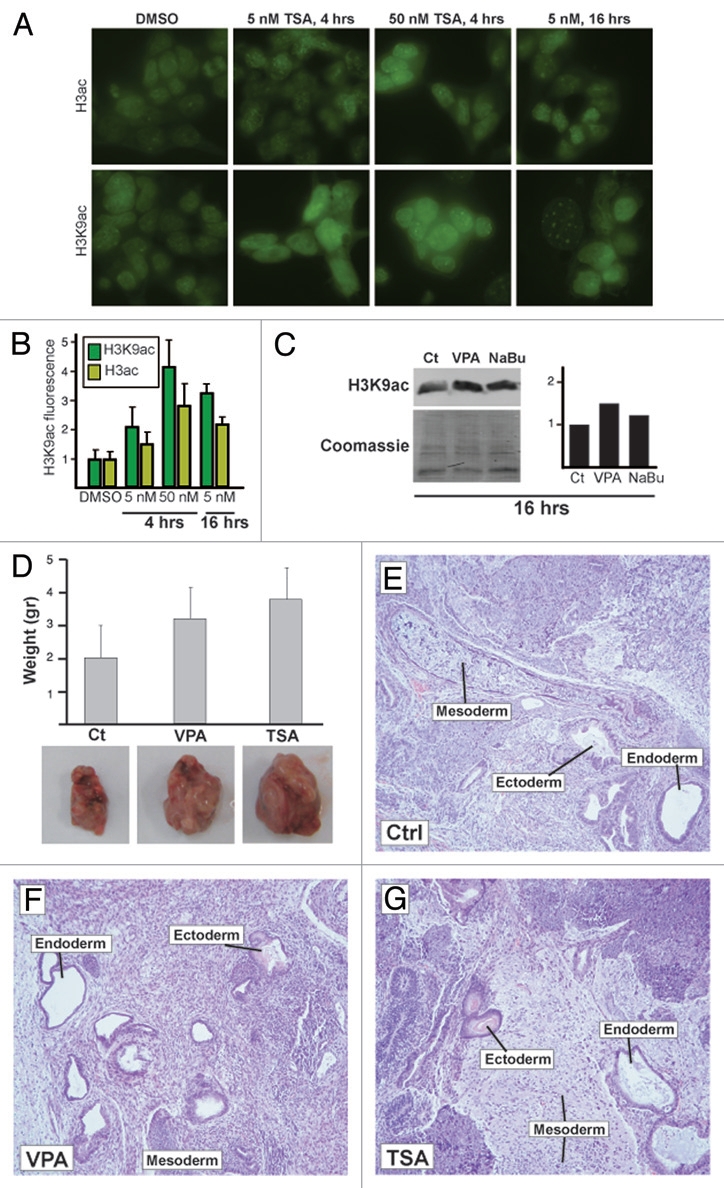
HDAC inhibitors increase acetylation and self-renewal. (A) E14 cells were treated with vehicle (DMSO, left) or with TSA for the indicated time, and histone acetylation levels were assayed using antibodies specific for pan-acetylated H3 (H3ac, top) and H3 acetylated on lysine 9 (H3K9ac, bottom). (B) Quantification of (A). (C) Western blots of H3K9ac in E14 ESCs treated with VPA or NaBu for 16 h. Quantification is shown on the right. (D) Increased acetylation of E14 ESC increases teratoma size in vivo. E14 ESC were treated with VPA (0.5 mM) or TSA (5 nM) for 14 h and transplanted under the kidney capsule in SCID mice. Teratomas were analyzed three weeks following transplantation. (E–G) Hematoxylin-Eosin staining of representative sections of teratomas formed by control E14 ESC (E), VPA-treated E14 ESC (F) and TSA-treated E14 ESC (G). All teratomas displayed evidence of endoderm, mesoderm and ectoderm.
Table 1B.
Summary of MEF/ESC fusion experiments
| Comparison of MEF/ESC hybrids formation before and after incubation with TSA | |||||||||
| Exp. 1 | Exp. 2 | Exp. 3 | |||||||
| ESC line | −TSA | +TSA | Ratio* | −TSA | +TSA | Ratio | −TSA | +TSA | Ratio |
| R1 | 16 | 16 | 1 | 22 | 24 | 1.1 | 18 | 20 | 1.1 |
| E14 | 4 | 24 | 6 | 6 | 21 | 3.5 | 4 | 19 | 4.75 |
Ratio of ESC before and after TSA treatment.
Numbers of GFP hybrid colonies/20 million MEFs.
We next tested the effects of HDACi on teratoma formation. We treated E14 ESCs with 5 nM TSA, 0.5 mM of VPA or DMSO for 14–16 h and injected the cells into the kidney capsule of SCID mice. We found that treatment of E14 ESCs with VPA and TSA caused an increase in the size of teratomas from 2.0 ± 0.9 gr to 3.2 ± 0.4 gr and 3.8 ± 0.97 respectively (p = 0.057 and 0.028, respectively, Mann-Whitney U-test; p < 0.05 when HDACi treated samples are regarded as one group). Histochemical studies demonstrated the presence of ectoderm, mesoderm and endoderm in TSA and VPA treated and untreated E14 ESC (Fig. 2D–G). This supports the notion that ESCs treated with low-level HDACi are more prone to self-renewal than differentiation.
To examine the molecular mechanism that underlies the increase in pluripotency following HDACi and subsequent increase in H3K9ac in E14 cells, we performed Affymetrix whole transcriptome microarrays before and after VPA in E14 cells (Fig. S3A). We used the same low level (0.5 mM) VPA treatment for 16 hrs and carried out the microarrays in duplicates. Unsupervised clustering showed that all samples appeared highly similar to each other (Fig. S3B), and only 36 genes overall displayed changes of over 1.5-fold or above (Fig. S3A, color dots). However, we nonetheless found a significant enrichment in the activation of genes associated with extracellular matrix (ECM) activity using GO classification (Fig. S3C). Among the upregulated genes we found Acan, Col11a1, Hmcn1, Itga8, Enpp1 and Plat. This suggests the intriguing possibility that ESCs secrete their own niche in the form of ECM to support their pluripotency and self-renewal. The microarray results were verified for several genes using qRT-PCR (Fig. S3D).
Genome-wide H3K9 acetylation profiles identifies the genomic regions associated with increased pluripotency.
Next, we performed chromatin immunoprecipitation (ChIP) followed by deep sequencing (ChIP-seq) of H3K9ac using SOLiD technology, before and after VPA (0.5 mM, 16 h). We sequenced similar amounts of input DNA as control. We used the CCAT algorithm,33 to identify enriched H3K9ac peaks in both untreated and VPA-treated E14 ESCs. A significant increase in the number of H3K9ac peaks was observed following HDACi. Interestingly, most of the peaks were aligned to non-annotated regions, suggesting a global increase in H3K9ac across the genome. However, out of ∼24,000 promoters, over 4,000 displayed significant enrichment in H3K9ac levels, suggesting that increased H3K9ac does not necessarily dictate a change in gene expression. Moreover, of the 36 genes which were significantly overexpressed following the 16 h incubation with VPA, only eight showed increased acetylation levels, once again, indicating that gene expression is not directly related to histone acetylation, at least after 16 h. Indirect mechanisms likely take place during this relatively prolonged period. Nonetheless, some of the ECM-related genes displayed both elevated RNA expression and elevated H3K9 acetylation (Fig. 3A), as well as elevated protein levels and cytoplasmic translocation observed by immunofluorescence in both the E14 and the MEF cells (Fig. S4). Additional examples of genes that gained H3K9 acetylation following VPA treatment (Fig. 3B) and intergenic regions with increased H3K9ac levels (Fig. 3C) are shown. These results delineate the genes and genomic regions in E14 cells that respond to HDACi by increasing H3K9ac. Since increased H3K9ac correlates with pluripotency, these regions may reflect the genomic regions that are involved with increased pluripotency.
Figure 3.
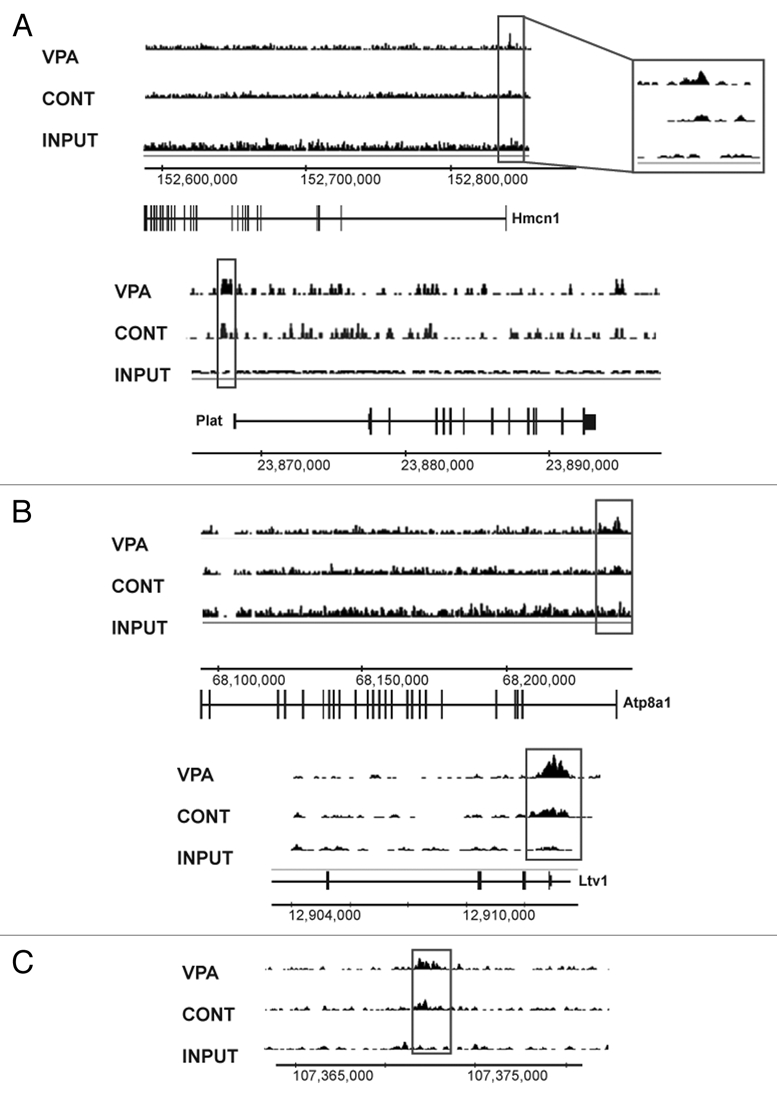
VPA increases H3K9ac across the genome. ChIP-seq for H3K9ac in E14 ESCs before (Cont, middle) and after (VPA, top) treatment of VPA (0.5 µM) for 16 h. Unprecipitated DNA was used as input control (Input, bottom). Chromosomal coordinates are shown at the bottom of each gene. Annotated genes above the coordinates denote left-to-right transcripts, annotation below the coordinates denote right-to-left transcripts. (A) H3K9ac profiles of two ECM-related overexpressed genes (Hmcn1, top; Plat, bottom) after VPA treatment. H3K9ac was increased around the transcription start sites of these two genes (gray boxes). (B) H3K9ac profiles for two genes (Atp8a1, top; Ltv1, bottom) which were among the most significantly enriched around the transcription start site for H3K9ac after VPA treatment. (C) H3K9ac profile for an intergenic region on mouse chromosome 17 which displayed a significant increase in H3K9 acetylation.
To test this hypothesis, we knocked down in R1 cells 2 ECM-related genes that displayed both elevated expression and elevated H3K9ac levels. We used Smartpools of siRNAs (Dharmacon) targeting Acan and Coll11a1 simultaneously. Intriguingly, we observed a mild but statistically significant decrease (p = 0.014) in OCT4 levels and a marked increase in NES (Nestin) levels (p < 0.01) in ∼50% of the colonies 54 h following knockdown compared with control Smartpool siRNAs (Fig. S5A), suggesting increased spontaneous differentiation. Real-time RT-PCR for Acan and Coll11a1 showed a 30–40% reduction in Acan and Coll11a1 RNA levels, likely reflecting an efficient knockdown of Acan and Coll11a1 in ∼50% of the colonies rather than a mild reduction in all colonies. We further repeated these experiments in the presence of VPA to test whether we would be able to rescue the spontaneous differentiation phenotype. In the presence of both siRNAs and VPA, ESCs also differentiated spontaneously although to a lesser extent (Fig. S5B). We further repeated these experiments in E14 cells and found a similar response to siRNA treatment of spontaneous differentiation (Fig. S5C). However, spontaneous differentiation was largely prevented when the E14 cells were also treated with VPA (Fig. S5D). These results imply that ESCs secrete their own pluripotency-promoting niche in the form of ECM, providing a potential explanation for the fact that ESCs remain largely undifferentiated when grown in compact colonies, whereas they exhibit spontaneous differentiation when grown individually or in sparse confluency.
H3K9 acetylation level correlates with Nanog expression.
Finally, in order to examine the relationship between H3K9ac and pluripotency markers, we examined H3K9ac and Nanog levels in R1 ESCs, in E14 ESCs and in fibroblasts (Fig. 4A). Double-staining demonstrated that high levels of H3K9ac predicted high levels of Nanog in the different ESCs tested. When H3K9ac and Nanog levels were correlated in the different cell types, we found a direct correlation between H3K9ac and Nanog (Fig. 4B). This may have important implications for therapy, when iPSCs become practical and for selecting superior ES and iPS cell lines for multiple purposes.
Figure 4.

H3K9ac predicts pluripotency. (A) R1 ESCs, E14 ESCs and MEFs were stained with DAPI (top), Nanog (green) and H3K9ac (red). The levels of Nanog and H3K9ac correlate well. (B) Correlation between Nanog levels (X-axis) and H3K9ac levels (Y-axis).
To sum, using an efficient fusion method that we developed, we found that ESC pluripotency is highly correlated with the acetylation state of these cells. Specifically, we showed that H3K9 histone acetylation level of ESC is directly correlated with the cells' reprogramming efficiency and pluripotency and that HDACi can restore pluripotency in lesser ESCs. Finally, we find that HDACi-increased pluripotency occurs through increased activity of ECM-related genes and we identified, using microarrays and ChIP-seq for H3K9 the genes and genomic regions that display increased acetylation when reverting to a more pluripotent state. Our data may help identify superior ESCs and iPSCs before cumbersome functional tests are conducted, may help restore lost pluripotency using histone-targeted drugs, and finally, may pave the way for the development of ECM-based optimal conditions to maintain pluripotency and self-renewal of ESCs and iPSCs.
Discussion
In this study, we established a method to produce hybrid cell lines by fusion of differentiated somatic cells (MEFs) with pluripotent ESC. By incubating the cells with activated baculovirus, we obtained high frequency fusion events resulting in a relatively high number of reprogrammed hybrids, which displayed expression and chromatin features of ESCs, as well as the capacity for multilineage differentiation.
The reprogramming potency of ESCs was evaluated by the number of hybrid colonies that were created after fusion. We showed that hybrid numbers from R1 ESC were about 5–10 fold higher than those from E14 cells. These significant differences suggest that the E14 ESC line lacks a certain capability that hinders its potency. Analysis of histone modifications demonstrated that low reprogramming potency was correlated exclusively with reduced H3 lysine 9 acetylation (H3K9ac) levels. Incubating E14 cells with HDAC inhibitors significantly increased H3K9ac levels, as well as their reprogramming capacity, to a level observed in R1 cells. Therefore the ability of ESCs to reprogram differentiated cells could reflect the pluripotency state of these cells and H3K9ac may serve as a marker for ESC pluripotency. The fact that different HDAC inhibitors (HDACi) resulted in a similar trend suggests that increased pluripotency is related to the elevated acetylation levels and not due to pleiotropic effects of the HDACi. By transplanting E14 treated cells with TSA or VPA into the kidney-capsule, we were able to show that high acetylation levels enable induction of ESC pluripotency in-vivo. The sizes of teratomas growth after HDACi treatments were significantly bigger than controls. These finding are in line with recent work by Huangfu et al. who showed that reprogramming of human fibroblasts can be increased by VPA treatment,37 and the finding of Ware et al. showing that TSA can support human ESC self-renewal.38 Supporting this notion, H3K9ac was markedly reduced upon differentiation of R1 ESC into neuronal progenitor cells (NPC) in a manner that was more pronounced than the reduction in the levels of pan-acetlyation of H3 and H4.31,35
The identification of an inferior ESC line with reduced H3K9 acetylation levels and our ability to rescue it with HDACi, allowed us to study the potential molecular mechanisms that underlie HDACi-mediated increased pluripotency. Interestingly, by gene ontology (GO) classification of our microarray data before and after low levels of HDACi, we found enrichment for extracellular matrix (ECM) activity. Although novel, these data are in line with several previous works that demonstrated ECM support of self-renewal and pluripotency of ESCs.39–41 Our findings suggest that ESCs themselves can secrete their own ‘self-renewing niche’. These findings may pave the way for improved conditions for self-renewal and pluripotency of ESCs and iPSCs.
Interestingly, using H3K9ac ChIP-seq before and after 16 h of low-level VPA treatment we were able to identify the genes and genomic regions that responded to HDAC inhibition. We found a pervasive, wide-spread increase in histone acetylation across the entire genome. Although gene expression changes were overall mild, increased histone acetylation may support pervasive low-level transcription across the entire genome as we previously observed in R1 ESCs.35 Whether pervasive transcription keeps an open chromatin conformation or whether it is a byproduct of open chromatin remains to be seen,42 but it may well go hand in hand with increased H3K9 acetylation level as observed in germ cells43 and human ESCs as well.44
Taken together, our data support the idea that pluripotency is associated with H3K9ac levels, and that HDACi may restore reprogramming efficiency and stem cell potency by promoting ECM activity.
Materials and Methods
Ethics statement.
The joint ethics committee (IACUC) of the Hebrew University and Hadassah Medical Center approved the study protocol for animal welfare. The Hebrew University is an AAALAC International accredited institute. All animal experiments were conducted in accordance with the Hebrew University's animal committee, ethical approval #IACUC:NS-09-11616-4.
Cells and cell culture.
In this study, we examined the reprogramming potency of several established murine ESC lines, including R1 (A. Nagy, Toronto, Canada), J1,28 C57BL/6 and E14 (E14Tg2a),29,30 all of which are male. E14 cells carry a mutated hypoxanthine/guanine phosphoribosyltransferase gene (HPRT). Selection medium for gain of HPRT function in hypoxanthine, aminopterin and thymidine (HAT) was used for the deficient derivative of E14 cells. ESC were grown on mitomycin-C treated, neomycin resistant primary embryonic fibroblasts (MEFs; Chemicon, Millipore Corporate), plated in plastic 60-mm tissue culture dishes (Millipore) and grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 15% fetal bovine serum (HyClone Laboratories), 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, 0.1 mM nonessential amino acids, 50 U/ml penicillin/50 µg/ml streptomycin (Gibco), and 1,000 U/ml leukemia inhibitory factor (LIF, Chemicon) (ESM).
MEFs were derived from 13.5 day-old Oct4/GFP transgene embryos (Hübner, 2003) × Rosa26; Pgk-neo (Jackson Laboratories) embryos (referred to as MEF-RoGoN) from which head, liver, heart, GI tract and limbs were removed. The remaining carcasses were washed three times in 1x Dulbecco's phosphate buffered saline (DPBS, Gibco), finely minced and incubated with 0.25% trypsin EDTA for 15 min at 37°C. Standard embryonic fibroblast media (EFM) consisting of Dulbecco's modified Eagle's medium (Gibco), 10% fetal bovine serum (HyClone Laboratories) and 50 U/ml penicillin, 50 µg/ml streptomycin (Gibco) was added and the cells were allowed to settle for 10–15 min in 37°C and plated on 100 mm dishes. The new lines of MEFs were passaged two to three times and then used for fusion experiments.
Fusion procedure.
One day prior to fusion, 5 × 106 ESC were plated on 100 mm tissue culture dishes previously coated with 0.1% gelatin (Chemicon) in ESM with 1 mg/ml of Nocodazole (Sigma; Sigma Aldrich). Sixteen hours later the drug was removed and 1 × 106 MEF's were plated. One hour later the medium was removed and the plates were washed with 1x Hanks Balanced Salt Solution (Cellgro; Mediatech Inc.). The cells were incubated with 2 ml of high titer Baculovirus (Invitrogen) for 5 min. 1x DPBS (pH 4.9) was added for 1 min prior to the addition of ESM. Twenty-four hours later selection medium containing G418 at a final concentration of 350 µg/ml (Cellgro) and hypoxanthine-aminopterin-thymidine (HATx1; Invitrogen) was supplemented. Fusion efficiency was determined by staining the ESC with fluorescent lipids PKH26 red (Sigma). The numbers of red-stained MEFs were recorded 10–15 min after fusion. For treatment with TSA (Biomol) and Valproic acid (VPA, Sigma Aldrich), ESC were incubated for 24 h at a final concentration of 5–25 nM and 0.5 mM, respectively. C57BL/6 MEF lines were made resistant to hygromycin by the introduction of the plasmid pDsRed-Hyg (Clontech, 632513) by electroporation using Nucleofector device (Amaxa Biosystems). Hybrid colonies expressing GFP emerged 8–12 d after fusion and were observed under the Leica DMI4000 B microscope (Leica Microsystems Inc.). These colonies were picked, briefly trypsinized with 0.25% trypsin EDTA (Gibco), seeded into 12 wells dish on mitomycin-C treated feeders and grown in ESM. Growing colonies were passaged, expanded and frozen according to standard ESC culture methods.
Embryoid body (EB) formation.
ES cells grown in 60 mm dishes were trypsinized, resuspended in ESM and plated again for 15–30 min, so that the majority of the feeders would attach. For preparation of EBs, 2 × 106 ES cells were plated onto fibroblast-free 90 mm low cell binding Petri dishes (Fisher scientific Inc., Pittsburgh, PA, www.fisherscientific.com) containing 10 ml of ESM in the absence of LIF (ATCC embryoid body formation protocol, www.atcc.org). The medium was replaced every day. EBs were collected after 5, 10 and 15 d for RT-PCR assays.
Karyotype analysis.
Cells were arrested in metaphase by adding colcemid (20 µg/ml, Gibco) in ESC medium for 2–4 h at 37°C, 5% CO2. The cells were dispersed by trypsin treatment and collected by centrifugation. The colcemid-treated cells were washed in DPBS, resuspended for 6 min at room temperature in 5 ml of 0.075 M KCl (Sigma) in water, added dropwise and fixed in 3:1 methanol/acetic acid. The fixed cells were dropped on pre-cleaned Superfrost/plus Microscopic slides (Fisher Scientific, 12-550-15) and air dried to create chromosome spreads. Slides were stained with DAPI (DAPI-Vectashield, Vector Laboratories Inc.) and were examined under an epi-fluorescent microscope.
Reverse transcription-polymerase chain reaction (RT-PCR).
Transgenic MEF lines of the Oct4/GFP/neoR/ROSA26-β-geo genotypes were identified by polymerase chain reaction (PCR) on DNA samples with the specific primers (see Table S1). Differentiation of ES, and hybrid cells through embryoid body (EB) formation was carried out following the published ATCC protocol. Total RNAs from ES, EBs and MEF cells was prepared as described in the RNeasy Mini Kit (Qiagen, Inc.). One microgram total RNA from each sample was used for Oligo(dT)20-primed reverse transcription (RT), carried out as described in the product protocol (SuperScript™ III First-Strand Synthesis System for RT-polymerase chain reaction; Invitrogen Corporation). The PCR reactions were performed using PCR Master Mix (Roche Diagnostics) under the following conditions: 95°C for 5 min, followed by 30–35 cycles of 95°C for 30 sec, 55–60°C for 30 sec and 72°C for 45 sec; and 1 cycle of 72°C for 7 min. RT-PCRs for somatic cells and ESC markers were preformed with the specific primer pairs (Table S1). RT-PCRs for EBs were preformed with specific primers for ectoderm (Fgf5, Pax6), mesoderm (PECAM, Myh6), endoderm (Sox17, α-FP) and GAPDH as control (Table S1).
Antibodies and immunofluorescence.
Antibodies (Millipore) used in this study include H4ac (06-598), H3K9ac (06-942), H3K4me3 (05-745), H3K27me3 (07-449), H3K9me3 (07-442), HP1α (05-689) and phosphoS5 of RNA Pol-II (Euromedex). For immunofluoresence, cells were grown on gelatinized coverslips and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were washed X3 with PBS, permeabilized with 0.5% Triton X-100 in PBS and washed in PBS. Coverslips were incubated with primary antibodies diluted 1:100–1:400 in PBS for 1 h at room temperature, followed by X3 washes in PBS and incubation with secondary antibodies diluted 1:200 in PBS. Detection was with anti-rabbit or anti-mouse antibodies conjugated to either Texas red or FITC (Jackson ImmunoResearch). Fluorescent images were taken with a NIKON Eclipse TE-2000-E inverted microscope with a 60x 1.4 NA lens. Image analysis and signal quantification was done using NIS-Elements software (Nikon).
Teratoma formation.
To test teratoma formation, severe immune-depleted mice (SCID) were anesthetized and cut open narrowly above the left kidney. The left kidney was exposed, the kidney capsule was punctured using a blunt needle and 300,000 cells, suspended in 100 µl of PBS, were injected into the kidney capsule. The cut was surgically stitched and mice were placed back in their original cages. Three weeks later, mice were sacrificed and teratomas were surgically removed.
Microscopy and photobleaching.
Photobleaching experiments were carried out using a spinning disk confocal microscope (Yokogawa Electric Corporation, CSU22) equipped with a photobleaching (FRAPPA) module and an iXon DU-897-BV monochrome EMCCD camera. A 50 mW solid-state 488 nm laser module at maximum power was used for photobleaching. FRAP and data analysis was performed as described in reference 31.
ChIP-seq.
Chromatin was cross-linked with 1% formaldehyde (10 min, 37°C), incubated with glycine (125 mM, 10 min, room temp.) and washed with cold PBS. Cells were then scraped in cold PBS and pelleted at 700 g at 4°C (5 min). Pellet was washed with buffer I (0.25% Triton, 10 mM EDTA pH 8, 0.5 mM EGTA pH 7.5, 10 mM Hepes pH 7.5), incubated on ice (10 min), washed with buffer II (0.2 M NaCl, 1 mM EDTA pH 8, 0.5 mM EGTA pH 7.5, 10 mM Hepes pH 7.5) and incubated on ice again (10 min). Pellet was then resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA pH 8, 50 mM Tris pH 8.1) at a concentration of 2 × 107 cells/ml, incubated in −20°C over night and sheared to 400–600 bp using a Bioruptor sonicator (Diagenode). Each sample was sonicated for 14 min on ice. Fifty microliters of the lysate was saved as “input” for later normalization. Equal amounts of sonicated chromatin were diluted 10-fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl) to a final volume of 1.5 ml. The chromatin solution was pre-cleared with 40 µl of salmon sperm DNA/protein A-agarose 50% gel slurry (Upstate, Millipore, 16-157) for 30 min at 4°C and immunoprecipitated overnight at 4°C with 1 µg of H3K9ac rabbit antibody (Upstate, 06-942). Chromatin-antibody complexes were isolated by incubation with 60 µl protein A-agarose beads while rocking at 4°C for 2 h. The beads were harvested and sequentially washed for 3 min on a rotating platform with 1 ml of each of the following buffers: low salt immune complex wash buffer (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris, pH 8.1, 15 mM NaCl), high salt immune complex buffer (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris, pH 8.1, 500 mM NaCl), LiCl immune complex buffer (0.25 M LiCl, 1% NP-40, 1% Deoxycholic acid, 1 mM EDTA, 10 mM Tris, pH 8.1) and twice with TE (10 mM Tris, 1 mM EDTA). Chromatin-antibody complexes were eluted from the protein A-agarose beads by the addition of 500 µl elution buffer (1% SDS, 0.1 M NaHCO3). Cross-linking was reversed by incubation of the eluted samples for 4 h at 65°C under high-salt conditions. Proteins were digested using proteinase K treatment for 1 h at 45°C. The DNA was extracted with phenol-chloroform, precipitated with ethanol and dissolved in TE. Preparation of sequence libraries and SOLiD sequencing were performed according to the manufacturer's instructions and kits (Applied Biosystems) at the Genomics Center at the Life Sciences Institute, The Hebrew University of Jerusalem. Sequences were aligned to the genome using Bowtie32 and peaks were calculated using CCAT.33
Accession numbers.
Microarray and ChIP-seq data have been deposited in Gene Expression Omnibus (GEO), accession numbers GSE23956 and GSE24211, respectively.
Acknowledgments
The authors would like to thank Asher Meshorer for histopathological analyses and Evgenia Leikina for technical assistant with the fusion protocols and for helpful comments. This work was supported by funds from the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development/NIH/HHS, the Israel Science Foundation (215/07 and 943/09) (to E.M.); the Israel Ministry of Health (6007) (to E.M.), the EU (IRG-206872 and 238176) (to E.M.), the ICRF (to A.M. and E.M.), The HUJI Internal Applicative Medical Grants (to E.M.), the Israeli Psychobiology Institute (to E.M.) and an Alon Fellowship (to E.M.). E.M. is a Joseph H. and Belle R. Braun Senior Lecturer in Life Sciences. H.H. is an Edmond J. Safra Fellow.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Han DW, Do JT, Gentile L, Stehling M, Lee HT, Scholer HR. Pluripotential reprogramming of the somatic genome in hybrid cells occurs with the first cell cycle. Stem Cells. 2008;26:445–454. doi: 10.1634/stemcells.2007-0553. [DOI] [PubMed] [Google Scholar]
- 2.Westphal H. Restoring stemness. Differentiation. 2005;73:447–451. doi: 10.1111/j.1432-0436.2005.00034.x. [DOI] [PubMed] [Google Scholar]
- 3.Gurdon JB, Elsdale TR, Fischberg M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 1958;182:64–65. doi: 10.1038/182064a0. [DOI] [PubMed] [Google Scholar]
- 4.Campbell PA, Perez-Iratxeta C, Andrade-Navarro MA, Rudnicki MA. Oct4 targets regulatory nodes to modulate stem cell function. PLoS ONE. 2007;2:553. doi: 10.1371/journal.pone.0000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 6.Jeanisch R, Eggan K, Humpherys D, Rideout W, Hochedlinger K. Nuclear cloning, stem cells and genomic reprogramming. Cloning Stem Cells. 2002;4:389–396. doi: 10.1089/153623002321025069. [DOI] [PubMed] [Google Scholar]
- 7.Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol. 2001;11:1553–1558. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 8.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–1373. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 9.Matveeva NM, Shilov AG, Kaftanovskaya EM, Maximovsky LP, Zhelezova AI, Golubitsa AN, et al. In vitro and in vivo study of pluripotency in intraspecific hybrid cells obtained by fusion of murine embryonic stem cells with splenocytes. Mol Reprod Dev. 1998;50:128–138. doi: 10.1002/(SICI)1098-2795(199806)50:2<128::AID-MRD2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 10.Pells S, Di Domenico AI, Gallagher EJ, McWhir J. Multipotentiality of neuronal cells after spontaneous fusion with embryonic stem cells and nuclear reprogramming in vitro. Cloning Stem Cells. 2002;4:331–338. doi: 10.1089/153623002321025005. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan S, Pells S, Hooper M, Gallagher E, McWhir J. Nuclear reprogramming of somatic cells by embryonic stem cells is affected by cell cycle stage. Cloning Stem Cells. 2006;8:174–188. doi: 10.1089/clo.2006.8.174. [DOI] [PubMed] [Google Scholar]
- 12.Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416:545–548. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- 13.Flasza M, Shering AF, Smith K, Andrews PW, Talley P, Johnson PA. Reprogramming in inter-species embryonal carcinoma-somatic cell hybrids induces expression of pluripotency and differentiation markers. Cloning Stem Cells. 2003;5:339–354. doi: 10.1089/153623003772032844. [DOI] [PubMed] [Google Scholar]
- 14.Kimura H, Tada M, Nakatsuji N, Tada T. Histone code modifications on pluripotential nuclei of reprogrammed somatic cells. Mol Cell Biol. 2004;24:5710–5720. doi: 10.1128/MCB.24.13.5710-5720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tada M, Morizane A, Kimura H, Kawasaki H, Ainscough JF, Sasai Y, et al. Pluripotency of reprogrammed somatic genomes in embryonic stem hybrid cells. Dev Dyn. 2003;227:504–510. doi: 10.1002/dvdy.10337. [DOI] [PubMed] [Google Scholar]
- 16.Yu J, Vodyanik MA, He P, Slukvin II, Thomson JA. Human embryonic stem cells reprogram myeloid precursors following cell-cell fusion. Stem Cells. 2006;24:168–176. doi: 10.1634/stemcells.2005-0292. [DOI] [PubMed] [Google Scholar]
- 17.Do JT, Scholer HR. Nuclei of embryonic stem cells reprogram somatic cells. Stem Cells. 2004;22:941–949. doi: 10.1634/stemcells.22-6-941. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keskintepe L, Norris K, Pacholczyk G, Dederscheck SM, Eroglu A. Derivation and comparison of C57BL/6 embryonic stem cells to a widely used 129 embryonic stem cell line. Transgenic Res. 2007;16:751–758. doi: 10.1007/s11248-007-9125-8. [DOI] [PubMed] [Google Scholar]
- 21.Wu H, Kim KJ, Mehta K, Paxia S, Sundstrom A, Anantharaman T, et al. Copy number variant analysis of human embryonic stem cells. Stem Cells. 2008;26:1484–1489. doi: 10.1634/stemcells.2007-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lachner M, O'Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 23.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goll MG, Bestor TH. Histone modification and replacement in chromatin activation. Genes Dev. 2002;16:1739–1742. doi: 10.1101/gad.1013902. [DOI] [PubMed] [Google Scholar]
- 25.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 26.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 27.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 28.Rideout WM, 3rd, Wakayama T, Wutz A, Eggan K, Jackson-Grusby L, Dausman J, et al. Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet. 2000;24:109–110. doi: 10.1038/72753. [DOI] [PubMed] [Google Scholar]
- 29.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 30.Thompson S, Clarke AR, Pow AM, Hooper ML, Melton DW. Germ line transmission and expression of a corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell. 1989;56:313–321. doi: 10.1016/0092-8674(89)90905-7. [DOI] [PubMed] [Google Scholar]
- 31.Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu H, Handoko L, Wei X, Ye C, Sheng J, Wei CL, et al. A signal-noise model for significance analysis of ChIP-seq with negative control. Bioinformatics. 2010;26:1199–1204. doi: 10.1093/bioinformatics/btq128. [DOI] [PubMed] [Google Scholar]
- 34.Shoji I, Aizaki H, Tani H, Ishii K, Chiba T, Saito I, et al. Efficient gene transfer into various mammalian cells, including non-hepatic cells, by baculovirus vectors. J Gen Virol. 1997;78:2657–2664. doi: 10.1099/0022-1317-78-10-2657. [DOI] [PubMed] [Google Scholar]
- 35.Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoto T, Saitoh N, Ichimura T, Niwa H, Nakao M. Nuclear and chromatin reorganization in the MHC-Oct3/4 locus at developmental phases of embryonic stem cell differentiation. Dev Biol. 2006;298:354–367. doi: 10.1016/j.ydbio.2006.04.450. [DOI] [PubMed] [Google Scholar]
- 37.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ware CB, Wang L, Mecham BH, Shen L, Nelson AM, Bar M, et al. Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell. 2009;4:359–369. doi: 10.1016/j.stem.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee ST, Yun JI, Jo YS, Mochizuki M, van der Vlies AJ, Kontos S, et al. Engineering integrin signaling for promoting embryonic stem cell self-renewal in a precisely defined niche. Biomaterials. 2010;31:1219–1226. doi: 10.1016/j.biomaterials.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 40.Meng G, Liu S, Li X, Krawetz R, Rancourt DE. Extracellular matrix isolated from foreskin fibroblasts supports long-term xeno-free human embryonic stem cell culture. Stem Cells Dev. 2010;19:547–556. doi: 10.1089/scd.2009.0303. [DOI] [PubMed] [Google Scholar]
- 41.Nur EKA, Ahmed I, Kamal J, Schindler M, Meiners S. Three-dimensional nanofibrillar surfaces promote self-renewal in mouse embryonic stem cells. Stem Cells. 2006;24:426–433. doi: 10.1634/stemcells.2005-0170. [DOI] [PubMed] [Google Scholar]
- 42.Efroni S, Melcer S, Nissim-Rafinia M, Meshorer E. Stem cells do play with dice: a statistical physics view of transcription. Cell Cycle. 2009;8:43–48. doi: 10.4161/cc.8.1.7216. [DOI] [PubMed] [Google Scholar]
- 43.Khromov T, Pantakani DV, Nolte J, Wolf M, Dressel R, Engel W, et al. Global and gene-specific histone modification profiles of mouse multipotent adult germline stem cells. Mol Hum Reprod. 2011;17:166–174. doi: 10.1093/molehr/gaq085. [DOI] [PubMed] [Google Scholar]
- 44.Krejci J, Uhlirova R, Galiova G, Kozubek S, Smigova J, Bartova E. Genome-wide reduction in H3K9 acetylation during human embryonic stem cell differentiation. J Cell Physiol. 2009;219:677–687. doi: 10.1002/jcp.21714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.