

Abstract
The aggregation of amyloid beta (Aβ) peptides plays an important role in the development of Alzheimer's disease. Despite extensive effort, it has been difficult to characterize the secondary and tertiary structure of the Aβ monomer, the starting point for aggregation, due to its hydrophobicity and high aggregation propensity. Here, we employ extensive molecular dynamics simulations with atomistic protein and water models to determine structural ensembles for Aβ42, Aβ40, and Aβ42-E22K (the Italian mutant) monomers in solution. Sampling of a total of >700 microseconds in all-atom detail with explicit solvent enables us to observe the effects of peptide length and a pathogenic mutation on the disordered Aβ monomer structural ensemble. Aβ42 and Aβ40 have crudely similar characteristics but reducing the peptide length from 42 to 40 residues reduces β-hairpin formation near the C-terminus. The pathogenic Italian E22K mutation induces helix formation in the region of residues 20–24. This structural alteration may increase helix-helix interactions between monomers, resulting in altered mechanism and kinetics of Aβ oligomerization.
Introduction
Alzheimer's disease (AD) is a progressive degenerative brain disorder and the most common cause for dementia (1). There is currently no cure for AD. Senile plaques composed of fibers formed primarily from amyloid beta (Aβ) peptides in the brains of AD patients are a pathological hallmark of AD (2, 3). The ∼4-kDa Aβ peptide in senile plaques is a proteolytic product of the Aβ precursor protein (APP). Due to the ability of the protease γ-secretase to cleave APP at multiple sites (at the C-terminus of Aβ), Aβ peptides are 39 to 43 amino acid residues in length, but Aβ40 and Aβ42 are the predominant species in vivo (4). In contrast, senile plaques in AD are composed primarily of Aβ42 and Aβ43 (5). Aβ42 and Aβ43 are more hydrophobic and aggregation-prone than the slightly shorter and more polar (but very abundant) Aβ40. A number of mutations in APP have been related to familial AD (6, 7). The mutations usually occur at or near the β- and γ-secretase cleavage sites, resulting in more production of Aβ peptides or an increased ratio of Aβ42/Aβ40 (7). In addition to the mutations at or near the secretase cleavage sites, there are five pathogenic mutations in the region of A21, E22, and D23 in the Aβ sequence: the Flemish A21G mutation, Arctic E22G mutation, Dutch E22Q mutation, Italian E22K mutation, and Iowa D23N mutation. These mutations modify the biochemistry of Aβ peptides and lead to AD, congophilic amyloid angiopathy, or hereditary cerebral hemorrhage with amyloid (7). Thus, these genetic studies support a central role for Aβ in AD.
The structure of amyloid plaques is rather well resolved. Amyloid plaques appear fibrillar under an electron microscope (8). X-ray diffraction patterns suggest the formation of cross-β-sheets in amyloid fibrils (9). Solid-state NMR results confirm that amyloid fibrils composed of Aβ42 are cross-β-sheets, arranged in-register in a parallel fashion. A three-dimensional structure has been proposed (10, 11). In contrast to the amyloid fibril, the three-dimensional structure of the Aβ monomer, the starting point for aggregation, remains elusive. The absence of high-resolution structural data for the Aβ monomer is particularly troublesome because it now appears likely that the toxic species in AD is not the fibril, but rather an early-stage toxic oligomer of controversial structure and size (12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23). Determining the possible structures of toxic oligomers, and developing means to avoid their formation, would be greatly aided by a detailed understanding of the conformational ensemble adopted by the Aβ monomer. The hydrophobicity and high aggregation propensity of the Aβ monomer thwart structure determination under physiological conditions by traditional methods, including x-ray crystallography and solution-phase NMR spectroscopy. NMR structural determination of full-length Aβ with either an organic cosolvent (for example, Aβ42 in 80% hexafluoroisopropanol (24)) or detergent (25) to prevent aggregation is possible, but the physiological relevance of these results is, at best, unclear. The structures of some short, monomeric Aβ fragments with enhanced water solubility have also been studied by NMR spectroscopy (26, 27, 28, 29, 30, 31). For example, Aβ21–30 monomer adopts a bend structure spanning residues 24–28 (30) and Aβ10–35 monomer forms a collapsed coil structure in water (28), but it is unclear how relevant these fragment conformations are to the structural ensemble adopted by full-length Aβ peptides (which have different biophysical properties such as solubility, fibril structure, and aggregation kinetics (11, 28, 32, 33, 34)).
Although it is difficult to determine a high-resolution structure for full-length Aβ in aqueous solution, some information on its structure has been inferred from NMR measurements (35, 36). For example, the nuclear Overhauser effect, amide-NH temperature coefficients and chemical shift indices of 1Hα, 13Cα, and 13Cβ suggest that both Aβ42 and Aβ40 adopt predominantly random chain structures (35), although a partially folded structure of Aβ40 has been reported recently from 2D NMR data (37). The Hα chemical shift indices suggest some tendency of residues 31–36 and 39–41 to populate β-strand conformations for Aβ42 and the absence of β-strand structure for the C-terminal residues of Aβ40 (35). Nonetheless, for conformationally flexible biomolecules it is not straightforward to interpret spectroscopic quantities to obtain representative structures (38, 39, 40). In fact, it has been shown that secondary structure assignment using NMR chemical shifts for intrinsically disordered peptides such as Aβ may be generally problematic (41). Another NMR study reported that Aβ42 and Aβ40 share similar relaxation rates and nuclear Overhauser effect values except at the C-terminus, indicating Aβ42 and Aβ40 monomers have similar global motions (36). The differences in spectral density function and order parameters suggest that the Aβ42 C-terminus is more rigid than the Aβ40 C-terminus (36).
Molecular dynamics (MD) simulations have been extensively used to provide atomistic-level information on protein structure and dynamics ever since the first protein MD simulation in 1977 (42), and have been applied to study Aβ peptides quite extensively. Below we briefly summarize simulation approaches to understand the structure of Aβ monomers.
To enable sufficient sampling of Aβ monomer configurations, implicit solvent models are often used, sometimes along with coarse-grained models for proteins (43, 44). For instance, discrete MD combined with a four-bead protein model and implicit solvent has been used to study the effects of the Arctic E22G mutation on full-length Aβ. The authors proposed that the mutation increased the average β-strand propensity and disrupted contacts in the A21–A30 region (44). Simulations of Aβ peptides using fully atomistic protein models and implicit solvent have also been reported (45, 46, 47, 48, 49). Aβ42 appeared disordered but showed some sequence-dependent propensity to form regular secondary structure (45, 48, 49). Compared to Aβ40, the two additional hydrophobic residues at the C-terminus increased the β-structure in Aβ42 (48, 49).
Using explicit solvent models (rather than implicit solvent models) to better describe the peptide-water interactions is likely to be beneficial in MD simulations of Aβ peptides, which are, at most, weakly structured and have very high solvent exposure. However, the use of explicit solvent models demands great computing power to obtain the amount of sampling that can be reached by implicit-solvent simulations. MD simulations implementing explicit solvent models have been performed on a number of Aβ fragments, whose sizes are usually of 10–20 residues, and provided some potential insights into Aβ structure (29, 31, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67). For instance, from simulations of the 10 residue long Aβ21–30, a bend structure between residues 24–28 was observed, consistent with the NMR study of the fragment (30). This bend motif was found to be retained in the E22 mutants of this Aβ fragment (52).
MD simulations implementing explicit solvent models have also been performed on full-length Aβ (41, 68, 69, 70, 71, 72, 73, 74, 75, 76). For example, a number of researchers have generated short trajectories (with a total simulation time <500 ns) beginning with a highly helical Aβ42 structure that was determined by NMR studies of Aβ42 in a water-organic solvent mixture (68, 69, 70, 71, 72). These trajectories usually showed a loss of helical structure after the helical Aβ42 was placed in water in the simulations. Several MD simulations using atomistic models for peptide and explicit water models have been performed to better characterize the structure of full-length Aβ in explicit water (41, 73, 74, 75, 76). Sgourakis et al. (73) performed replica exchange MD to investigate structural differences between Aβ42 and Aβ40 monomer. By analyzing data from 52 100-ns replicas, a number of representative conformations were identified and the formation of a β-hairpin was observed near the C-terminus of Aβ42, but not of Aβ40. More recently, Sgourakis et al. performed replica exchange MD of Aβ42 using an improved simulation force field (77) and obtained an aggregation simulation time of 11.7 μs, during which the peptide sampled a highly diverse range of conformations (74). These results provided useful information on the solution structure of Aβ monomer, but we note that because Aβ monomer may be intrinsically not well structured and can clearly adopt numerous configurations, it is crucial to characterize the structural ensemble, rather than uncovering a handful of low free energy states. Furthermore, there has been little attempt to perform extensive MD simulations using explicit solvent models on pathogenic (or other) mutants of Aβ42, which could provide essential information regarding how the structural ensemble of monomeric Aβ can influence disease progression (75).
Here, by using distributed computing and Markov state model (MSM) analysis (78, 79), we are able to sample on an exceptional submillisecond timescale (>200 μs for each system) using an explicit water model and thereby characterize the structural ensembles of full length Aβ42 and Aβ40 monomers. In addition to the effects of peptide length we report the effects of a pathogenic mutation, the Italian mutation E22K, on Aβ monomer structures. Our data provide insights into how this pathogenic mutation can alter the structural ensemble and, potentially, the oligomerization of Aβ.
Methods
MD simulations
Details of the MD simulations regarding system setup, simulation parameters, simulation lengths, etc. can be found in the Supporting Material. For each system, the total number of trajectories used for analysis was ∼8000, the average simulation length of each trajectory was ∼30 ns, and the total accumulative simulation time was >200 μs. A number of convergence checks were performed and the results can be found in the Supporting Material.
Markov state model analysis
MSM analysis was performed to identify representative conformations and compute their equilibrium populations. For reviews and detailed description on MSMs, please refer to (78, 79, 80). In brief, the MD trajectories were first subsampled every 10 steps. The k-center clustering algorithm (81) with the root mean-square deviation (RMSD) metric (RMSD of heavy atoms common among all three systems in this study; however, heavy atoms in the side chains that are torsionally degenerate, for example, CD1 and CD2 in leucines, were excluded) was then performed on the subsampled data to identify cluster centers and partition the configurations into different states. The clustering procedure was performed until the newly identified cluster center had a RMSD <5 Å from the previously identified representatives (convergence was reached). Using these cluster centers, state indices were assigned to the full trajectories and transitions between states were recorded at a lag time of 20 ns. With this transition matrix, the master equations were solved and equilibrium populations for each state (representative conformation) deduced.
Results and Discussion
Characterizing the conformational ensemble for intrinsically disordered full-length Aβ peptides
Approximately 30,000 representative conformations were identified in the Aβ42 conformational ensemble by comparing structural similarity (see Methods). The Aβ42 monomer conformational ensemble displays great diversity and no native state that is particularly populated relative to the others could be identified. We performed principal component analysis to analyze the RMSD of the peptide heavy atoms (82, 83). The free energy landscape of Aβ42 is shown as a function of the first and the second principal components in Fig. 1. A single large cluster with no well-separated basins is observed. In Fig. S4 in the Supporting Material the free energy profile of Aβ42 is projected on the peptide radius of gyration and the peptide RMSD from the Aβ fibril structure (backbone of residues 17–42) (11) and shows a rather featureless contour as well. Each grid point also encompasses structures that show significant diversity. This is consistent with the idea that Aβ peptides are intrinsically disordered (we specifically note that there has been a very recent report on Aβ42 structure using both NMR experiments and MD simulations, in which Aβ42 was found to sample a highly heterogeneous structural ensemble (76)). In Fig. 2, we show the conformations of the six most populated representative conformations in the Aβ42 ensemble (84). They have populations of only 0.17%, 0.16%, 0.13%, 0.11%, 0.11%, and 0.10%, respectively. Thus, it is not appropriate to describe Aβ42 using a number of representative configurations. Instead, below we report the structural characteristics of Aβ42 by averaging over the full conformational ensemble. Note that in Fig. 1 and Fig. S4, we show how the results of principal component analysis and free energy landscape projection can depend on a number of independent trajectories in the simulations. Although with all data sets a single large cluster with no well-separated basins was observed (top panels in Fig. 1 and Fig. S4), separated basins could be (mis)identified when fewer trajectories were used (bottom panels in Fig. 1 and Fig. S4).
Figure 1.

Top: Free energy landscape (in kT) of Aβ42 as a function of the first and the second principal components from the principal component analysis using the root mean-square deviations among conformations. Bottom: Dependence of the principal component analysis results on the number of independent trajectories during the second-stage sampling. Upper left, upper right, lower left, and lower right are the results from 10, 50, 100, and 500 independent trajectories, respectively.
Figure 2.
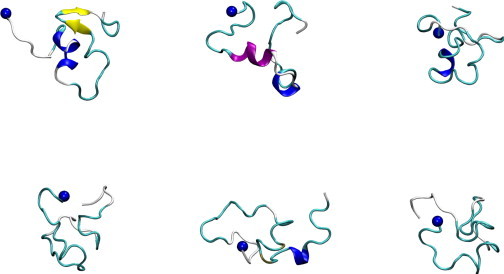
Conformations of the six most populated representative conformations in the Aβ42 ensemble. Secondary structure visualizations were generated using VMD (84). The blue spheres in the figure represent the N atoms in the N-terminal amino groups. α-helices are shown in purple, 310-helices in blue, extended β in yellow, isolated β bridges in tan, turns in cyan, and loops/irregulars in white. The secondary structure assignment was generated using the STRIDE program implemented in VMD.
Aβ42 forms short helix segments and hairpins
To investigate the local structural segments in the Aβ42 ensemble, the secondary structures for each representative conformation were assigned using the DSSP program (85). To compute the ensemble-averaged secondary structure, the contribution from each representative conformation was then weighted by its MSM population (see Methods).
In Fig. 3, we report the ensemble-averaged population of each residue in Aβ42 adopting one of the DSSP secondary structures. The peptide conformations appear to be dominated by the unstructured bend, turn, and loop/irregular, which is rather consistent with the earlier NMR study of Aβ42 (35). However, we note that the region of residues 24–26 has a very high bend propensity. Bends are regions with high curvature and a bend at residue i in the DSSP definition means the bond angle formed by the Cα atoms of residues i – 2, i, and i + 2 is at least 70° (85). This high bend propensity in the region of residues 24–26 is consistent with previous experiment and simulation results on Aβ fragments (28, 30, 52, 60, 86).
Figure 3.
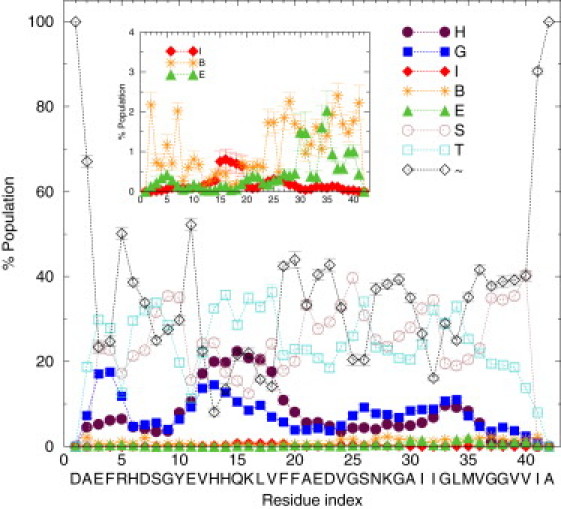
Ensemble-averaged %population of the secondary structures based on the DSSP analysis for Aβ42. The eight DSSP assignments are α-helix (H), 310-helix (G), π-helix (I), isolated bridge (B), extended β (E), bend (S), turn (T), or loop/irregular (∼). Inset: Zoom in on the π-helix (I), isolated bridge (B), and extended β (E) curves. The 1-σ error bars were calculated by dividing the total simulation data into 10 sets.
We observed some preferential formation of α- and 310-helices, especially in the region of residues 10–20. Through further length analysis of the helix segments as described in Fig. S5, we found that in most conformations only short α-helix segments involving four residues were formed. This is noteworthy because a structural tool with weak sensitivity to short α-helices such as circular dichroism spectroscopy (87), may not detect the short helix segments in the Aβ42 monomer ensemble. Enhanced 310-helix structure was also observed near the N-terminus. On average, there was a very low tendency to form π-helices (<1%), isolated bridges (<3%), and extended β (<3%) for all the residues in Aβ42. Nonetheless, some β-hairpins were indeed observed, mostly containing a glycine (G9, G25, G29, G33, G37, G38) as part of their turns. Two β-hairpins were particularly populated: a β-hairpin composed of residues I32 and G33, stabilized by antiparallel bridges between A30···M35 and I31···L34 and a second β-hairpin composed of residues G37 and G38, stabilized by antiparallel bridges M35···V40 and V36···V39, as can be seen in the green E curve in the Fig. 3 inset.
Aβ42 decomposes into three tertiary regions
We analyzed the tertiary structure in the Aβ42 ensemble by calculating the ensemble-averaged contact map (plotted in log scale in Fig. 4). Contacts between two residues are defined by an 8 Å cutoff for their Cα-Cα distances (88). Strong (i,i+3) and (i,i+4) contacts indicating helix formation are observed around residues 11–17, in accordance with the secondary structure trend shown in Fig. 3. It also appears the Aβ42 monomer has three distinct tertiary regions with substantial intraregion contacts: (I) an N-terminal region around residues 1–15, (II) a central region ranging approximately from residues 16–23, and (III) a C-terminal region expanding around residues 24–42. Notably, we observed few contacts between regions II and III and even fewer contacts between regions I and II.
Figure 4.
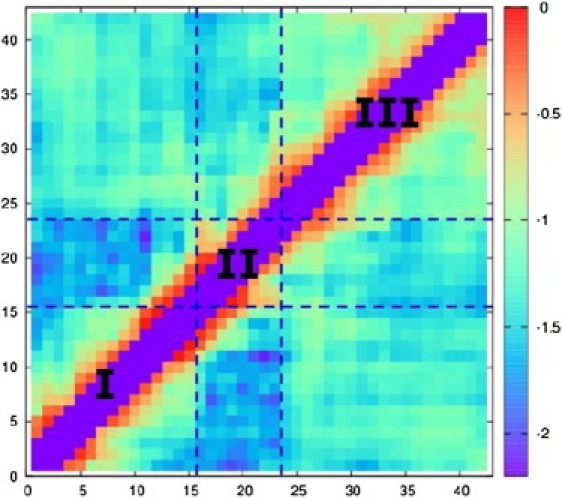
Ensemble-averaged contact map for Aβ42 (in log scale). Two residues are considered in contact when their Cα-Cα distance is <8 Å (88). Note the (i,i), (i,i±1), and (i,i±2) contacts are not computed and result in the purple diagonal shown in the figure.
Region I (∼residues 1–15 with sequence DAEFR HDSGY EVHHQ) has four residues with negatively charged side chains, one residue with a positively charged side chain, and three histidines; region II (∼residues 16–23 with sequence KLVFF AED) has a stretch of five hydrophobic residues (often referred to as the central hydrophobic cluster) capped by a residue with a positively charged side chain on one end (K16), and by two residues with negatively charged side chains on the other (E22 and D23); region III (∼residues 24–42 with sequence VG SNKGA IIGLM VGGVV IA) is mostly uncharged and hydrophobic, and is glycine-rich. The lack of contacts between region I and region II may be attributed to 1), the incompatibility between the charged residues in region I and the hydrophobic side chains of residues 17–21 in region II and 2), the repulsion between the four negatively charged residues in region I and the negatively charged E22 and D23 in region II.
Aβ42 and Aβ40 have crudely similar characteristics but Aβ40 has decreased β-propensity near the C-terminus
To investigate the effects of peptide length on the Aβ monomer structure, MD simulations were performed for Aβ40. In Fig. S6, we plot the distribution of radii of gyration (Rg) for Aβ42 and Aβ40. Both Aβ42 and Aβ40 appear rather compact and the similar Rg profiles suggest that the change in length has small effects on the size distribution of the peptide. The significance of the E22–K28 and D23–K28 salt bridges have been discussed and debated in Aβ fragment studies (30, 31, 46, 51, 52, 64, 66, 86). In Fig. S7, we plot the distribution of the E22–K28 and D23–K28 salt bridge distances. The similar distance distributions suggest that the change in length has small effects on E22–K28 and D23–K28 salt-bridge formation. In Fig. 5 (top panel), we compare the residue-dependent tendency to adopt a α-helix conformation of Aβ42 (black curve with sphere symbol) and Aβ40 (red curve with square symbol). Both Aβ42 and Aβ40 have similar residue dependence on their α-helix propensity.
Figure 5.

Ensemble-averaged %population of α-helix (H) (top panel) and extended β (E) (bottom panel) based on the DSSP analysis for Aβ42 (black circle), Aβ40 (red square), and Aβ42-E22K (green diamond).
Previous NMR studies have indicated β-hairpin formation near the C-terminus of Aβ42 but not Aβ40 (35). To verify this finding in our simulations, in Fig. 5 we compare the residue-dependent tendency to adopt a β-hairpin conformation (bottom panel) of Aβ42 (black curve with sphere symbol) and Aβ40 (red curve with square symbol). Although the β-hairpin propensity appears low for both Aβ42 and Aβ40 (with quite significant error bars), notably, the likelihood to form a β-hairpin by 30AIIGLM35 and that by 35MVGGVV40 is significantly lower for Aβ40. The simulation result that peptide length does not strongly perturb the α-helix propensity but reduces β-hairpin formation near the C-terminus is consistent with the earlier observation from NMR studies that suggested Aβ42 and Aβ40 have similar global motions but that there is a tendency for residues 31–36 and 39–41 to populate β-strand conformations for Aβ42 and no such tendency for the C-terminal residues of Aβ40 (35, 36). Although the absolute difference of extended β-propensity near the C-terminus between the two peptides in our simulations is a few percent (bottom panel of Fig. 5), the β-hairpin formation near the C-terminus is ∼threefold higher in Aβ42 relative to Aβ40. This difference at the molecular level may be critical for facilitating oligomerization and could account for the faster aggregation of Aβ42 relative to Aβ40 (89).
We note a partially folded structure of Aβ40 has been reported recently using 2D NMR data (37). In this NMR structure Aβ40 forms a 310-helix from H13 to D23, and in our simulations we indeed observed some preferential formation of 310-helices (and α-helices) in the region of residues 10–20 (for both Aβ42 and Aβ40). Nonetheless, in our simulations no single native state could be identified and our results are more compatible with the view that full-length Aβ peptides are intrinsically disordered with highly heterogeneous structural ensembles (76).
Comparison to previous Aβ42/Aβ40 simulation results
MD simulation results of Aβ42 and Aβ40 using implicit solvent models suggest that residues 30–36 have more β-structure in Aβ42 than in Aβ40, and residues 39 and 40 have some β-structure in Aβ42 but none in Aβ40. Although our findings in Fig. 5 are qualitatively consistent with these results, the β-propensity from our explicit-solvent MD simulations (<3% as seen in the bottom panel of Fig. 5) is much lower than that reported from the implicit solvent simulations (∼30% in Fig. 6 of (44), ∼6% in Fig. 3 of (48), and ∼15% in Fig. 5 of (49)).
The structural similarity of Aβ42 and Aβ40 has been predicted by implicit-solvent simulation studies (44, 48, 49) and deduced from experiments (35, 36). The similarity between the two peptides, however, has not been observed in previous explicit-solvent simulations (73), possibly due to insufficient sampling (74). In our explicit-solvent simulations Aβ42 and Aβ40 have crudely similar characteristics. Nonetheless, differences of β-propensity between the two peptides were observed. It is important to fully characterize the structural ensemble of intrinsically disordered peptides, such as Aβ, to accurately describe significant structural differences.
Italian E22K mutation increases helix propensity in the region of residues 20–24
A number of mutations in APP have been related to familial AD (6, 7). The mutations usually occur at or near the β- and γ-secretase cleavage sites, besides which there are five pathogenic mutations in the region of A21, E22, and D23. We performed simulations on the Italian mutant of Aβ42, Aβ42-E22K, which aggregates at least twice as fast as Aβ42 (89), to investigate the effects of the E22K substitution on the monomer conformation ensemble.
The E22K mutation has minimal effects on Aβ42's radius of gyration and on salt-bridge formation between D23–K28 (see Fig. S6 and Fig. S7). Thus, in Fig. 5 we compare the residue-dependent tendency to form an α-helix (top panel) or a β-hairpin (bottom panel) in Aβ42-E22K (green curve with diamond symbol) to that in wild-type (WT) Aβ42 (black curve with sphere symbol). The difference contact map for Aβ42-E22K compared to the WT Aβ42 is reported and discussed in the Supporting Material. As shown in Fig. 5, Aβ42-E22K showed significant α-helix propensity in the region of residues 20–24, which is absent in Aβ42, and showed decreased helix propensity in the region of residues 30–35, compared to Aβ42 (top panel of Fig. 5). This may be attributed to 1), the electrostatic repulsion between E22 and D23 in Aβ42, which may destabilize any helices formed in this region, is eliminated in Aβ42-E22K, and/or 2), lysine has a longer aliphatic chain than glutamate, which may provide a more favorable hydrophobic interaction with (i-4) V18 to stabilize a α-helix.
It is interesting to note that there have been studies reporting a α-helical kinetic intermediate during the oligomerization of Aβ (90). Such a α-helical intermediate may play a general role in amyloid formation (91). Here, we found that E22K induces helix formation in the region of residues 20–24 in Aβ42-E22K, which is known to aggregate at least twice as fast as the WT Aβ42 (89). It should still be noted that because this mutation replaces a negatively charged glutamate by a positively charged lysine, it does change the net charge of the Aβ peptide from –3 to –1, which may decrease Coulombic repulsion between monomers. However, besides the change of the peptide net charge we also note that the induced helix fragments in the region of residues 20–24 in Aβ42-E22K may increase helix-helix interactions between monomers and could lead to alignment of unstructured regions nearby the helices, thus promoting oligomerization.
It is interesting to note that within the Aβ sequence, A21, E22, and D23 are most frequently mutated in familial AD (7). Here, our simulation results on Aβ42-E22K indicate that the electrostatic repulsion between E22 and D23 may prevent helix formation in the region of residues 20–24 and substitution of E22 by a positively charged lysine that increases the helix propensity in this region. In WT Aβ42, the repulsion between these two negatively charged residues (E22 and D23) and those near the N-terminus may prevent substantial contacts between these two regions. The mutation E22K is able to alter the structural ensemble of Aβ monomer by decreasing these electrostatic repulsions. We note that in a pH dependence study of two Aβ fragments (Aβ1–28 and Aβ10–42), it was suggested that elimination of charge from one of the E22 and D23 and replacement by G, Q, or N would likely promote formation of a turn at residues 23–26 (46). In our Aβ42-E22K simulation, the E22K mutation not only eliminates the negative charge of E22 but also introduces a positive charge of K22, and this promotes helix formation at residues 20–24 instead of turn formation at residues 23–26. We also note that the study here and the previous pH dependence study use different force fields (AMBER ff99sb + tip3p vs. CHARMM22 + GBSW) and although in our simulations the protonation state of the peptide stays unchanged, constant pH MD was implemented in the pH dependence study.
Conclusions
Using distributed computing and Markov state model analysis, we report simulation results on a submillisecond time scale for Aβ42, Aβ40, and Aβ42-E22K. These simulations enabled us to characterize the structural ensemble of Aβ monomer, the starting point for Aβ aggregation, and investigate how the ensemble structure is affected both by the peptide length and by mutation. We observed many similarities in the structural ensembles adopted by the Aβ variants studied here, but we also discovered some significant differences in specific regions of the monomers. Aβ42, Aβ40, and Aβ42-E22K monomers are largely not well structured, with a slight tendency to form short α and 310-helix segments. The helix-forming tendency is strongest in the region of residues 10–20 for Aβ42 and Aβ40. Some formation of β-hairpins was also observed, mostly employing a glycine in their turns. β-hairpins between residues 30, 31 and residues 34, 35 (using I32 and G33 as its turn) and between residues 35, 36 and residues 39, 40 (using G37 and G38 as its turn) are most populated in Aβ42. The formation of both these β-hairpins is substantially decreased in Aβ40. The pathogenic Aβ42-E22K forms β-hairpins to a similar extent and at the same locations as the WT Aβ42, but notably has increased α-helix formation in the region of residues 20–24. These induced helix fragments in the region of residues 20–24 may increase helix-helix interactions between Aβ42-E22K monomers and could lead to alignment of unstructured regions nearby the helices, thus promoting oligomerization. Future MD simulations of other familial mutants with mutations at A21, E22, and D23 will provide even more insights into the role of this region in Aβ structure.
Acknowledgments
The project described was supported by Award No. PN2EY016525 from the National Eye Institute. Y.S.L. was supported by the Bio-X postdoctoral fellowship at Stanford University. G.R.B. was funded by the Berry Foundation and Miller Institute. K.A.B. was funded by a Stanford graduate fellowship. The authors acknowledge the following award for providing computing resources that have contributed to the research results reported within this paper: MRI-R2: Acquisition of a Hybrid CPU/GPU and Visualization Cluster for Multidisciplinary Studies in Transport Physics with Uncertainty Quantification. This award is funded under the American Recovery and Reinvestment Act of 2009 (Public Law 111-5). The authors thank the valued contributors of the Folding@home project.
Editor: Michael Feig.
Footnotes
Details of the MD simulations, free energy profile of Aβ42 as a function of radius of gyration and RMSD, results of the helix length analysis for Aβ42, figures of the radius of gyration and salt bridges distributions for Aβ42, Aβ40, and Aβ42-E22K, difference contact map for Aβ42-E22K compared to Aβ42, and references (92, 93, 94, 95, 96, 97, 98, 99, 100, 101) are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(11)05365-3.
Supporting Material
References
- 1.Querfurth H.W., LaFerla F.M. Alzheimer's disease. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer A. On a peculiar disease of the cerebral cortex. Neurologische Centralblatt. 1906;23:1129–1136. [Google Scholar]
- 3.Glenner G.G., Wong C.W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 4.Teplow D.B. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 5.Iwatsubo T., Odaka A., et al. Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 6.Morishima-Kawashima M., Ihara Y. Alzheimer's disease: β-Amyloid protein and tau. J. Neurosci. Res. 2002;70:392–401. doi: 10.1002/jnr.10355. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D.J., Podlisny M.B. Deciphering the genetic basis of Alzheimer's disease. Annu. Rev. Genomics Hum. Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- 8.Terry R.D., Gonatas N.K., Weiss M. Ultrastructural studies in Alzheimer's presenile dementia. Am. J. Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 9.Kirschner D.A., Abraham C., Selkoe D.J. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc. Natl. Acad. Sci. USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antzutkin O.N., Leapman R.D., et al. Tycko R. Supramolecular structural constraints on Alzheimer's β-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 11.Lührs T., Ritter C., et al. Riek R. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lue L.-F., Kuo Y.-M., et al. Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Näslund J., Haroutunian V., et al. Buxbaum J.D. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 14.Lesné S., Koh M.T., et al. Ashe K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 15.Lambert M.P., Barlow A.K., et al. Klein W.L. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo Y.-M., Emmerling M.R., et al. Roher A.E. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 17.Klein W.L., Krafft G.A., Finch C.E. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 18.Watson D., Castaño E., et al. Roher A.E. Physicochemical characteristics of soluble oligomeric Aβ and their pathologic role in Alzheimer's disease. Neurol. Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 19.Cole G.M., Frautschy S.A. Alzheimer's amyloid story finds its star. Trends Mol. Med. 2006;12:395–396. doi: 10.1016/j.molmed.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 20.De Felice F.G., Wu D., et al. Klein W.L. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shankar G.M., Li S., et al. Selkoe D.J. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barghorn S., Nimmrich V., et al. Hillen H. Globular amyloid β-peptide1–42 oligomer — a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 23.Bernstein S.L., Dupuis N.F., et al. Bowers M.T. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crescenzi O., Tomaselli S., et al. Picone D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002;269:5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 25.Shao H., Jao S., et al. Zagorski M.G. Solution structures of micelle-bound amyloid β-(1–40) and β-(1–42) peptides of Alzheimer's disease. J. Mol. Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.P., Stimson E.R., et al. Maggio J.E. 1H NMR of Aβ amyloid peptide congeners in water solution. Conformational changes correlate with plaque competence. Biochemistry. 1995;34:5191–5200. doi: 10.1021/bi00015a033. [DOI] [PubMed] [Google Scholar]
- 27.Zhang S., Casey N., Lee J.P. Residual structure in the Alzheimer's disease peptide: probing the origin of a central hydrophobic cluster. Fold. Des. 1998;3:413–422. doi: 10.1016/S1359-0278(98)00054-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S., Iwata K., et al. Lee J.P. The Alzheimer's peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 2000;130:130–141. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- 29.Massi F., Peng J.W., et al. Straub J.E. Simulation study of the structure and dynamics of the Alzheimer's amyloid peptide congener in solution. Biophys. J. 2001;80:31–44. doi: 10.1016/S0006-3495(01)75993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazo N.D., Grant M.A., et al. Teplow D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fawzi N.L., Phillips A.H., et al. Head-Gordon T. Structure and dynamics of the Aβ21–30 peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 2008;130:6145–6158. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benzinger T.L.S., Gregory D.M., et al. Meredith S.C. Two-dimensional structure of β-amyloid(10–35) fibrils. Biochemistry. 2000;39:3491–3499. doi: 10.1021/bi991527v. [DOI] [PubMed] [Google Scholar]
- 33.Petkova A.T., Ishii Y., et al. Tycko R. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarrett J.T., Berger E.P., Lansbury P.T., Jr. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 35.Hou L., Shao H., et al. Zagorski M.G. Solution NMR studies of the Aβ(1–40) and Aβ(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 36.Yan Y., Wang C. Aβ42 is more rigid than Aβ40 at the C terminus: implications for Aβ aggregation and toxicity. J. Mol. Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 37.Vivekanandan S., Brender J.R., et al. Ramamoorthy A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glättli A., van Gunsteren W.F. Are NMR-derived model structures for β-peptides representative for the ensemble of structures adopted in solution? Angew. Chem. Int. Ed. Engl. 2004;43:6312–6316. doi: 10.1002/anie.200460384. [DOI] [PubMed] [Google Scholar]
- 39.Trzesniak D., Glättli A., et al. van Gunsteren W.F. Interpreting NMR data for β-peptides using molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:14320–14329. doi: 10.1021/ja044285h. [DOI] [PubMed] [Google Scholar]
- 40.van Gunsteren W.F., Dolenc J., Mark A.E. Molecular simulation as an aid to experimentalists. Curr. Opin. Struct. Biol. 2008;18:149–153. doi: 10.1016/j.sbi.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Wood G.P.F., Rothlisberger U. Secondary structure assignment of amyloid-β peptide using chemical shifts. J. Chem. Theory Comput. 2011;7:1552–1563. doi: 10.1021/ct200156e. [DOI] [PubMed] [Google Scholar]
- 42.McCammon J.A., Gelin B.R., Karplus M. Dynamics of folded proteins. Nature. 1977;267:585–590. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- 43.Borreguero J.M., Urbanc B., et al. Stanley H.E. Folding events in the 21–30 region of amyloid β-protein (Aβ) studied in silico. Proc. Natl. Acad. Sci. USA. 2005;102:6015–6020. doi: 10.1073/pnas.0502006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lam A.R., Teplow D.B., et al. Urbanc B. Effects of the Arctic (E22→G) mutation on amyloid β-protein folding: discrete molecular dynamics study. J. Am. Chem. Soc. 2008;130:17413–17422. doi: 10.1021/ja804984h. [DOI] [PubMed] [Google Scholar]
- 45.Baumketner A., Bernstein S.L., et al. Shea J.E. Amyloid β-protein monomer structure: a computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khandogin J., Brooks C.L., 3rd Linking folding with aggregation in Alzheimer's β-amyloid peptides. Proc. Natl. Acad. Sci. USA. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W., Zhang J., et al. Wang W. Effects of zinc binding on the conformational distribution of the amyloid-β peptide based on molecular dynamics simulations. J. Phys. Chem. B. 2007;111:13814–13821. doi: 10.1021/jp076213t. [DOI] [PubMed] [Google Scholar]
- 48.Yang M., Teplow D.B. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vitalis A., Caflisch A. Micelle-like architecture of the monomer ensemble of Alzheimer's amyloid-β peptide in aqueous solution and its implications for Aβ aggregation. J. Mol. Biol. 2010;403:148–165. doi: 10.1016/j.jmb.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Cruz L., Urbanc B., et al. Stanley H.E. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc. Natl. Acad. Sci. USA. 2005;102:18258–18263. doi: 10.1073/pnas.0509276102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baumketner A., Bernstein S.L., et al. Shea J.E. Structure of the 21–30 fragment of amyloid β-protein. Protein Sci. 2006;15:1239–1247. doi: 10.1110/ps.062076806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krone M.G., Baumketner A., et al. Shea J.E. Effects of familial Alzheimer's disease mutations on the folding nucleation of the amyloid β-protein. J. Mol. Biol. 2008;381:221–228. doi: 10.1016/j.jmb.2008.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei G., Shea J.-E. Effects of solvent on the structure of the Alzheimer amyloid-β(25–35) peptide. Biophys. J. 2006;91:1638–1647. doi: 10.1529/biophysj.105.079186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito M., Johansson J., et al. Nilsson L. Unfolding of the amyloid β-peptide central helix: mechanistic insights from molecular dynamics simulations. PLoS ONE. 2011;6:e17587. doi: 10.1371/journal.pone.0017587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baumketner A., Krone M.G., Shea J.-E. Role of the familial Dutch mutation E22Q in the folding and aggregation of the 15–28 fragment of the Alzheimer amyloid-β protein. Proc. Natl. Acad. Sci. USA. 2008;105:6027–6032. doi: 10.1073/pnas.0708193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daidone I., Simona F., et al. Di Nola A. β-hairpin conformation of fibrillogenic peptides: structure and α-β transition mechanism revealed by molecular dynamics simulations. Proteins. 2004;57:198–204. doi: 10.1002/prot.20178. [DOI] [PubMed] [Google Scholar]
- 57.Baumketner A., Shea J.-E. Folding landscapes of the Alzheimer amyloid-β(12–28) peptide. J. Mol. Biol. 2006;362:567–579. doi: 10.1016/j.jmb.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 58.Kamiya N., Mitomo D., et al. Higo J. Folding of the 25 residue Aβ(12–36) peptide in TFE/water: temperature-dependent transition from a funneled free-energy landscape to a rugged one. J. Phys. Chem. B. 2007;111:5351–5356. doi: 10.1021/jp067075v. [DOI] [PubMed] [Google Scholar]
- 59.Ikebe J., Kamiya N., et al. Higo J. Simulation study on the disordered state of an Alzheimer's β amyloid peptide Aβ(12–36) in water consisting of random-structural, β-structural, and helical clusters. Protein Sci. 2007;16:1596–1608. doi: 10.1110/ps.062721907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massi F., Straub J.E. Probing the origins of increased activity of the E22Q “Dutch” mutant Alzheimer's β-amyloid peptide. Biophys. J. 2001;81:697–709. doi: 10.1016/S0006-3495(01)75734-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massi F., Klimov D., et al. Straub J.E. Charge states rather than propensity for β-structure determine enhanced fibrillogenesis in wild-type Alzheimer's β-amyloid peptide compared to E22Q Dutch mutant. Protein Sci. 2002;11:1639–1647. doi: 10.1110/ps.3150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Massi F., Straub J.E. Structural and dynamical analysis of the hydration of the Alzheimer's β-amyloid peptide. J. Comput. Chem. 2003;24:143–153. doi: 10.1002/jcc.10101. [DOI] [PubMed] [Google Scholar]
- 63.Han W., Wu Y.-D. A strand-loop-strand structure is a possible intermediate in fibril elongation: long time simulations of amyloid-β peptide (10–35) J. Am. Chem. Soc. 2005;127:15408–15416. doi: 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]
- 64.Tarus B., Straub J.E., Thirumalai D. Dynamics of Asp23-Lys28 salt-bridge formation in Aβ10–35 monomers. J. Am. Chem. Soc. 2006;128:16159–16168. doi: 10.1021/ja064872y. [DOI] [PubMed] [Google Scholar]
- 65.Baumketner A., Shea J.-E. The structure of the Alzheimer amyloid β 10–35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007;366:275–285. doi: 10.1016/j.jmb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 66.Reddy G., Straub J.E., Thirumalai D. Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Aβ peptides with implications for rates of fibril formation. J. Phys. Chem. B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu C., Murray M.M., et al. Bowers M.T. The structure of Aβ42 C-terminal fragments probed by a combined experimental and theoretical study. J. Mol. Biol. 2009;387:492–501. doi: 10.1016/j.jmb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flöck D., Colacino S., et al. Di Nola A. Misfolding of the amyloid β-protein: a molecular dynamics study. Proteins. 2006;62:183–192. doi: 10.1002/prot.20683. [DOI] [PubMed] [Google Scholar]
- 69.Tomaselli S., Esposito V., et al. Picone D. The α-to-β conformational transition of Alzheimer's Aβ-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of β conformation seeding. Chem.Bio.Chem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 70.Triguero L., Singh R., Prabhakar R. Molecular dynamics study to investigate the effect of chemical substitutions of methionine 35 on the secondary structure of the amyloid β (Aβ(1–42)) monomer in aqueous solution. J. Phys. Chem. B. 2008;112:2159–2167. doi: 10.1021/jp0771872. [DOI] [PubMed] [Google Scholar]
- 71.Yang C., Li J., et al. Zhu X. The effect of solvents on the conformations of Amyloid β-peptide (1–42) studied by molecular dynamics simulation. J. Mol. Struct. THEOCHEM. 2009;895:1–8. [Google Scholar]
- 72.Lee C., Ham S. Characterizing amyloid-beta protein misfolding from molecular dynamics simulations with explicit water. J. Comput. Chem. 2011;32:349–355. doi: 10.1002/jcc.21628. [DOI] [PubMed] [Google Scholar]
- 73.Sgourakis N.G., Yan Y., et al. Garcia A.E. The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sgourakis N.G., Merced-Serrano M., et al. Garcia A.E. Atomic-level characterization of the ensemble of the Aβ(1–42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J. Mol. Biol. 2011;405:570–583. doi: 10.1016/j.jmb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Velez-Vega C., Escobedo F.A. Characterizing the structural behavior of selected Aβ-42 monomers with different solubilities. J. Phys. Chem. B. 2011;115:4900–4910. doi: 10.1021/jp1086575. [DOI] [PubMed] [Google Scholar]
- 76.Ball K.A., Phillips A.H., et al. Head-Gordon T. Homogeneous and heterogeneous tertiary structure ensembles of amyloid-β peptides. Biochemistry. 2011;50:7612–7628. doi: 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hornak V., Abel R., et al. Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pande V.S., Beauchamp K.A., Bowman G.R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods. 2010;52:99–105. doi: 10.1016/j.ymeth.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beauchamp K.A., Bowman G.R., et al. Pande V.S. MSMBuilder2: modeling conformational dynamics on the picosecond to millisecond scale. J. Chem. Theory Comput. 2011;7:3412–3419. doi: 10.1021/ct200463m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Noé F., Fischer S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008;18:154–162. doi: 10.1016/j.sbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 81.Gonzalez T.F. Clustering to minimize the maximum intercluster distance. Theor. Comput. Sci. 1985;38:293–306. [Google Scholar]
- 82.Kitao A., Hirata F., Gō N. The effects of solvent on the conformation and the collective motions of protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 1991;158:447–472. [Google Scholar]
- 83.Kitao A., Gō N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 84.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 85.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 86.Tarus B., Straub J.E., Thirumalai D. Structures and free-energy landscapes of the wild type and mutants of the Aβ21–30 peptide are determined by an interplay between intrapeptide electrostatic and hydrophobic interactions. J. Mol. Biol. 2008;379:815–829. doi: 10.1016/j.jmb.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manning M.C., Illangasekare M., Woody R.W. Circular dichroism studies of distorted α-helices, twisted β-sheets, and β turns. Biophys. Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- 88.Voelz V.A., Shell M.S., Dill K.A. Predicting peptide structures in native proteins from physical simulations of fragments. PLOS Comput. Biol. 2009;5:e1000281. doi: 10.1371/journal.pcbi.1000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murakami K., Irie K., et al. Shirasawa T. Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy. J. Biol. Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 90.Kirkitadze M.D., Condron M.M., Teplow D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 91.Abedini A., Raleigh D.P. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 2009;6:015005. doi: 10.1088/1478-3975/6/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wood S.J., Maleeff B., et al. Wetzel R. Physical, morphological and functional differences between ph 5.8 and 7.4 aggregates of the Alzheimer's amyloid peptide Aβ. J. Mol. Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 93.McLaurin J., Yang D.-S., et al. Fraser P.E. Review: modulating factors in amyloid-β fibril formation. J. Struct. Biol. 2000;130:259–270. doi: 10.1006/jsbi.2000.4289. [DOI] [PubMed] [Google Scholar]
- 94.Khandogin J., Brooks C.L., 3rd Constant pH molecular dynamics with proton tautomerism. Biophys. J. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jorgensen W.L., Chandrasekhar J., et al. Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 96.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 97.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 98.Klement K., Wieligmann K., et al. Fändrich M. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer's Aβ(1–40) amyloid fibrils. J. Mol. Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 99.Kubelka J., Hofrichter J., Eaton W.A. The protein folding ‘speed limit’. Curr. Opin. Struct. Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 100.Ghosh K., Ozkan S.B., Dill K.A. The ultimate speed limit to protein folding is conformational searching. J. Am. Chem. Soc. 2007;129:11920–11927. doi: 10.1021/ja066785b. [DOI] [PubMed] [Google Scholar]
- 101.Vuister G.W., Bax A. Quantitative J correlation: a new approach for measuring homonuclear three-bond J(HNHα)coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 1993;115:7772–7777. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.