

Abstract
In non-dividing cells, the nuclear pore complex provides the major route for viruses and viral genomes to enter the nucleus. However, SV40 infection of non-dividing cells is very inefficient suggesting that the nuclear envelope prevents most viral genomes from entering the nucleus. Surprisingly, we observed that following infection of quiescent CV-1 cells with SV40, the nuclear envelope was dramatically deformed, as seen by immunohistochemistry detection of lamins A/C, B1, B2 and the nuclear pore complexes. Accompanying deformation of the nuclear envelope, we also observed fluctuations in the levels of lamin A/C, dephosphorylation of an unknown epitope on lamin A/C and accumulation of lamin A in the cytoplasm. The nuclear envelope deformations occured just prior to and during nuclear entry of the viral genome and were transient and the spherical structure of the nuclear envelope was restored subsequent to nuclear entry. Nuclear envelope deformation and lamin A/C dephosphorylation required caspase-6 cleavage of a small fraction of lamin A/C. Taken together the results suggest that virus-induced alterations of the nuclear lamina, are involved in the nuclear entry of the SV40 genome in non-dividing cells. We propose that SV40 utilize this unique, previously unknown mechanism for direct trafficking of its genome from the ER to the nucleus.
Keywords: nuclear envelope, lamina, lamin A/C, SV40, nuclear entry, caspase-6
Introduction
Viruses have adopted multiple mechanisms to insert their genomes into the nuclei of infected cells. Some, like retroviruses, infect only dividing cells. Their reverse-transcribed DNA associates with cellular chromatin during mitosis when the nuclear envelope (NE) is disassembled. The viral chromosome of other viruses, including Herpes, adeno and hepatitis viruses use the nuclear pore complex for nuclear entry, while the viral capsid remains in the cytoplasm. The route of nuclear entry of polyomaviruses, following their disassembly in the ER, is mostly unknown.
Three members of the polyomavirus family are known human pathogens: JC virus, BK virus and the recently identified Merkel cell polyomavirus, MCPyV.1 JC may cause progressive multifocal leukoencephalopathy,2 BK activation during renal transplant leads to rejection of the transplanted kidney3 and MCPyV is suspected as an emerging pathogen in Merkel cell carcinoma.1,4 The related polyomavirus, SV40, has been a valuable tool for molecular genetics and cell biology research for many years and has been a model for investigations into mammalian gene regulation, DNA replication and transcription.
Polyomaviruses are capable of infecting dividing as well as non-dividing cells. They share an unusual cell entry mechanism via binding to glycolipid molecules in caveolar/lipid raft domains at the plasma membrane.5,6 During the first 3–6 h of infection, the polyomavirus traffics via the endosomal pathway to the ER,7 where disassembly occurs at 5–6 h post-infection. The viruses utilize the cellular machinery for intracellular trafficking,8 and ER chaperones for disassembly.6,9 The 5–5.5 kb viral genome enters the nucleus complexed with nucleosomes in a chromatin-like structure termed the minichromosome. It has been suggested that the viral inner capsid proteins VP2/3 accompanies the viral genome to the nucleus by classical nucleocytoplasmic transport, via the nuclear pore.10 Following DNA entry into the nucleus, transcription of the early region is initiated, leading to the appearance of T-antigen at 12-14 h post infection.11 Infection is rather inefficient with only 1 in 100–200 DNA-containing virion particles able to lead to a productive infection12 (and our data). We have recently observed that efficiency of infection varies between different cell types.13 This may be due to variations in cellular factors employed by the infecting virus.
The NE is a complex organelle that forms a semi-permeable barrier between the cytoplasm and nucleus. Macromolecular traffic across the NE occurs through large proteinaceous channels called nuclear pore complexes (NPCs), which occupy pores formed by fusion of the outer and inner nuclear membranes. The NPCs are anchored to a protein meshwork that underlies the inner membrane composed of the type V intermediate filament proteins, nuclear lamins. The lamins, which are classified into two types, the A-type (lamins A and C) and B-type lamins (lamins B1 and B2), maintain the mechanical stability and shape of the nucleus.14 Each type of lamin forms a separate meshwork in the lamina, but the A- and B-type meshworks also extensively interact.15 In addition to their role in nuclear membrane structure, lamins play important functions in DNA replication, RNA transcription and epigenetic modification of chromatin.16 Lamins are also key factors in the disassembly of the nucleus when cells enter mitosis, in the assembly of the mitotic spindle17 and in reassembly of daughter nuclei following cell division.18
In the present study we found that infection of non-dividing cells leads to extensive deformation of NE structure. The changes in NE structure occur just prior and during nuclear entry of the viral genome and are coincident with changes in the phosphorylation state of the A-type lamins and their structural integrity, suggesting that nuclear trafficking of the polyomavirus genome is more complex than previously recognized.
Results
The nuclear envelope restricts genome entry to the nucleus.
In order to gain a better understanding of SV40 minichromosome entry into the nucleus, we applied Fluorescence In Situ Hybridization (FISH) analysis to localize viral DNA at various times following infection. CV1 cells were infected at a multiplicity of infection (moi) of 2 plaque-forming units (pfu) per cell to avoid high DNA background. It has previously been reported that there are 100–200 DNA-containing virus particles for each pfu.12 In our own experiments, comparing infective units of purified viral preparations to DNA content (measured by quantitative PCR), we measured 300–500 DNA-containing particles per pfu in different virus stocks. A representative experiment using a fluorescently labeled DNA probe is shown in Figure 1A. Most of the viral DNA was found outside the nucleus and remained so at 36 h post infection, well into the late phase of the viral life cycle. These results were supported by probing with Qdot-streptavidin conjugates (Fig. 1B) that provided higher resolution and sensitivity. Q dots are too large to penetrate the cell, therefore, nuclei were isolated by hypotonic lysis in 0.5% KCl in the absence of detergents to preserve integrity of the outer nuclear membrane and ER. An moi of 0.5 pfu/cell was used to reduce the DNA background. The increased sensitivity and higher resolution of this procedure revealed SV40 replication foci in the nucleus at 20 h post infection and assembly foci at 55 h (Fig. 1B). We concluded from these results that the NE hinders nuclear entry of the viral genome.
Figure 1.

The nuclear envelope is a barrier to SV40 infection. (A) CV1 infected cells (moi 2) were fixed and hybridized with a dUTP-biotin labeled probe at the designated time after infection. FISH detection was performed with avidin conjugated to FITC and viewed by fluorescence microscopy. (B) Nuclei were isolated from cells infected at moi 0.5, the dUTP-biotin labeled SV40 probe was detected with streptavidin-conjugated Qdot 565 and viewed by fluorescence microscopy. Each experiment was repeated twice. No hybridization of labeled probe was detected in mock-infected cells. The images were adjusted using Adobe Photoshop CS2, increasing brightness by 15%. (C) Logarithmic (at 40–60% confluence, dashed line) and quiescent (at 100% confluence, solid line) cultures of CV1 cells were infected at several moi's. The cells were harvested 48 h later, fixed, stained for T-ag and analyzed by FACS for the percent of T-ag positive cells. The graphs represent averages of 4 experiments and the error bars represent standard deviation. (D) HEK-293 cells with ectopically expressed Lamin A-GFP were treated with lamin-specific shRNA, as designated. The protein gel blot shows the degree of silencing of both ectopic and endogenous lamin A [antibody 323, RD Goldman]. (E) HEK-293 cells (lane 5) and cells with ectopically expressed lamin A-GFP (lanes 1–4) were infected with SV40 (lane 2–5). Infections were performed following treatment with lamin A/C shRNA (lane 4) or with scrambled shRNA (lane 3). After 24 h, total extracts were analyzed by protein gel blotting for T-ag expression. The experiment was repeated twice.
If the NE were a barrier to nuclear entry, one would expect that proliferating cells would be more susceptible to SV40 infection than quiescent cells. Indeed we consistently found that proliferating cells were infected at a significantly higher efficiency than quiescent cells as assessed by the percent of T-antigen expressing cells (Fig. 1C), an established assay for SV40 infectivity.13 Supporting results were obtained by protein gel blotting analysis of T-ag level (not shown).
In another approach we used HEK-293 cells, which express very low levels of lamin A/C. The level of lamin A may be increased in these cells by overexpression from a cDNA and decreased by shRNA silencing. CV1 cells were not appropriate for these experiments because lamin A/C silencing with the human targeted shRNA was not effective, presumably due to species difference. In HEK-293 cells, both ectopically expressed GFP-tagged lamin A and endogenous lamin A were silenced (Fig. 1D). Silencing efficiency was highest (80–90%) at 5 d post transfection, at which time the cells were infected with SV40. T-ag expression increased dramatically, up to ∼7 fold, following lamin A/C silencing, as estimated by quantification of the signal by densitometry (Fig. 1E, lane 4). The control cells treated with scrambled shRNA sequence (lane 3) showed the same level of T-ag as cells overexpressing lamin A (lane 2). T-ag expression was diminished ∼2 fold in cells with lamin A overexpression (Fig. 1E, compare lane 5 to lane 2). These data demonstrate that the NE is a barrier to nuclear entry of the SV40 minichromosome and that this barrier function is mediated, in part, by lamin A/C levels.
Viral DNA enters the nucleus at 8 h post infection.
Although the NE is a barrier to viral minichromosome entry into the nucleus, non-dividing quiescent cells are still susceptible to infection. This suggests the existence of a viral nuclear entry mechanism that does not require NE breakdown. In order to investigate this mechanism, it was necessary to establish the timing of viral genome entry into the nucleus. To that end, we measured the amounts of viral DNA in nuclei isolated from infected cells by quantitative PCR. For these experiments we have used the COS-1 cell-line, a CV1 derivative that expresses the viral T-antigen constitutively,19 since these cells support SV40 DNA replication as soon as the viral minichromosome enters the nucleus.
To synchronize the infection, virus adsorption was performed at 4°C in serum-free medium, for 1 h. Medium supplemented with serum was then added to the culture (time 0) and infection was carried on at 37°C. Using this assay SV40 DNA was detected in the nuclei around 8 h after infection (Fig. 2), followed by a sharp increase in its level. The increase was due to DNA replication, as judged by its sensitivity to the α-DNA polymerase inhibitor aphidicolin20 (Fig. S1). The timing of nuclear entry of the genome at ∼8 h is consistent with studies showing that SV40 disassembly occurs at 5–6 h post infection9 and T-ag is first seen after 12 h.11
Figure 2.
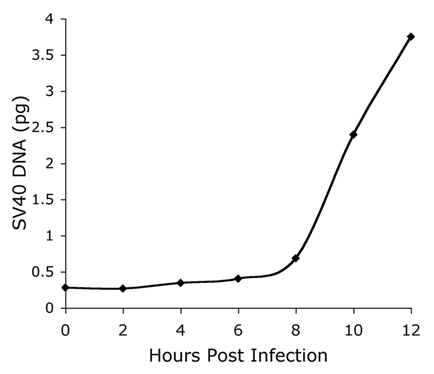
Timing nuclear entry of the SV40 genome. Nuclei of SV40-infected COS-1 cells (at a moi 50) were collected at the indicated time points after infection. The cells were washed and nuclei were isolated as described in Materials and Methods. The nuclear pellet was sonicated and the amount of SV40 DNA was determined by quantitative real-time PCR with SV40-specific primers. SV40 DNA served as a known standard for quantification.
Fluctuations in lamin A/C level following SV40 infection of quiescent CV1 cells.
Protein gel blot analyses of lamins in SV40-infected quiescent CV1 cells showed a transient reduction in the level of both lamins A and C at 6 h post-infection. The reduction in lamin A was observed with 4 antibodies, 2 monoclonal and 2 polyclonal (Fig. 3A). Similar results were obtained in experiments with additional antibodies, one monoclonal and one polyclonal (not shown).
Figure 3.
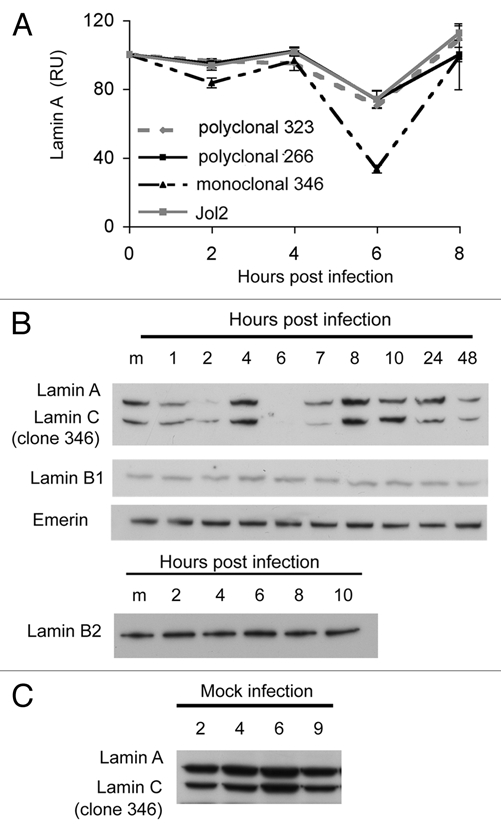
Dynamic fluctuations of lamin A/C level following SV40 infection. Total cell extracts of infected quiescent CV1 cells, infected at a moi of 10 were analyzed by protein gel blotting. (A) Lamin A/C was analyzed by protein gel blotting and detected with four different antibodies, as designated. Intensity of the bands was quantified by densitometry using Kodak Molecular Imaging software and normalized according to intensity of the lamin B1 signal in each lane. Each line in the graph represents an average of at least three experiments. (B) Protein gel detection was performed with the following antibodies: monoclonal lamin A/C (346), lamin B1 (M-20), lamin B2 and emerin. Lamin B1 and emerin were detected on the same membrane as lamin A/C. (C) Mock infected cells treated in parallel to the infection. As mammalian lamins A and C are derived from a single gene (LMNA) by alternative splicing,46 many antibodies recognize both isoforms.
The fluctuations in protein level were specific to lamins A and C and were not observed for lamin B1, lamin B2 and emerin (Fig. 3B). Interestingly, the most dramatic decrease in lamin A/C levels was detected by a monoclonal antibody (antibody 346—against an unknown epitope) during the first 10 h following virus adsorption (Fig. 3B). The epitope detected by this antibody was reduced at 2 h, returned to a normal level at 4 h, completely disappeared at 6 h (just prior to nuclear entry of the SV40 DNA) and reappeared at 8 h. These results suggested that in addition to fluctuations in the protein level, detected by the 2 polyclonal and one monoclonal antibodies (Fig. 3A), the virus altered the post-translational modification of lamins A and C. Further experiments (see below) demonstrated that antibody 346 detects phosphorylation of a specific, as yet unidentified, epitope. These changes in lamin A/C level or modification were not seen in mock-infected cells (Fig. 3C), confirming that they were induced by the infecting virus.
In contrast to these results, no changes in lamin levels were observed during infections of proliferating CV1 cells (Fig. S2). We conclude that the changes in lamin proteins are specific to lamins A and C and to SV40 infection of G1-arrested, quiescent cells. In subsequent studies we have exclusively used quiescent cells.
Fluctuations in lamin A/C levels correlate with changes in nuclear morphology.
Examination of nuclear structure by immunofluorescence with antibodies to lamins and nucleoporins revealed dramatic changes in nuclear shape during the first 8 h of infection. As early as 2 h post adsorption, the nucleus became misshapen with one or more areas of indentation or folding of the nuclear envelope (Fig. 4A). Examination of reconstructed Z-stack images showed that the folds were comprised of small invaginations (Fig. S3). Staining for lamin A/C, lamin B1 and lamin B2 were consistently similar, following the same contours of deformation (Fig. S4A). As an additional marker for the NE, and to eliminate the possibility that the deformations were driven by depletion or migration of NPCs, we localized NPCs with MAb414, which detects a family of O-glycosylated nucleoporins. NPCs followed the same pattern as the lamins at the peak of nuclear deformation (Fig. 5B and C). The extent of nuclear deformation peaked at 6 h post infection in parallel with the disappearance of the lamin A/C epitope detected by antibody 346 (Fig. 3A and B). In many experiments, the nuclei also appeared larger during the first hours of infection, which may be due to flattening of the nuclei.
Figure 4.
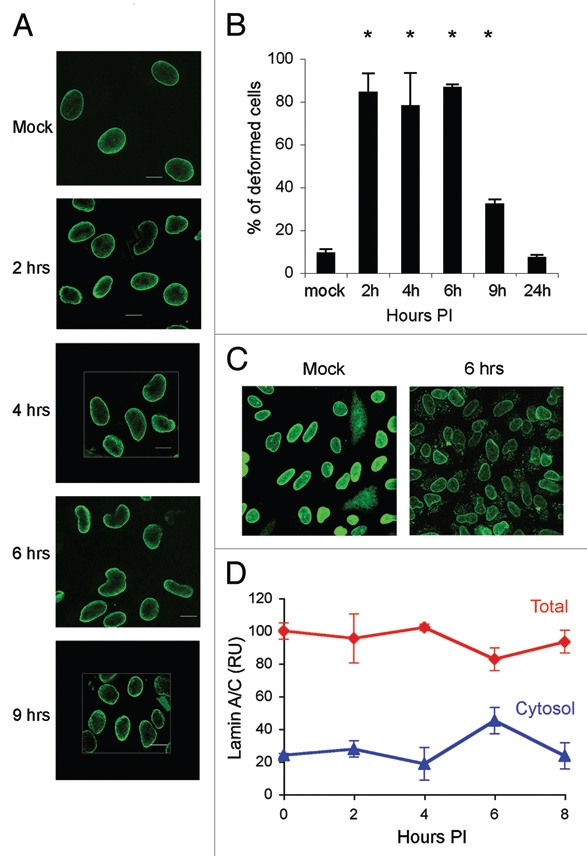
Fluctuations in lamin A/C levels studied by immunofluorescence. (A) Confocal microscopy images of CV1 cells at 2, 4 and 6 or 9 h following SV40 infection. The cells were fixed and stained with monoclonal lamin A/C antibody (Jol2). Cross section images were taken with a confocal Carl Zeiss Laser Scanning Systems LSM 510, magnification X60, zoom 2. (B) Frequency of cells with deformed nuclei. Cells were scored, double-blind, in 5 different experiments. In two experiments the cells were stained with the lamin A/C monoclonal antibody Jol2 and in the other three with the polyclonal antibody 266. Statistical analysis using the 1-tailed t-test indicated that the number of cells with deformed nuclei in the infected cell at 2, 4, 6 and 9 h was significant at p ≤ 0.05. (C) Lamin A/C detected in the cytoplasm following SV40 infection. Infected cells were fixed and stained with polyclonal lamin A/C (sc-26081). To enhance the fluorescence signal, both images were adjusted together, increasing brightness by 60% and contrast by 20% using Adobe Photoshop CS2. (D) Cytosolic fractions of CV1 infected cells from three different experiments were analyzed by protein gel blotting and detected with several lamin A/C antibodies. The graph presents average of three experiments and standard errors of lamin A/C detected by the polyclonal antibody 266. Total lamin A/C and cytosolic lamin A/C are shown as percent of the level of total lamin A/C in mock infected cells.
Figure 5.
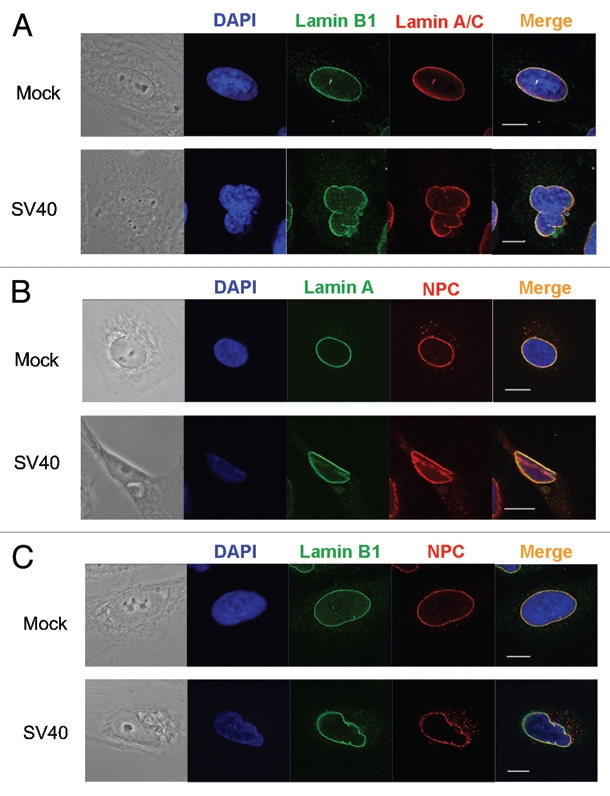
Deformation of the nuclear envelope. Images of CV1 cells taken at 6 h post infection. Top parts: Mock infected cells. Bottom parts: SV40 infected cells. (A) Lamin A/C (red) was detected with mouse monoclonal antibody (Jol2); lamin B1 (green) with a lamin B1 specific polyclonal antibody. In the mock part, contrast was increased in the red and green channels by 20%, using ZEN 2009 light edition. (B) Lamin A/C (green) detected with mouse monoclonal antibody (Jol2); NPC (red) with mouse monoclonal antibody clone 414. In the mock part contrast was increased in the green channel by 25% and in the red channel contrast was increased by 15% and brightness was decreased by 5% (C) Lamin B1 (green); NPC (red). In the mock part contrast was increased by 25% in the green channel and in the red channel contrast was decreased by 6% and brightness decreased by 3%. In the SV40 infected part, the contrast of the blue channel was increased by 38% and the brightness decreased by 3% and the red channel contrast was increased by 20%. Images were taken by Carl Zeiss Laser Scanning Systems LSM 510, magnification 4.
To evaluate the extent and specificity of these observations, we counted the number of cells with deformed nuclei in five independent infection experiments. Of a total of 2,530 cells scored at 6 h post infection, 2,194 (86.7%) showed NE deformation as detected by antibodies to lamin A/C. Detection of lamin A/C with the monoclonal antibody Jol2 or the lamin A alone with the polyclonal antibody 323 showed similar percentages of cells with NE deformation. In comparison, in the mock infected cells, only 9.3% of the cells (173 of 1,853 cells scored) showed NE deformation (Fig. 4B). In these experiments, 97% (965 of 998 cells scored) were positive for T-ag expression, suggesting that not every infected cell underwent NE deformation or that the changes in some cells were too subtle to detect. Alternatively, it is possible that the infection process was not absolutely synchronized and that the transient deformations occurred in different cells at different times.
We also detected an accumulation of lamin A/C in the cytoplasm with several antibodies to lamin A/C (polyclonal antibody sc-26081 shown in Fig. 4C) at 6 h post adsorption. These findings were supported by cell fractionation experiments (Fig. 4D). No increase of lamin A/C in the soluble fraction was observed in mock infected cells. At 8 h no cytoplasmic lamin A/C was observed (not shown). These results suggest that SV40 infection induces alterations in NE structure, specifically changes in lamin A/C levels and/or modification, which lead to deformation of the nucleus.
Lamin transcription following infection.
The fluctuations in lamin A/C levels (Fig. 3) suggested that SV40 infection also induced transcription of LMNA. Indeed, lamin A/C mRNA increased ∼3 fold at 6 h, and then decreased to 1/6 of the level of mock-infected cells at 9 h (Fig. S5). Over the same 9 h following infection, the levels of lamin B1 and lamin B2 mRNA remained unchanged, similar to their levels in mock-infected cells.
Fluctuations in lamin A/C levels are dependent on caspase-6 activity.
The release of lamin A/C from the NE to the cytoplasm has been described as a part of apoptosis and lamin A/C are known substrates of caspase-6.21,22 We have previously found that caspase-6 is activated by the infecting SV40 and remains active during the first 8 h of infection.11 We have also demonstrated that caspase-6 activity is required for SV40 infection to proceed since inhibition of caspase-6 for 1 h before infection blocks T-ag production.11 In order to determine if caspase-6 cleavage of lamin A/C is involved in the nuclear deformation of infected cells, we immunoblotted cell lysates to detect cleaved lamins during the first hours of infection. Indeed, we detected caspase-6 cleavage of lamin A/C, albeit at a very low level, between 6 and 10 h post infection (Fig. S6).
In light of these findings, we next asked if caspase-6 was involved in the fluctuations in lamin A/C levels described above. Addition of a caspase-6 specific inhibitor (Ac-VEID-CHO), or the pan-caspase inhibitor Z-VAD-FMK, eliminated the changes in the levels of the lamin epitope detected by antibody 346 (Fig. 6A). Furthermore, NE deformation was abolished by inhibitors of caspase-6 and -10 (during SV40 infection caspase-10 activates caspase-6 11) and by a pan-caspase inhibitor, as seen in the images of representative cells (Fig. 6B) and by the significant reduction in the number of deformed cells (Fig. 6C). A similar reduction in the number of deformed cells was obtained with an additional caspase-6 inhibitor, sc-3081 (not shown). Control experiment demonstrated that caspase-6 inhibitor (sc-3081) inhibited lamin A/C cleavage. These results indicate that changes in the lamin epitope recognized by antibody 346 require a previous caspase-6 dependent event, possibly cleavage of a fraction of lamin A/C. Furthermore, they suggest that the NE deformations during infection are caused by alterations of the lamin A/C networks.
Figure 6.
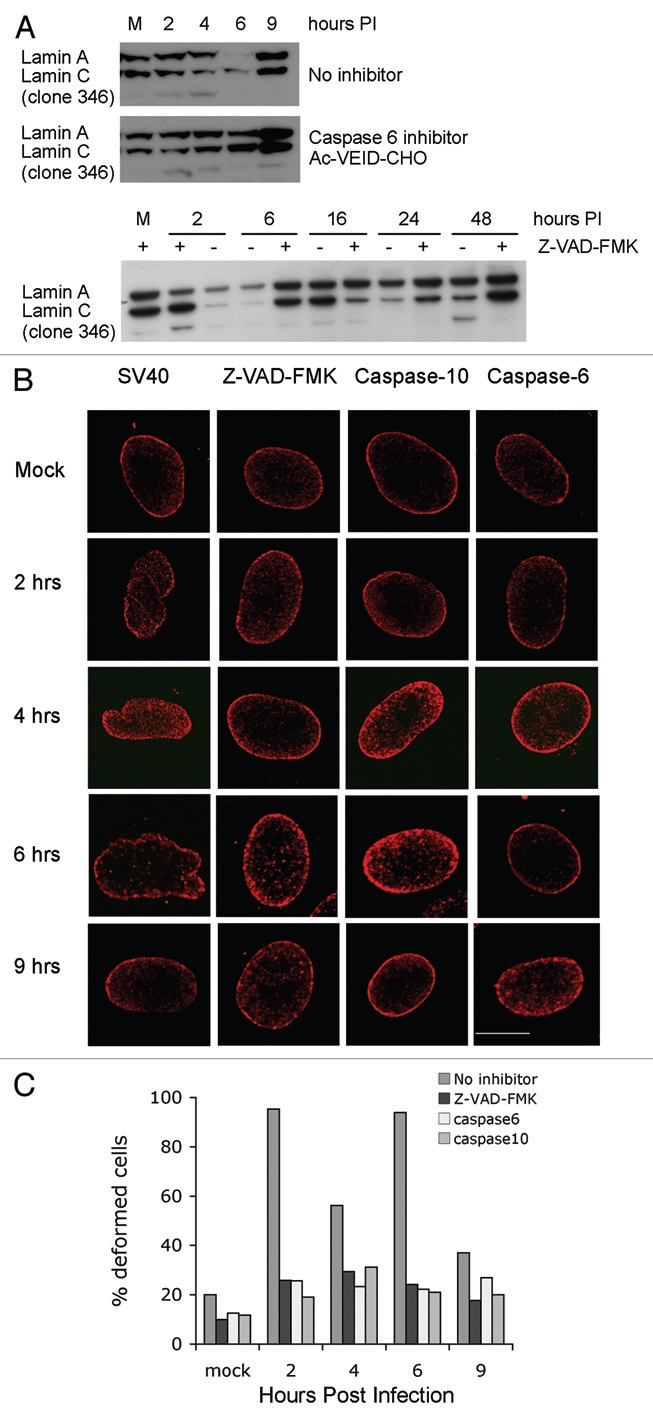
Caspase-6 is required for the fluctuations in laminA/C levels. (A) Infection was performed in the absence of inhibitors (top part), following the addition of caspase-6 inhibitor Ac-VEID-CHO (middle part) or pan-caspase inhibitor Z-VAD-FMK (bottom part). Total cell extracts were harvested at different time points and analyzed by protein gel blotting with lamin A/C monoclonal antibody clone 346. (B) CV1 cells were grown on cover slips and treated for 1 h with a caspase inhibitor immediately prior to SV40 infection. Z-VAD-FMK, a pan-caspase inhibitor was applied at a concentration of 70 µM; caspase-6—20 µM Ac-VEID-CHO; caspase-10—5 µM Z-AEVD-FMK. At the designated time the cells were fixed and stained with polyclonal lamin A/C antibody. Images were taken with confocal Carl Zeiss Laser Scanning Systems LSM 510, magnification X100, zoom 2. (C) The histogram presents the percent of cells with deformed nuclei with each inhibitor.
Mechanism of deformation of the nuclear envelope.
Since only a small fraction of lamin A/C was cleaved by caspase-6, we hypothesized that other changes to the lamina or NE are required for the dramatic NE deformations seen after infection. Lamins are phosphorylated at multiple sites and phosphorylation at Ser22 and Ser392 23 is required for their disassembly during mitotic prophase.24 Based upon these findings, we carried out a series of experiments to determine if altered lamin phosphorylation played a role in NE deformation during SV40 infection.
As mentioned above, we predicted that the fluctuations in lamin A detected with antibody 346 reflect changes in post-translational modification. To determine if this epitope was phosphorylated, we treated lysates of infected CV1 cells with Calf Intestinal Alkaline Phosphatase, CIAP (Fig. 7A). Indeed, the antibody 346 signal was partially decreased by CIAP treatment, suggesting that the antibody detected a phosphorylated epitope. Phosphorylation of the epitope was confirmed by applying CIAP treatment to preparations of intermediate filaments, which are enriched for lamins. As seen in Figure 7B, detection of lamin A by antibody 346 signal was completely eliminated by phosphatase treatment.
Figure 7.
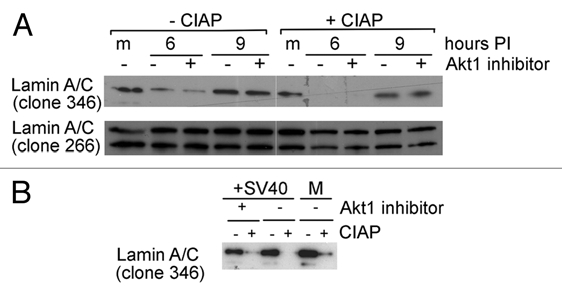
Dephosphorylation of lamin A/C following SV40 infection. (A) CV1 cells were infected at a moi of 10. Akt1 inhibitor (Akt1/2 kinase inhibitor, Sigma, A6730) was added as designated, to 58 nM, a concentration that inhibits only Akt1 and not Akt2 or 3. The inhibitor was added at 1 h prior to the infection, and was re-added after the adsorption period, thus it was present throughout the experiment. Total cell lysates were prepared at 6 and 9 h post infection. Each lysate was divided into 2 aliquots, one treated with CIAP (right) and one served as untreated control (left). CIAP concentration in the reaction was 10 units/µl. The antibodies used for detection are shown on the left. (B) The experiment was performed as in (A). Cells were harvested at 6 h post infection, IF were prepared and analyzed by protein gel blotting with monoclonal anti-lamin A/C antibody 346. The lower signal in presence of the Akt-1 inhibitor may be due to different lamin A recovery in the different IF preparations.
Our previous study in reference 11, showed that SV40 infection elicits a complex signaling network, including the activation of Akt-1 starting at 1 h post infection. Since Akt-1 is known to phosphorylate lamin A/C, we tested if an Akt-1 inhibitor could affect the epitope detected by antibody 346. As seen in Figure 7A and B, inhibition of Akt-1 had a minimal effect, if any, on detection of the specific epitope. We next tested if Akt-1 phosphorylation of lamin A/C on serine 404,25 was modulated during infection, using monoclonal antibody against S404 phosphorylated lamin A/C. Using this antibody, we did not detect any changes in S404 phosphorylation during the first 8 h of infection (Fig. S7A), indicating that lamin A phosphorylation is not directly affected by Akt-1. We concluded that Akt-1 activation in SV40 infection does not function directly in NE deformations. Supporting evidence was provided by repeated experiments that showed that addition of Akt-1 inhibitor had a minimal and inconsistent effect in reducing nuclear deformations.
The signaling network elicited by SV40 infection is robustly balanced, leading neither to apoptosis nor to proliferation.11 Nevertheless, we considered the possibility that the changes in lamin A/C levels during infection could represent the initiation of mitotic signaling and partial NE disruption. The NE disassembles during mitotic prophase removing the diffusion barrier between the cytoplasm and the nucleus. We applied AlexaFluor 488-labeled BSA to digitonin permeabilized cells to investigate if NE permeability increased during infection. The BSA was seen as aggregates excluded from the nuclei of both mock-infected cells and virus-infected cells (Fig. S7B). These results indicate that SV40 does not confer general permeability to the nuclear membrane as might be expected if there were a partial disruption of the NE diffusion barrier.
The VP1 pentamer is sufficient to induce the signals leading to fluctuations in lamin A/C levels.
As the changes in lamin A/C levels occurred in the first hours post-infection, prior to nuclear entry of the viral genome at 8 h and the beginning of viral gene expression, we postulated that they were triggered by the viral capsid. The outer shell of the SV40 capsid is composed of a single protein, VP1. Recombinant VP1 produced in insect cell assembles spontaneously into Virus Like Particles (VLPs). As seen in Figure 8, treatment of CV1 cells with VLPs led to dephosphorylation of the 346 espitope that was very similar both in timing and in intensity to that induced by the wild type virus (Fig. 8). Furthermore, similar fluctuations were also induced by VP1ΔC, a mutant VP1 with a deletion (in the carboxy-terminal arm) that abrogates capsid assembly.26,27 It appears that the VP1 pentamer is sufficient to induce the signals leading to fluctuations in lamin A/C levels during SV40 infection.
Figure 8.
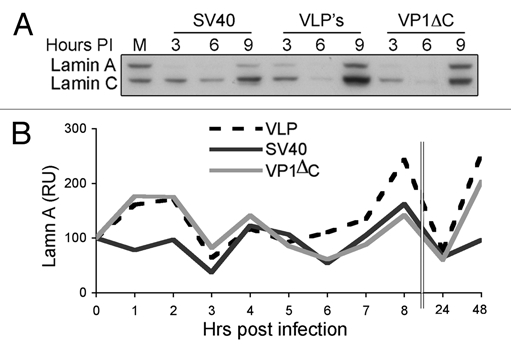
VLPs and VP1ΔC induce fluctuations in lamin A/C levels. (A) G1-arrested CV1 cells were infected with SV40 at a moi of 10 or treated with 5 ng of either VLPs or VP1ΔC per 105 cells. Lamin A/C was detected with monoclonal lamin A/C antibody 346. (B) Densitometry analysis was performed with Tina 2.09 software. The level of lamin A/C was normalized to the level of lamin B1 that was detected on the same membrane.
Discussion
SV40 infection of permissive primate cells is quite inefficient, with fewer than 1% of virus particles leading to productive infection.12 This block to SV40 infection could occur at several levels: at the cell membrane, in transport to the ER, at minichromosome entry into the nucleus and at the initiation of T-antigen transcription. In the present study we found that the NE is a major barrier to infection: the majority of the viral DNA molecules that enter the cell do not enter the nucleus and seem to be retained in the ER. Furthermore, proliferating cultures, in which some of the cells are undergoing mitosis at any given time during the long cell entry period, are infected at a 3 fold higher rate than quiescent cells. In addition, the experiments with HEK-293 cells demonstrated that ectopic expression of lamin A decreased infection by ∼2 fold, while shRNA silencing of lamin A/C increased the infection by ∼7 fold. Our data strongly support a model in which the NE, and in particular lamin A/C, plays a major role in obstructing nuclear entry of the SV40 minichromosome. This finding challenges the previously accepted model that the SV40 genome enters the nucleus via the NPC as a karyopherin cargo.28
Our experimental results identified changes to the NE that began shortly after virus entry to the cell and continued through nuclear entry of the viral minichromosome at 8 h post infection. The most obvious of these changes were deformations in the NE including folds and invaginations of the NE into the nuclear interior. These alterations in NE structure were accompanied by fluctuations in lamin A/C protein amounts and alterations in specific lamin antibody epitopes. The deformations of the NE and fluctuations in lamin protein levels were abrogated by caspase inhibitors, suggesting that cleavage of a small fraction of lamin A/C or other proteins was required for these changes to occur. Along with the changes in lamin levels, we also observed a transient accumulation of lamin A in the cytoplasm at 6 h post infection. The appearance of lamin A in the cytoplasm was coincident with both increased transcription of the lamin A/C gene and caspase cleavage of lamin. Therefore, we cannot differentiate between release of lamins from the lamina and leakage to the cytoplasm or accumulation of newly translated lamin in the cytoplasm.
The climax of the NE changes is at 6–8 h, just prior and during nuclear entry of the viral genome at around 8 h. This temporal correlation implicates alterations in the NE in nuclear entry of the viral genome, suggesting a model of direct minichromosome transport from the ER to the nucleus. Notably, our results indicate that the NE deformations and fluctuations in lamin A/C levels are triggered by the infecting virus. Since we did not observe such changes during infection of proliferating cells, we propose that they are specifically induced by the virus to facilitate its entry into quiescent cells. The temporal correlation of the NE changes with nuclear entry of the viral DNA supports this model. Viral DNA entry into the nucleus is unlikely to occur via general disruption of the NE permeability barrier, since we did not find increased permeability of the NE to BSA following infection. Note that in nature the in vivo habitat of SV40 and some of the other polyomaviruses is the kidney and brain, where the cells are non-proliferating. Furthermore, SV40 infection does not induce cell division.11 Similar alterations to the NE upon genome entry into the nucleus have not been reported for any other virus, and may be unique to SV40 and probably other polyomaviruses.
Daniels et al. have proposed that the SV40 genome exits from the ER lumen via a pore formed in the ER membrane by oligomers of VP3, and perhaps also VP2. VP3 pore formation appears to require capsid disassembly, as it was found to be negatively controlled by VP1.29 Similarly, Rainey-Barger et al. demonstrated that polyomavirus VP2 can integrate into and perforate the ER membrane. Accordingly, VP2 and/or VP3 viroporins could allow exit of the viral genome from the ER into the cytoplasm or directly into the nucleus, bypassing the cytoplasm. In another study (Kopatz et al., manuscript in preparation) we have found that the viral DNA is hardly ever present in the cytoplasm of infected cells. In fact, most of the viral DNA, as well as VP2 and VP3 were associated with membrane fractions throughout the entry period. All these findings taken together support a model whereby the SV40 genome enters the nucleus directly from the ER, bypassing the cytoplasm. We suggest that the NE deformations reported here reflect the mechanism of this unusual mode of transport.
To date, no mechanisms for viral entry into the nucleus have been described that suggest destabilization of the NE as a required component. However, both herpes simplex virus type 1,31 type 2,32 and human cytomegalovirus33 induce local phosphorylation of the lamins and lamina-associated proteins and structural changes in the lamina as part of the mechanism of viral capsid egress from the nucleus. In addition, the human immunodeficiency virus-1 VPR protein has been suggested to cause transient breaks in the NE of G2 phase cells leading to a G2 arrest in infected cells.34 In this study, we found that NE deformations during SV40 infection correlate with dephosphorylation of a specific epitope in lamin A/C. While we were unable to identify the specific epitope involved, it is likely that this epitope is involved in potential structural changes in the lamina that lead to alterations in NE structure.
We have previously reported that SV40 infection triggers activation of pro-apoptotic signaling, which is arrested by a survival pathway.11 In this context we found that SV40 activates caspase-3 and caspase-10, which in turn activates caspase-6, by an as yet unknown mechanism.11 It has been shown that lamin A is the nuclear target for caspase-6,21 and that its cleavage by caspase-6 is essential for the chromosomal DNA to undergo complete condensation during apoptotic execution.22 We have shown by inhibitors that caspase-3, known to cleave lamin associated proteins Lap2alpha,35 NuMA36 and Nup153,37 is not required for the infection. On the other hand inhibition of capase-10 and -6 abolished T-ag expression. Here we demonstrate that nuclear deformations during SV40 infection, as well as dephosphorylation of the antibody 346 epitope, are caspase-6 dependent. These findings strongly suggest that the two events are linked and are associated with productive infection. The timing of these events implies that they facilitate nuclear entry of the viral genome. Our experiments show that only a small fraction of lamin A is cleaved by caspase-6. It is possible that lamin cleavage is not the direct cause of NE deformation, since other proteins required for NE structure could be caspase-6 targets. Caspase 3-dependent cleavage of lamins and transient disruption of the NE has also been suggested as a requirement for the nuclear entry for the parvovirus Minute virus of mice.38
The requirement for caspase activity is intriguing. Activation of caspases, as part of the apoptotic pathway, is a frequent cell defense mechanism against infecting pathogens.39 Individual cells commonly respond to pathogen infection by apoptosis, an “altruistic” measure to facilitate protection of the organism from spread of the infection. However the activation of caspase-10 and 6 during SV40 cell entry appears to play a completely different role, facilitating the viral infection by promoting nuclear entry of the viral genome. Nevertheless, the mechanism is rather inefficient and the NE remains a hurdle to SV40 infection. It appears that the virus compensates for the inefficiency of nuclear entry by robust DNA replication, yielding thousands of progeny from every infected cell.
Polyomaviruses have been found to share an unusual cell binding and entry route. The remarkable similarity to SV40 of BK and °C, and the homology of MCPyV, suggests that nuclear entry of their genomes will follow a mechanism that is similar to the one investigated here. This unique nuclear entry pathway may provide a specific target for drug therapy, for example by inhibition of caspase activity, found to be required for productive infection of SV40.
Materials and Methods
Cell line and culture conditions.
African Green Monkey CV1 cells (ATCC #CCL-70) and human embryonic kidney cells HEK-293 (ATCC #CRL-1573) were obtained from the American Type Culture Collection. COS-1 are a CV1 derivative with constitutive expression of SV40 T-ag.19 COS-1 cells were obtained from Y. Gluzman. CV1-PD is a derivative of CV1, obtained from J. Mertz. HEK-293 stably expressing GFP-lamin were prepared by transfection of HEK-293 cells with pEGFP-lamin A and selection for stable clones. Mammalian cells were cultured in high glucose Dulbecco's modified Eagle's medium containing glutamine, penicillin, streptomycin and 10% FBS. CV1 cells grow as monolayers and are contact inhibited at confluence. Spodoptera frugiperda (Sf9) cells (ATCC #CRL-1711) were grown at 27°C in serum-free Bio-insect medium containing glutamine, penicillin, streptomycin and amphotericin.
Antibodies.
The following antibodies were purchased from Santa Cruz: SV40 T-antigen, monoclonal; monoclonal lamin A/C clone 346 (sc-7293); polyclonal lamin A/C (sc-26081); polyclonal lamin B1 (M-20); polyclonal emerin. The polyclonal antibodies lamin A/C 266 and lamin A 323, lamin A/C mouse monoclonal and lamin B1/B2 mouse monoclonal are from RD Goldman's collection; Lamin B1/B2 polyclonal was kindly provided by R. Moir. Monoclonal lamin A/C antibody Jol2 is from ABCAM. Mouse monoclonal antibody against the NPC (MAb414) is from ABCAM. The antibody against Lamin A/C phosphorylated at Ser 404 was a gift from S. Marmiroli. For immunofluorescence staining and microscopy we used Alexa fluor 488 and Alexa fluor 648 secondary antibodies purchased from Invitrogen.
SV40 production and purification.
SV40 was propagated on CV1-PD cells. Cells were harvested on the 5th day post-infection by the di-detergent method.40 Triton X-100 and deoxycholate were added to the culture medium to final concentrations of 1% and 0.5%, respectively. The cell suspension was centrifuged at 9,500 rpm (10,000x g) for 30 min at 4°C to remove cell debris. The virus was sedimented by centrifugation at 80,000x g for 4 h at 4°C. The virus pellet was resuspended in PBS overnight at 4°C, sonicated and centrifuged to clarify the virus suspension. Titration was performed by scoring for replication centers in CV1-PD cells infected at different dilutions. Replication centers were scored two days post infection, at the peak of viral DNA replication, by in situ hybridization with SV40 DNA labeled with [α-32P]dCTP.
Production and purification of VLPs and VP1ΔC.
Recombinant baculovirus expressing VP1 (Swiss-Prot P03087, PDB 1SVA) from the polyhedrin promoter were used for production of VLPs as previously described in reference 41. The VLPs were harvested from the medium of baculovirus-infected Sf9 cells. At 5 d post infection, cells were lysed and intact cells and cell debris were removed by centrifugation at 6,000x g for 10 min. The supernatant was further clarified at 17,000x g for 20 min. VLPs were pelleted at 80,000x g for 3 h. The VLP pellet was suspended in 0.5 M NaCl, purified by ultrafiltration and stored at −20°. VP1ΔC was purified as previously described in reference 26.
SV40, VLP and VP1ΔC infection experiments.
SV40 at a moi of 10, or as designated in the particular experiment, was added in a small volume of serum-free medium to confluent CV1 monolayers11 grown in 10 cm diameter tissue-culture dishes. Viral adsorption was performed at 4°C, in the absence of serum, in order to synchronize the infection, which started with the addition of serum-containing medium and transfer to 37°C. Mock-infected cells were similarly treated, including cold incubation in the absence of serum, but without virus. Independent infection experiments were performed using different virus batches.
Alternatively, 5 ng VLPs or 5 ng VP1ΔC were added per 105 cells in 12-well plates, approximately equivalent to a moi of 10 capsids per cell. Molecular weight of a VP1 capsid is ∼15 MDa, thus 5 ng represent ∼2 × 108 capsids; as ∼1 in 200 particles in SV40 stocks are infectious,12 2 × 108 SV40 virions contain 1 × 106 infectious particles, or moi 10 when applied to 105 cells.
Confluent CV1 monolayers were washed twice with PBS. To synchronize the infection, the virus or VLPs were allowed to adsorb to the cells for 40 min at 4°C on a gyratory shaker at 20 rpm. Non-adsorbed virus or VLPs were washed twice with SFM, followed by addition of DMEM + 10% FCS and the cells were transferred to 37°C until harvest. This point was considered as 0 time.
Inhibition studies.
The following caspase inhibitors were from Alexis: Pan-caspase inhibitor Z-VAD-FMK; caspase-6 inhibitor Ac-VEID-CHO; caspase-10 inhibitor Z-AEVD-FMK. Caspase-6 inhibitor sc-3081 was from Santa Cruz Biotechnology. The inhibitors were used at non-toxic levels, as determined by preliminary assays. The inhibitors were added to the cells 1 h before adsorption of virus. Infection was carried out as described below.
Protein analyses.
Total cell lysates were prepared by lysis in a solution containing 0.6% SDS, 10 mM Tris pH-7.4 and boiling for 10 min. Samples containing 20 µg protein were loaded on each lane. PAGE and protein gel blot analyses were performed using NuPAGE 4–12% Bis-Tris-Gel (Invitrogen), transferred to Immobilon (Millipore) and detected with appropriate antibodies.
Fractionation. Cells were treated with hypotonic buffer: 10 mM HEPES-K+ at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.2 mM PMSF and 1x protease inhibitor cocktail (Roche). Cytoplasmic and nuclear fractions were separated by centrifugation following treatment with 0.5% NP-40. Nuclear extracts were prepared as previously described in reference 41.
Intermediate filament preparation.
Intermediate filaments (IF) were prepared as previously described in reference 42. Briefly, cells were lysed with IF extraction buffer containing 0.5 M NaCl, 100 mM Hepes, pH 7.4, 60 mM Pipes, 1.0% Triton X-100 and Complete-EDTA free protease inhibitors, 1 tablet/10 ml buffer. After 5 min, cells were collected and homogenized, and DNaseI (3 units/µl) was then added to the solution. IF-enriched cytoskeletons were then pelleted by centrifugation (3,210 g; Beckman Allegra 5R centrifuge) and washed three times with the IF extraction buffer. The final pellet was sonicated into Laemmli buffer43 and 5–10 µg of protein from each time point was loaded per lane and analyzed by SDS-PAGE and immunoblotting.
Dephosphorylation experiments.
Cells were harvested after 6 h of infection. Total cell lysates or IF extracts were prepared as described above. An aliquot of each sample was treated for 30 min with 10 units of Calf Intestinal Alkaline Phosphatase (Sigma) and another aliquot was mock-treated in parallel. The samples of 10 µg/lane were then resolved by SDS-PAGE.
Immunostaining.
Confluent CV1 cells, grown on cover slips and infected with SV40 were fixed in 4% formaldehyde for 15 min at RT or with 100% methanol for 7 min at −20°, and permeabilized by treatment with 0.5% Triton X-100 for 3 min 3 times sequentially. The cells were incubated with the appropriate antibody for 45–60 min at 25°C (RT). Staining with secondary antibody conjugated to Alexa fluor 488 or Alexa fluor 648 (Invitrogen) was carried out for 1 h at RT. Images were obtained using either a Carl Zeiss Laser Scanning Systems LSM 510, or Olympus confocal microscope. To ascertain that cross section images were collected on similar planes, Z-sectioning was performed before images were taken. For planner images we used AxioVision LE (Zeiss).
FISH experiments.
Cells grown on cover slips were infected with SV40 at moi 2. At the time of harvest the cells were fixed with 4% formaldehyde for 20 min at RT. Alternatively nuclei were isolated from CV1 cells infected at a moi of 0.5 by treatment with hypotonic buffer (0.5% KCl) for 15 min. The nuclei were fixed with methanol:acetic acid (3:1) with three repeats and dropped on a slide.
The probe for both experiments was pUC19-SV40 that contains the complete SV40 DNA (total size 8.3 kDa). The probe was labeled with dUTP-biotin using DNA polymerase I (5 u per reaction) following pretreatment with DNAse I, 1 ng/µl. The labeling procedure was performed at 15°C for 30 min. Labeled probe was separated from unincorporated nucleotide by ethanol precipitation. Hybridization was performed as follows: denaturation in 70% formamide and 2x SSC for 2 min at 72°C, sequential dehydration in ice-cold ethanol 70%, 90% and 100%, for 5 min each, followed by hybridization with the probe for 16 h (ON) at 37°C. The probe was detected by incubation with Avidin-FITC for 30–45 min at 37°, or with streptavidin-conjugated Qdot 565 with excitation at 488 nm (Invitrogen). The detection reaction was carried out for 45 min at 37°C.
DNA determination by real-time PCR.
The quantitative PCR reaction was performed in LightCycler 2.1 instrument (Roche) in 20 µl reactions, as previously described in reference 44. The primers span the SV40 PolyA region:
5′-ACA TTG ATG AGT TTG GAC AAA CCA C-3′ (SV40 coordinates 2,533 to 2,577) and 5′-CCC CTG AAC CTG AAA CAT AAA ATG-3′ (SV40 coordinates 2,710 to 2,687).
RNA determination by quantitative RT-PCR.
CV1 cells were infected with SV40 at a moi of 10. Total RNA was extracted using Trizol reagent (Applied Biosystems) and subjected to DNAse I treatment (3 u/reaction) followed by additional Trizol precipitation. RT-PCR reaction was done using SuperSkript VILO cDNA Synthesis kit (Invitrogen). For quantitative RT-PCR reaction we used Qiagen QuantiFast Multiplex RT-PCR Kit. The primers for lamin A/C, lamin B1, lamin B2 and GAPDH (QuantiTect Primer Assays kits) were obtained from Qiagen. The data was analyzed as described in Schefe et al. including multiple runs.
FACS analysis of the percent of T-ag expressing cells.
We followed the experimental protocols previously described in reference 13. Briefly, cells were collected by trypsinization, washed and re-suspended in 100% methanol by drop-wise addition to the cell pellet while vortexing. The cells were fixed at −20°C and re-hydrated in cold PBS. Next, the cells were stained for T-ag (Pab 101, mouse monoclonal, Santa Cruz) followed by AlexaFluor 488 goat anti-mouse (A11001, Invitrogen) secondary antibody. Fluorescence was measured using the FL-1 sensor of a BD FACScan™.
Silencing experimental procedure.
HEK-293 cells stably expressing GFP-lamin A were electroporated (220 V 960 mF) with plasmids (20 µg DNA for 105 cells) encoding lamin A/C shRNA or scrambled sequence shRNA.15 Expression of the endogenous and GFP-lamin A was followed for 5 d. On the fifth day, the cells were infected with SV40 at an moi of 10. T-ag level was assayed 24 h later by protein gel blotting as described above.
Acknowledgements
We thank Sandra Marmiroli for the kind gift of antibody against phosphorylated-S404 lamin A/C. This study was supported by the Israel Science Foundation grant number 604/07.
Abbreviations
- ER
endoplasmic reticulum
- FACS
fluorescence activated cell sorting
- FISH
fluorescence in situ hybridization
- moi
multiplicity of infection
- NE
nuclear envelope
- NPC
nuclear pore complex
- pfu
plaque-forming units
- PI
post infection
- T-ag
T-antigen
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger JR, Major EO. Progressive multifocal leukoencephalopathy. Semin Neurol. 1999;19:193–200. doi: 10.1055/s-2008-1040837. [DOI] [PubMed] [Google Scholar]
- 3.Drachenberg CB, Beskow CO, Cangro CB, Bourquin PM, Simsir A, Fink J, et al. Human polyoma virus in renal allograft biopsies: morphological findings and correlation with urine cytology. Hum Pathol. 1999;30:970–977. doi: 10.1016/s0046-8177(99)90252-6. [DOI] [PubMed] [Google Scholar]
- 4.zur Hausen H. Novel human polyomaviruses—re-emergence of a well known virus family as possible human carcinogens. Int J Cancer. 2008;123:247–250. doi: 10.1002/ijc.23620. [DOI] [PubMed] [Google Scholar]
- 5.Smith AE, Lilie H, Helenius A. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett. 2003;555:199–203. doi: 10.1016/s0014-5793(03)01220-1. Campanero-Rhodes MA, Smith A, Chai W, Sonnino S, Mauri L, Childs RA, et al. N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J Virol 2007; 81:12846–58. [DOI] [PubMed] [Google Scholar]
- 6.Tsai B, Qian M. Cellular Entry of Polyoma viruses. Curr Top Microbiol Immunol. 2010;343:177–194. doi: 10.1007/82_2010_38. [DOI] [PubMed] [Google Scholar]
- 7.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, et al. The role of endosomes in SV40 entry and infection. J Virol. 2011;85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, Krausz E, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 9.Norkin LC, Anderson HA, Wolfrom SA, Oppenheim A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J Virol. 2002;76:5156–5166. doi: 10.1128/JVI.76.10.5156-5166.2002. Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, et al. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 2007; 131:516–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakanishi A, Itoh N, Li PP, Handa H, Liddington RC, Kasamatsu H. Minor capsid proteins of simian virus 40 are dispensable for nucleocapsid assembly and cell entry but are required for nuclear entry of the viral genome. J Virol. 2007;81:3778–3785. doi: 10.1128/JVI.02664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butin-Israeli V, Drayman N, Oppenheim A. Simian virus 40 infection triggers a balanced network that includes apoptotic, survival and stress pathways. J Virol. 2010;84:7. doi: 10.1128/JVI.01735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Black PH, Crawford EM, Crawford LV. The Purification of simian virus 40. Virology. 1964;24:381–387. doi: 10.1016/0042-6822(64)90175-8. [DOI] [PubMed] [Google Scholar]
- 13.Drayman N, Kler S, Ben-nun-Shaul O, Oppenheim A. Rapid method for SV40 titration. J Virol Methods. 2010;164:145–147. doi: 10.1016/j.jviromet.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Wilson KL, Berk JM. The nuclear envelope at a glance. J Cell Sci. 123:1973–1978. doi: 10.1242/jcs.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22:3409–3421. doi: 10.1101/gad.1735208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dechat T, Adam SA, Goldman RD. Nuclear lamins and chromatin: when structure meets function. Adv Enzyme Regul. 2009;49:157–166. doi: 10.1016/j.advenzreg.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y, Tsai MY. The mitotic spindle matrix: a fibro-membranous lamin connection. Cell Cycle. 2006;5:2345–2347. doi: 10.4161/cc.5.20.3365. [DOI] [PubMed] [Google Scholar]
- 18.Gant TM, Harris CA, Wilson KL. Roles of LAP2 proteins in nuclear assembly and DNA replication: truncated LAP2beta proteins alter lamina assembly, envelope formation, nuclear size and DNA replication efficiency in Xenopus laevis extracts. J Cell Biol. 1999;144:1083–1096. doi: 10.1083/jcb.144.6.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gluzman Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 20.Ikegami S, Taguchi T, Ohashi M, Oguro M, Nagano H, Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, et al. Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruchaud S, Korfali N, Villa P, Kottke TJ, Dingwall C, Kaufmann SH, et al. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 2002;21:1967–1977. doi: 10.1093/emboj/21.8.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward GE, Kirschner MW. Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell. 1990;61:561–577. doi: 10.1016/0092-8674(90)90469-u. Heald R, McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990; 61:579–89. [DOI] [PubMed] [Google Scholar]
- 24.Glass JR, Gerace L. Lamins A and C bind and assemble at the surface of mitotic chromosomes. J Cell Biol. 1990;111:1047–1057. doi: 10.1083/jcb.111.3.1047. Fields AP, Thompson LJ. The regulation of mitotic nuclear envelope breakdown: a role for multiple lamin kinases. Prog Cell Cycle Res 1995; 1:271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marmiroli S, Bertacchini J, Beretti F, Cenni V, Guida M, De Pol A, et al. A-type lamins and signaling: the PI 3-kinase/Akt pathway moves forward. J Cell Physiol. 2009;220:553–561. doi: 10.1002/jcp.21807. [DOI] [PubMed] [Google Scholar]
- 26.Roitman-Shemer V, Stokrova J, Forstova J, Oppenheim A. Assemblages of simian virus 40 capsid proteins and viral DNA visualized by electron microscopy. Biochem Biophys Res Commun. 2007;353:424–430. doi: 10.1016/j.bbrc.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 27.Mukherjee S, Abd-El-Latif M, Bronstein M, Ben-nun-Shaul O, Kler S, Oppenheim A. High cooperativity of the SV40 major capsid protein VP1 in virus assembly. PLoS ONE. 2007;2:765. doi: 10.1371/journal.pone.0000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakanishi A, Shum D, Morioka H, Otsuka E, Kasamatsu H. Interaction of the Vp3 nuclear localization signal with the importin alpha2/beta heterodimer directs nuclear entry of infecting simian virus 40. J Virol. 2002;76:9368–9377. doi: 10.1128/JVI.76.18.9368-9377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniels R, Rusan NM, Wadsworth P, Hebert DN. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell. 2006;24:955–966. doi: 10.1016/j.molcel.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Rainey-Barger EK, Magnuson B, Tsai B. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J Virol. 2007;81:12996–13004. doi: 10.1128/JVI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reynolds AE, Liang L, Baines JD. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J Virol. 2004;78:5564–5575. doi: 10.1128/JVI.78.11.5564-5575.2004. Park R, Baines JD. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 2006; 80:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cano-Monreal GL, Wylie KM, Cao F, Tavis JE, Morrison LA. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology. 2009;392:137–147. doi: 10.1016/j.virol.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radsak KD, Brucher KH, Georgatos SD. Focal nuclear envelope lesions and specific nuclear lamin A/C dephosphorylation during infection with human cytomegalovirus. Eur J Cell Biol. 1991;54:299–304. [PubMed] [Google Scholar]
- 34.de Noronha CM, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, et al. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294:1105–1108. doi: 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- 35.Gotzmann J, Vlcek S, Foisner R. Caspase-mediated cleavage of the chromosome-binding domain of lamina-associated polypeptide 2alpha. J Cell Sci. 2000;113:3769–3780. doi: 10.1242/jcs.113.21.3769. [DOI] [PubMed] [Google Scholar]
- 36.Taimen P, Berghall H, Vainionpaa R, Kallajoki M. NuMA and nuclear lamins are cleaved during viral infection—inhibition of caspase activity prevents cleavage and rescues HeLa cells from measles virus-induced but not from rhinovirus 1B-induced cell death. Virology. 2004;320:85–98. doi: 10.1016/j.virol.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 37.Buendia B, Santa-Maria A, Courvalin JC. Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci. 1999;112:1743–1753. doi: 10.1242/jcs.112.11.1743. [DOI] [PubMed] [Google Scholar]
- 38.Cohen S, Behzad AR, Carroll JB, Pante N. Parvoviral nuclear import: bypassing the host nuclear-transport machinery. J Gen Virol. 2006;87:3209–3213. doi: 10.1099/vir.0.82232-0. Cohen S, Marr AK, Garcin P, Pante N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J Virol 2011; 85:4863–74. [DOI] [PubMed] [Google Scholar]
- 39.Best SM. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol. 2008;62:171–192. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg B, Deutch J, Unger G. Growth and purification of SV40 virus for biochemical studies. J Virol Methods. 1981;3:167–176. doi: 10.1016/0166-0934(81)90051-3. [DOI] [PubMed] [Google Scholar]
- 41.Sandalon Z, Oppenheim A. Self assembly and protein-protein interactions between the SV40 capsid proteins produced in insect cells. Virol. 1997;237:414–421. doi: 10.1006/viro.1997.8796. [DOI] [PubMed] [Google Scholar]
- 42.Helfand BT, Mendez MG, Pugh J, Delsert C, Goldman RD. A role for intermediate filaments in determining and maintaining the shape of nerve cells. Mol Biol Cell. 2003;14:5069–5081. doi: 10.1091/mbc.E03-06-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 44.Ben-nun-Shaul O, Bronfeld H, Reshef D, Schueler-Furman O, Oppenheim A. The SV40 capsid is stabilized by a conserved pentapeptide hinge of the major capsid protein VP1. J Mol Biol. 2009;386:1382–1391. doi: 10.1016/j.jmb.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J Mol Med. 2006;84:901–910. doi: 10.1007/s00109-006-0097-6. [DOI] [PubMed] [Google Scholar]
- 46.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–16326. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.