

Abstract
Confocal Raman spectroscopy is a noninvasive alternative to established cell imaging methods because it does not require chemical fixation, the use of fluorescent markers, or genetic engineering. In particular, single live-cell, high-resolution imaging by confocal Raman microscopy is desirable because it allows further experiments concerning the individually investigated cells. However, to derive meaningful images from the spectroscopic data, one must identify cell components within the dataset. Using immunofluorescence images as a reference, we derive Raman spectral signatures by means of information measures to identify cell components such as the nucleus, the endoplasmic reticulum, the Golgi apparatus, and mitochondria. The extracted signatures allow us to generate representations equivalent to conventional (immuno)fluorescence images with more than three cell components at a time, exploiting the Raman spectral information alone.
Introduction
Intracellular compartmentalization is the major basis for our understanding of functional processes in eukaryotic cells. Therefore, the identification and characterization of cellular architecture is an important issue in cell biology.
Most well-established methods for intracellular imaging, such as electron microscopy, cryoelectron microscopy, and (immuno)fluorescence (IF) microscopy, are invasive due to the necessity of fixation or freezing and the use of dyes or biomarkers. All of these techniques are destructive or at least harmful to the examined cells. Hence, label-free and nondestructive imaging techniques have been established for biochemical investigations, such as coherent anti-Stokes Raman (CARS), multiphoton microscopy, and confocal Raman microscopy (1, 2, 3). Applications range from the discrimination of tissues (4, 5) to single-cell biology (6, 7, 8, 9, 10, 11, 12). Because a confocal Raman system is based on a standard confocal light microscope setup, its resolution is restricted only by the diffraction limit. Furthermore, acquisition times have become short enough to enable visualization of cellular compartments of living cells without fixation or drying (11, 13, 14). Matthäus et al. (15) demonstrated live-cell imaging by depicting mitochondria at a spatial resolution comparable to that of fluorescence microscopy. Although high-resolution Raman imaging was mainly conducted with near-infrared laser excitation to minimize tissue degeneration and avoid (auto)fluorescence of the biological specimen, the applicable spectral range has been expanded further into the visible range (15, 16). However, interpretation of the Raman data is often confounded by the intrinsic similarity of the respective biological materials in the cells. Although many biochemical assignments of individual Raman bands are available in the literature, they are mostly based on specimens extracted from cells and not imaged in situ (11, 16, 17, 18, 19, 20, 21, 22). For some selected bands, however, direct correlations have been observed. For example, Pully and Otto (21) reported that the 1602 cm−1 band in yeast cells is dependent on Ca2+ uptake. Therefore, a comprehensive method to distinguish cellular constituents in situ should be based on the assembly of spectral information at various wavenumbers rather than focused on single spectral lines.
Labeling methods such as IF allow highly selective identification of previously specified cell compartments through targeted linkers. In contrast, Raman spectral data reflect the local chemical composition for a given optical focus. Thus, the potential information content is likely to be more encompassing than that obtained by other labeling methods, because Raman spectroscopy offers unbiased biochemical diagnostic information about all subcellular structures (11, 13, 14). Algorithms for data processing (e.g., principal component analysis (PCA)) and clustering procedures (e.g., K-means clustering) can be used to analyze this information (21, 23, 24, 25, 26).
In this work, we report on an approach to decipher confocal Raman microscopic data based on reasoning from IF imaging. We investigated single living cells of a human glioma cell line (LN-18) under cell culture conditions using both techniques. We then registered the data from Raman spectroscopy to the IF data using nonlinear methods to identify different subcellular compartments and to derive multichannel IF image equivalences from Raman datasets (hereafter called artificial IF (aIF) images). In the future, our approach may enable investigators to distinguish between healthy and infected or cancerous cells without having to use staining procedures.
Materials and Methods
Sample preparation
LN-18 is a human malignant glioma cell line (27). It was established from a fragment of a temporal tumor whose glial nature has been assessed by ultrastructural examination. The cells have a bipolar or stellate structure and show pleomorphic nuclei (70–80 chromosomes, modal number 78, XXYY). The doubling time of LN-18 cells is ∼72 h at a plating efficiency of 3%. They synthesize large quantities of cytoplasmic fibronectin. We grew LN-18 cells on commercially available object slides structured with a coordinate grid sized in the range of the field of view of the light microscope (200 μm fields in a coordinate system, England Finders; Plano Elektronenmikroskopie, Wetzlar, Germany). The England Finders were positioned in Quadriperm culture flasks (Greiner Bio-One GmbH, Frickenhausen, Germany) in which cells were incubated with 6 ml of Dulbecco's modified Eagle's medium (DMEM cell culture; Gibco-Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mMol/ml glutamine (all from Biochrom, Berlin, Germany) at 37°C in a humidified atmosphere of 5% CO2. After 48–72 h of incubation, the England Finders were washed in phosphate-buffered saline (PBS; PAA Laboratories GmbH, Pasching, Austria) at 37°C to remove all DMEM residuals before the Raman measurements were obtained.
Raman experimental setup
We recorded Raman spectra by raster-scanning the sample under a confocal Raman microscope (alpha300 R; WITec GmbH, Ulm, Germany). The system was equipped with an SHG Nd:YAG laser (532 nm, 22.5 mW) and a lens-based spectrometer with a CCD-camera (1024 × 128 pixel, Peltier cooled to −65°C). A 60× water immersion microscope objective (working distance 2.0 mm, NA 1.0) was used in the experiments. Spectra were recorded at each image pixel with an integration time of 70–80 ms at a laser power of 10.0 mW measured at the microscope objective. For confocal imaging, the 50 μm core of a multimode fiber served as the pinhole, leading to a focal depth of ∼1 μm. The diffraction-limited focus resulted in a lateral resolution of ∼500 nm. Reflected and elastically scattered photons were rejected by an edge filter. The nominal spectral resolution was ∼3 cm−1 per CCD pixel (600 mm−1 grating).
The object slides were clamped to an aluminum petri dish and immersed in PBS during the measurements. In this way, the cells could be kept in good condition for longer than the measurement time of an image scan.
When mapping the composition of adherent cells, one must ensure that the contribution of the substrate to the signal is minimal but the Raman scattering from the cells is maximal. Therefore, we initially acquired x,z maps (depth scans) to determine the correct distance above the substrate for x,y imaging (typically at z ≈ 2 μm). The chosen spatial resolution of the spectral recording was 2–4 spectra/μm. To check the condition of the cells, we acquired light microscopy images before and after each scan.
After a maximum of two data acquisitions per slide, which required a maximum measurement time of ∼45–60 min per scan, the samples were washed in PBS, fixed in 4% phosphate-buffered formalin solution (Carl Roth GmbH & Co KG, Karlsruhe, Germany) for 20 min at room temperature, washed again in PBS, and stored at 4°C. To assess potential fixation artifacts, we scanned the identical cells again after fixation for several measurements. After the Raman data were acquired, the slides were stored in PBS at 4°C.
IF staining and imaging
In addition to staining of the nucleus and the cytoskeleton, which are the most evident intracellular structures, we used one of the following three antibodies at a time to visualize an additional intracellular compartment: anti-COX-IV for the mitochondria, anti-Calnexin for the endoplasmic reticulum, and anti-Syntaxin-6-antibody for the Golgi apparatus (all from New England Biolabs, Frankfurt, Germany).
IF staining was performed on formalin-fixed cells at 4°C overnight, followed by an incubation with secondary antibodies conjugated to FITC (Zymed Laboratories, San Francisco, CA). Rhodamin-conjugated phalloidin (Sigma-Aldrich, Heidelberg, Germany) was used to stain actin filaments and thus indicate the cell borders. The nucleus was stained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Heidelberg, Germany).
We conducted fluorescence imaging on the triply-stained samples using a Zeiss AxioImager microscope with standard AxioVision 4.5 software (Zeiss, Jena, Germany) with standard filter sets. All channels imaged gave a distinct signal, and the shutter times were chosen carefully to avoid overexposure.
The cells previously investigated by Raman microscopy were retrieved at the beginning of fluorescence imaging with the help of a coordinate grid on the object slides. To facilitate an optimal match with Raman data, in many cases stacks of eight to 16 fluorescence images per cell with a z-spacing of 0.5 μm were acquired.
Data analysis
The data analysis aims to identify cellular components from selected spectral characteristics without the need for a labeling procedure. To obtain these characteristics for each cellular component, we derive spectral signatures from an information-based comparison of Raman maps and IF images of a training sample. The resulting label-free cell-imaging technique avoids the necessity for laborious sample treatment (e.g., fixation and staining) and overcomes the practical restriction of IF imaging to a small number of fluorescence labels per sample at a time. The different steps of data preparation and image analysis are described below.
Data preparation
A Raman dataset consists of a single spectrum acquired at each pixel of the imaged area. We applied a spectral minimum subtraction for each image pixel to correct for the CCD dark counts. Due to the relatively long scanning times and the use of array detectors (i.e., CCD camera), Raman spectral maps can be contaminated by a number of cosmic rays. Typically, only single pixels or very few adjacent pixels along the scan direction are contaminated. Because of this preferential direction of the cosmic ray effect, an anisotropic kernel filtering with a threshold based on a simple variance criterion is most suitable for detecting intensity outliers.
After removing the major effects of cosmic rays, we performed a PCA to reduce the effects of further random noise and to remove other kinds of artifacts and pollution. Typically, in the field of Raman spectroscopy, one arranges a Raman dataset in an array of k, L × M-dimensional vectors, where k is the number of measured wavelengths and L × M is the size of the image in pixel units. The PCA components are, by definition, ordered according to the level of explained variances. However, some remaining artifacts carry high variances and display structures that are predominantly oriented along the scan direction, which may be due to temperature fluctuations. To increase image quality in a manner similar to PCA, other investigators considered approaches in which the resulting principal components are ordered with respect to the signal/noise ratio (28, 29). These methods require knowledge of the nature of the noise, which in practical situations can only be estimated. In our approach, we enhance image quality by just eliminating the undesirable PCA components using a structural filter. Using an information-based procedure, we rearrange the k principal components with respect to their anisotropic structural content (see Fig. 1). Numerical experiments showed that back-transformation using only the first 10 rank-selected PCA components yielded considerably improved Raman images.
Figure 1.

Structural filtering of the PCA components of a Raman spectroscopic image. The upper row shows the components in the usual rank order according to their explained variances. The lower panels are reordered with respect to the anisotropic structural information content. Most prominently, components 1–5 remained in their original order, while components 6–8 were rejected due to the high proportion of artificial structural elements (i.e., systematic errors).
Registration procedure
The registration procedure aims to achieve an optimized mapping of the two different image modalities (i.e., Raman and IF images of the same cell) for quantitative comparison. We tested several registration techniques based on both linear and nonlinear methods. Registration approaches that rely on the concept of mutual information proved to be superior to linear cross-correlation measures (e.g., methods based on a Fourier transform of the single images), which is due to the highly different imaging modalities used.
With our experimental setup and imaging protocol, there is no need to account for (local and/or higher-order) distortions or rotations. Therefore, we found it was sufficient to restrict ourselves to the linear transformations, namely, global scaling (or magnification f) and translation (spatial offsets x0 and y0).
Registration by maximizing the normalized mutual information
The mutual information M is a well established similarity measure that is derived from principles of information theory (30, 31). In the case of a registration of two image modalities I1 and I2 with image pixels i1(x,y) and i2(x′,y′), the mutual information M between both datasets is given by
where H(I1) and H(I2) denote the marginal entropies, i.e., the Shannon information entropy H applied to the density distribution p(i) of the intensities in the images I1 and I2, respectively: . The mixed term H(I1, I2) describes the joint entropy of the density distribution in the I1 versus I2 plane.
In our case, the marginal entropies are given by the density distributions of the three-dimensional color distribution, say r, g, and b, of the IF image with p = p(r,g,b) and the density of the n-dimensional intensity distribution of the first n PCA components c of the Raman spectroscopic dataset q = q(c1, c2,…, cn). Consequently, the mixed term is given by H(I1, I2) = H(p, q) = H([r,g,b],[c1, c2,…, cn]), corresponding to a data representation in a (3+n)-dimensional state space.
Our aim was to achieve a registration of the multimodal images in a completely unsupervised manner by scanning the plausible ranges of magnification f and spatial offsets x0 and y0 automatically. We figured out that the best results are obtained by maximizing the normalized mutual information . The normalization of mutual information makes the solution more robust, because scaling and translation affect not only the joint entropy but also the marginal entropies of the single image sections under consideration. Fig. 2 shows an overlay of the integrated Raman data (80–3040 cm−1) and the corresponding IF image.
Figure 2.
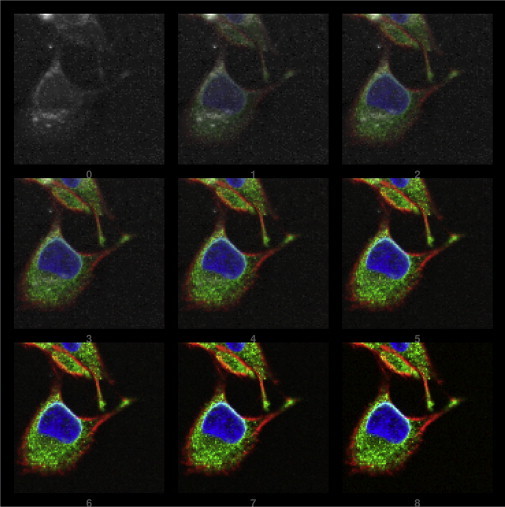
Matching sequence of a Raman image (by integration over Raman intensities, 80–3040 cm−1) and the corresponding IF data (RGB image). Panel 0 (top row, left) shows the integrated Raman, and panel 8 (bottom row, right) shows the IF image. Panels 1–7 show stepwise overlays demonstrating the quality of image registration achieved by successively changing the respective image contribution. For the registration procedure, the first three (rearranged) PCA components were considered.
As described above, we acquired IF image stacks. These z-stacks have a twofold advantage: 1), they facilitate the construction of IF images with an artificial extended focus; and 2), they enable a fine adjustment of the registration with respect to some differences in focal characteristics of both image modalities.
Decomposition of the Raman spectroscopic images
In the following, we describe a methodology that allows a decomposition of the Raman spectroscopic datasets and provides information about cellular compartments similar to that obtained by IF imaging.
After the preparatory steps listed above are completed, the IF images and the Raman images perfectly match in such a way that each color pixel of the IF image has its counterpart in terms of a Raman spectrum. Each single spectrum consists of >700 contributing intensities at the set of wavenumbers Λ (80–3040 cm−1).
To generate aIF images from the Raman spectra while avoiding overfitting, we must select a limited number of relevant wavenumbers for an IF image reconstruction as the first step. To achieve this, we developed an approach based on a redundancy analysis of both data modalities. Redundancy can be interpreted as a generalization of the concept of mutual information to multidimensional distributions.
Assume that IC,l is the lth color layer of an IF image, representing a specific cell component, and are Raman intensity maps, i.e., spectroscopic image sections at wavenumbers . Then, the marginal redundancy Rm is given by
where H(Ii,..,Ii+n−1) is the Shannon information entropy of an n-dimensional distribution.
The marginal redundancy yields the information content of in and vice versa. Thus, it quantifies the redundant information content in both distributions.
To find a set S of appropriate spectral lines in the Raman spectra for image reconstruction, we apply the following scheme, aiming to select spectral lines that carry a maximum of information with respect to the considered IF image layer:
Starting with n = 1, we search for the spectral line at which R1m (the agreement between the channel l of the IF image and the intensity map of this spectral line) is maximal:
This is equivalent to finding the Raman spectroscopic image section, i.e., the map of Raman intensity at a given Raman shift, which maximizes the mutual information.
For n > 1, we iteratively determine the wavenumber k for which
We end up with a set S of wavenumbers: , which are subsequently used for the construction of an aIF image. It should be noticed that one can show that the set S does not necessarily represent the best subset of wavenumbers for the (re)construction but yields at least a good approximation.
Once a set of representatives is found, appropriate coefficients ad=0..2, l=0..2, j=1..n for mapping from the Raman subspace to different IF image layers are calculated by simple polynomial least-square fitting of second degree:
where the indices of a refer to the degree d, the color layer l, and the selected Raman line j, respectively.
We obtain this set of fit parameters for each combination of IF stainings (red, green, and blue (RGB)). Fig. 3 a displays for every cellular compartment (i.e., cytoskeleton, endoplasmic reticulum, mitochondria, Golgi apparatus, and nucleus) the weighted average of the most prominent spectra. We selected and weighted these spectra using the channel information of each aIF image. It is striking that even spectral positions without apparent Raman scattering intensity contribute strongly to the IF-to-Raman fitting. This shows that only the simultaneous information of the set S of spectral features is able to reproduce the IF images.
Figure 3.
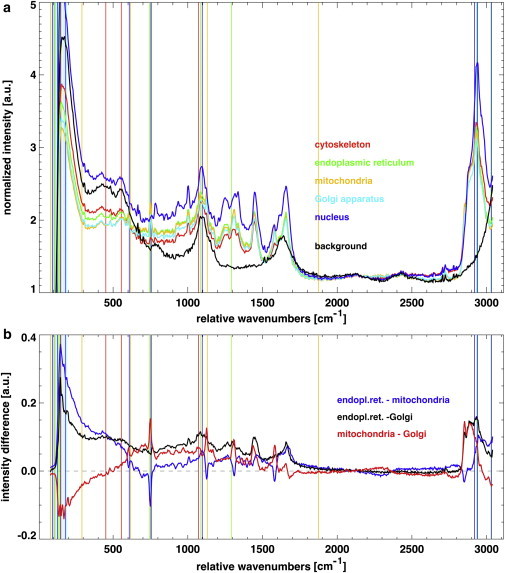
(a) The five different cellular compartments can be distinguished by sets of Raman intensities at selected wavenumbers determined by an information-based approach (for more details, see text). The average spectrum of each respective compartment is shown together with its characteristic barcode. The broad band observed at ∼1100 cm−1 originates from the glass substrate, and the one at 1600 cm−1 originates from the buffer solution (PBS). (b) The curves show the difference between the average spectra of the organelles displayed in panel a. The corresponding barcodes have also been included for the sake of clarity. (In color online.)
Due to the biological colocalization of the organelles, the spatial regions associated with these three cellular compartments as visualized in aIF images may overlap. This is revealed in Fig. 3 a, which shows that the average spectra corresponding to these organelles (endoplasmic reticulum, mitochondria, and Golgi apparatus) are more similar to each other than to those of the nucleus or cytoskeleton. Nonetheless, we observe considerable differentiations in the aIF, which are unveiled by the spectral differences between the organelles displayed in Fig. 3 b. The different curves clearly indicate the singular and common spectral features of the considered cellular compartments.
Results
IF imaging of cells is a standard technique in the life sciences that enables a visualization of selected intracellular structures. In our experimental approach, IF staining is performed subsequent to Raman data acquisition. The lifetime of the cells was much longer than the acquisition times of the Raman recordings.
The approach to generate aIF images of living cells from Raman spectroscopic data is based on the following two assumptions: First, in the training phase, where spectral characteristics are extracted, the location of the intracellular organelles must not change significantly (i.e., in a statistical sense) between the acquisitions of the two image modalities. Second, the fixation process itself does not essentially affect the Raman spectroscopic data at the wavenumbers of relevance. To corroborate our assumptions, we included in our trainings data set both living and fixed samples. For a few cases, we compared the Raman spectroscopic data before and after fixation. In Fig. 4 the aIF images of the living (a) and fixed (c) cells based on Raman spectral data are shown together with the IF image (b).
Figure 4.
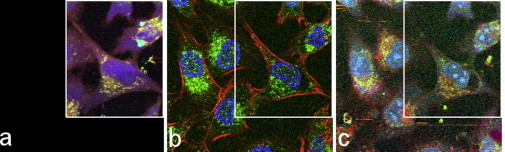
Comparison of aIF images of living (a) and fixed (c) LN18 cells with the corresponding IF image (b). The green channel represents the Golgi apparatus. The overall shape of the cell (cytoskeleton) and the bulk distribution of the specific organelles (Golgi apparatus) are preserved between Raman scanning before and after fixation. The positions of the nucleus and nucleoli in both aIF images are almost unchanged. However, what is probably a fixation artifact can be observed in b and c, affecting the cell in the center. The Raman recording in panel a covers a smaller area compared with b and c. The frame indicates the common part of all images.
Thus, we may assume that both imaging procedures are applied to a practically unchanged object. A comparison of IF and Raman images as shown in Figure 2, Figure 5 confirms that the prominent structures are also revealed by Raman imaging, a fact already exploited by the registration procedure.
Figure 5.
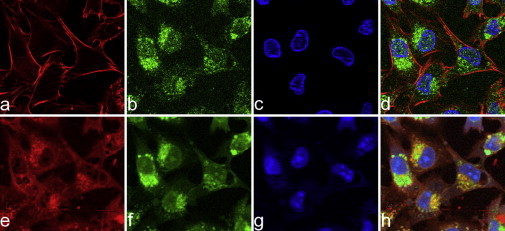
Comparison of original IF images (a–d) and (Raman-based) aIF images (e–h). Generally, the images show high similarities. However, the red channel corresponding to the cytoskeleton differs in that panel a shows high localization, and panel e reveals that actin is actually present throughout the cells. Thus, the correlation between a and e is only 0.36. Another obvious difference is observed between c and g (blue channel): In the IF image the nucleus is inhomogeneously stained, whereas the aIF image reveals the presence of nucleoli at these sites. Yet the correlation is 0.74. (b and f) The images of the Golgi apparatus (green channel) yield a correlation of 0.71. Thus, the RGB overlays in d and h are in great agreement.
Antibodies against COX-IV, Calnexin, and Syntaxin-6 revealed specific information about the subcellular structure and its exact location in LN-18 glioma cells.
Rhodamin-conjugated phalloidin depicts the cellular boundaries through a specific binding to the cytoskeleton, whereas DAPI stains the nucleus, sparing the nucleoli. Fig. 5 d shows a three-channel IF image of a specimen from the LN-18 cell line considered in this work. One can clearly identify the described compartments, i.e., the nucleus (blue), actin filaments (red), and Golgi apparatus (green channel). Fig. 5, a–c, show the intensities in the individual RGB channels separately. To demonstrate the feasibility of our approach, we generated aIF images by evaluating the pairwise relations between Raman spectra and the intensity in the corresponding IF image according to the methodology presented above. An example of such an aIF image construction from Raman data and the corresponding original IF image are shown in Fig. 5, h and d, respectively. A visual inspection reveals that the corresponding channels (columns in Fig. 5) share a great deal of information. Fig. 5, e–g, show the resulting aIF images based on the Raman data. We observe that within the region of the nucleus, circumscribed regions show strong signals in the Raman data (Fig. 5 g), whereas there is only low intensity in the IF image (Fig. 5 c). We assign these regions to nucleoli. Note that we chose the settings for fluorescence imaging such that saturation effects were minimized. The nuclei of cells are reliably observed by both imaging approaches. This is also reflected by the correlation coefficient of ∼0.74 calculated beween these two images.
When considering the red channels of IF and aIF images (Fig. 5, a and e), one can see an intrinsic difference in the pictures. The targeting molecule of the red color staining of the cells relies on the binding of phalloidin to actin. Most actin is arranged as microfilaments in the cytoskeleton of the cell. Filamentous actin can easily be depicted in IF microscopy. The Raman signals from actin monomers floating inside the cells are highly similar to those from actin polymers bundled together in the cytoskeleton. This largely explains why we obtain a reddish shading of the entire cell body in the red channel of the aIF image. Because the localization of the cytoskeleton signals is not identical due to the differences in imaging modalities, the correlation coefficient accordingly drops to 0.36.
For the green channel, we chose three targets: the endoplasmic reticulum, mitochondria, and the Golgi apparatus. These compartments represent the major components of the cell and form the structural basis for fundamental cellular functions. Therefore, their localization and investigation on an ultrastructural level are important for a functional understanding of intracellular processes. The chosen antibodies specifically depict the distribution of the discussed organelles. This in turn allows us to allocate and exactly map them in the aIF images. Fig. 5, b and f, show the reconstruction of the Golgi apparatus. In this case, the correlation coefficient between the images is 0.71.
This approach, however, bears the risk of overfitting when accidental relations in pairs of images dominate the fitting procedure. To eliminate this risk of overfitting, a leave-one-out technique is applied. For each generation of an aIF image, the training sample is formed by all images except the one dataset considered. Moreover, this allows us to create aIF images with more than three channels (cell compartments) as long as the training set comprises the required information. Once the rules (fitting parameters) are established, this approach can be applied to any Raman spectral dataset. Fig. 6 illustrates three examples of this kind of construction. Note that this approach (unlike the IF images) allows us to compute image constructions of all five cellular components available in the training-sample pool, namely, the cytoskeleton (always shown in red), the nucleus (always blue), the endoplasmic reticulum (green I), mitochondria (green II), and Golgi apparatus (green III) at the same time. The leftmost column in Fig. 6 shows the original Raman data. For comparison with the originally stained organelles, see the image pairs [A2, A5], [B3, B5], and [C4, C5], respectively.
Figure 6.
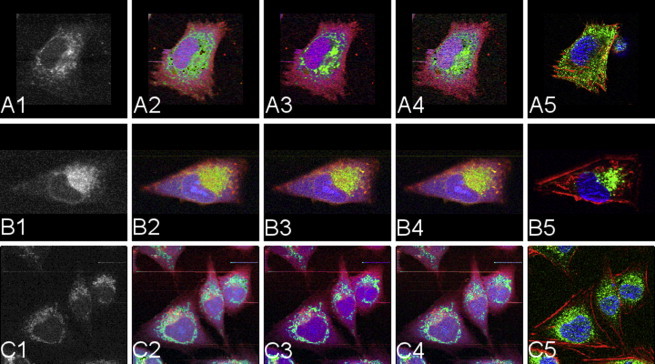
IF and aIF images from Raman data. The leftmost column 1 shows the integrated Raman images. Columns 2–4 display the aIF images , which are sensitive to the signal of the endoplasmic reticulum, mitochondria, and the Golgi apparatus (green channel of the RGB images, from left to right). Among these reconstructions, pictures A2, B3, and C4 should be compared with the respective RGB overlay of IF images in the rightmost column 5. Rows A–C correspond to the three different targets for the green fluorophores. The red (blue) channel of the constructions and the IF images represents the signal of the cytoskeleton (nucleus).
The IF images and the aIF images both show the expected cellular structure of LN-18. These cells are of a bipolar or stellate shape, dominated by a prominent, plemorphic nucleus that shows inhomogeneous absorption of the fluorescent marker revealing the position of the nucleolus. The endoplasmic reticulum is attached to the nuclear membrane and spreads out through the cytoplasm of the cell. mRNA crossing this membrane encodes proteins in the endoplasmic reticulum, and these proteins are then processed further in the Golgi apparatus. Thus, we expect a high density of endoplasmic reticulum close to the nucleus, which then transitions to the regions of the Golgi system. It is known that highly proliferative LN-18 cells express high numbers of mitochondria that occupy a major part of the cytoplasm. In our data, the mitochondria display a distinct and strong signal, and are typically observed in the periphery of the nucleus.
We performed a number of scans with corresponding IF images to repeat the trained algorithm and confirm the accuracy of our approach. A first impression of the precision is given by images that show more than one single cell (Fig. 5). Here, the features are detectable in all cells shown in this field of view.
Discussion
Live-cell imaging has become an important tool in cell biology. It allows the investigation of cellular functions down to the molecular level. To date, such techniques have depended on recombinant alterations or the intravital labeling of cells, and the majority of protocols allow the registration of only a small number of markers (usually one marker per experiment). Alternatively, cells must be fixed for labeling with multiple markers. Confocal Raman spectroscopy together with our methodology to construct aIF images offers a technology for comprehensive assessment of functional processes on a label-free and live-cell basis.
Because our algorithm is trained for five different stainings, it allows the composition of five-color-channel images from the Raman spectral datasets instead of the mere RGB information obtained from fluorescence imaging, and, most importantly, without any directed, preselected staining. This is only possible because there is a higher amount of biochemical information on the scanned section of a cell in the Raman data, whereas IF is restricted to specific, predetermined parts associated with the three practicable channels. However, we have to consider that there are other sources of signal besides those of biochemical origin, such as the medium, glass, or possible autofluorescence. We tried to minimize the influence of the medium and the object slide on the Raman data by means of our experimental protocol. Autofluorescence was not observed in any measurement.
A close observation of the features shown in Fig. 6 demonstrates that the aIF images mostly match their counterparts on the IF side. Moreover, the solutions are plausible when an IF counterpart for the same organelle is not available. The mitochondria are spread throughout the cytoplasm, whereas the endoplasmic reticulum tends to show higher concentration around the nucleus accompanied by the Golgi apparatus. Because of their biological colocalization, the latter two have to appear at similar positions in the IF and aIF images. To assess the quality of the computed representations (aIF images), one can compare (in Fig. 6) panels A2 and A5 for endoplasmic reticulum, B3 and B5 for mitochondria, and C4 and C5 for the Golgi apparatus. Correlation of the Raman-based aIF images with the gold standard of IF imaging proves that specific organelles match not only by visual judgement but also by quantitative evaluation.
We note that in all of the images analyzed in this work, phalloidin yields a very distinct, line-like appearance of the actin structures of the cytoskeleton in the IF images, whereas they seem rather blurred in the aIF images. This may be due to the functional properties of phalloidin, which prefentially binds much more tightly to actin filaments than to actin monomers. The constructed aIF channel shows us that the globular subunits of actin (G-actin) in addition to the filamentous F-actins are relatively widely spread throughout the cell, but of course are mostly condensed in the cytoskeleton. This is in agreement with the biochemical picture of the cytoskeleton as a network that extends between the cellular structures.
The aIF channel constructed for the nucleus fits very well with the corresponding IF image, but also reveals an additional feature. The aIF channel shows a strong signal for the nucleoli, which is absent in the IF images. In the nucleus the DAPI stain attaches to the minor grooves of the DNA helices. The nucleolus, however, is formed by a combination of the nucleolus organizing regions of five chromosomes into a very compact structure where ribosomal RNA is produced. Thus, the structure is highly compact and the DNA no longer provides the adsorption sites for DAPI. This is why DAPI cannot stain the nucleolus while the spectroscopic signature in the Raman spectra is maintained. Hence, the fitting procedure shows a higher density of nucleus-related material in the areas of the nucleoli.
Overall, the localization of the main organelles is feasible through the exact overlaying and specific mapping performed with the IF images. Images of more than one cell give an impression of the reproducibility of the results obtained (see Fig. 5).
When selecting the five different cellular components, we took care to cover a great proportion of the cells' volume. Because we did not consider other organelles explicitly, their specific contribution to the spectra cannot be distinguished. However, because we averaged over an extensive dataset while training the algorithm, the contribution of such additional organelles should average out in a statistical sense.
One of the major aspects of this work is illustrated by the barcode visualization in Fig. 3. It is difficult to interpret Raman data of living cells in terms of the cellular compartments based on single spectral lines. Instead, we interpret the barcode as a combination of spectral characteristics representing the different structures and organelles. The information content is tied to certain peaks but also exploits other spectral features. Because the biochemical composition of a cellular compartment is very complex, it seems to be more promising for a quantitative analysis to evaluate the collective information content of a set of lines than to consider single line properties independently.
Conclusions
We have presented a method for evaluating the biochemical information given in Raman datasets of living cells. IF staining and imaging of the cells previously investigated by Raman microscopy allowed an ultrastuctural mapping of cellular compartments and the registration with Raman spectra at these positions. The successful IF reconstructions demonstrate that our method is able to capture the specific Raman spectral characteristics of the different cellular compartments in addition to its single constituents (i.e., proteins, carbohydrates, and fatty acids). Raman spectroscopy is of great interest in molecular pathology because it may facilitate the identification of cellular alterations in various human diseases, including tumors, inflammation, and neurodegenerative diseases. Moreover, the method is label-free and avoids technical bias due to fixation and staining procedures. It is also applicable to living cells and therefore clearly extends the spectrum of experimental protocols in molecular medicine.
Acknowledgments
We thank Ingrid Höpner and Guido Piontek for assisting with the cell culture, and Michael Bauer for fruitful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft Cluster of Excellence “Nanosystems Initiative Munich” and grant DFG-SFB-824 (Project B6: Imaging for Selection, Monitoring and Individualization of Cancer Therapies).
Editor: Leonid S. Brown.
References
- 1.Carey P.R. Raman crystallography and other biochemical applications of Raman microscopy. Annu. Rev. Phys. Chem. 2006;57:527–554. doi: 10.1146/annurev.physchem.57.032905.104521. [DOI] [PubMed] [Google Scholar]
- 2.Krafft C., Dietzek B., Popp J. Raman and CARS microspectroscopy of cells and tissues. Analyst (Lond.) 2009;134:1046–1057. doi: 10.1039/b822354h. [DOI] [PubMed] [Google Scholar]
- 3.Owen C.A., Notingher I., et al. Hench L.L. Progress in Raman spectroscopy in the fields of tissue engineering, diagnostics and toxicological testing. J. Mater. Sci. Mater. Med. 2006;17:1019–1023. doi: 10.1007/s10856-006-0438-6. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths J. Raman spectroscopy for medical diagnosis. Anal. Chem. 2007;79:3975–3978. doi: 10.1021/ac071917k. [DOI] [PubMed] [Google Scholar]
- 5.Krafft C., Sobottka S.B., et al. Salzer R. Near infrared Raman spectroscopic mapping of native brain tissue and intracranial tumors. Analyst (Lond.) 2005;130:1070–1077. doi: 10.1039/b419232j. [DOI] [PubMed] [Google Scholar]
- 6.Draux F., Jeannesson P., et al. Sockalingum G.D. Raman spectral imaging of single living cancer cells: a preliminary study. Analyst (Lond.) 2009;134:542–548. doi: 10.1039/b812610k. [DOI] [PubMed] [Google Scholar]
- 7.Fang H., Qiu L., et al. Perelman L.T. Confocal light absorption and scattering spectroscopic microscopy. Appl. Opt. 2007;46:1760–1769. doi: 10.1364/ao.46.001760. [DOI] [PubMed] [Google Scholar]
- 8.Hamada K., Fujita K., et al. Kawata S. Raman microscopy for dynamic molecular imaging of living cells. J. Biomed. Opt. 2008;13:044027. doi: 10.1117/1.2952192. [DOI] [PubMed] [Google Scholar]
- 9.Krafft C. Bioanalytical applications of Raman spectroscopy. Anal. Bioanal. Chem. 2004;378:60–62. doi: 10.1007/s00216-003-2266-6. [DOI] [PubMed] [Google Scholar]
- 10.Notingher I., Bisson I., et al. Hench L.L. In situ spectral monitoring of mRNA translation in embryonic stem cells during differentiation in vitro. Anal. Chem. 2004;76:3185–3193. doi: 10.1021/ac0498720. [DOI] [PubMed] [Google Scholar]
- 11.Notingher I., Hench L.L. Raman microspectroscopy: a noninvasive tool for studies of individual living cells in vitro. Expert Rev. Med. Devices. 2006;3:215–234. doi: 10.1586/17434440.3.2.215. [DOI] [PubMed] [Google Scholar]
- 12.Matthäus C., Kale A., et al. Diem M. New ways of imaging uptake and intracellular fate of liposomal drug carrier systems inside individual cells, based on Raman microscopy. Mol. Pharm. 2008;5:287–293. doi: 10.1021/mp7001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krafft C., Knetschke T., et al. Salzer R. Studies on stress-induced changes at the subcellular level by Raman microspectroscopic mapping. Anal. Chem. 2006;78:4424–4429. doi: 10.1021/ac060205b. [DOI] [PubMed] [Google Scholar]
- 14.Swain R.J., Stevens M.M. Raman microspectroscopy for non-invasive biochemical analysis of single cells. Biochem. Soc. Trans. 2007;35:544–549. doi: 10.1042/BST0350544. [DOI] [PubMed] [Google Scholar]
- 15.Matthäus C., Chernenko T., et al. Diem M. Label-free detection of mitochondrial distribution in cells by nonresonant Raman microspectroscopy. Biophys. J. 2007;93:668–673. doi: 10.1529/biophysj.106.102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthäus C., Boydston-White S., et al. Diem M. Raman and infrared microspectral imaging of mitotic cells. Appl. Spectrosc. 2006;60:1–8. doi: 10.1366/000370206775382758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Gelder J., De Gussem K., et al. Moens L. Methods for extracting biochemical information from bacterial Raman spectra: focus on a group of structurally similar biomolecules—fatty acids. Anal. Chim. Acta. 2007;603:167–175. doi: 10.1016/j.aca.2007.09.049. [DOI] [PubMed] [Google Scholar]
- 18.Jess P.R., Smith D.D., et al. Herrington C.S. Early detection of cervical neoplasia by Raman spectroscopy. Int. J. Cancer. 2007;121:2723–2728. doi: 10.1002/ijc.23046. [DOI] [PubMed] [Google Scholar]
- 19.Puppels G.J., Garritsen H.S., et al. Greve J. Raman microspectroscopic approach to the study of human granulocytes. Biophys. J. 1991;60:1046–1056. doi: 10.1016/S0006-3495(91)82142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strehle M.A., Rosch P., et al. Popp J. A Raman spectroscopic study of the adsorption of fibronectin and fibrinogen on titanium dioxide nanoparticles. Phys. Chem. Chem. Phys. 2004;6:5232–5236. [Google Scholar]
- 21.Pully V.V., Otto C. The intensity of the 1602 cm−1 band in human cells is related to mitochondrial activity. J Raman Spectrosc. 2009;40:473–475. [Google Scholar]
- 22.Reference deleted in proof.
- 23.Bonnier F., Knief P., et al. Byrne H.J. Imaging live cells grown on a three dimensional collagen matrix using Raman microspectroscopy. Analyst (Lond.) 2010;135:3169–3177. doi: 10.1039/c0an00539h. [DOI] [PubMed] [Google Scholar]
- 24.Pully V.V., Lenferink A.T.M., Otto C. Time-lapse Raman imaging of single live lymphocytes. J Raman Spectrosc. 2011;42:167–173. [Google Scholar]
- 25.Hartigan J.A. John Wiley & Sons; New York: 1975. Clustering Algorithms. [Google Scholar]
- 26.Jolliffe I.T. Springer; New York: 2002. Principal Component Analysis. [Google Scholar]
- 27.Diserens A.C., de Tribolet N., et al. Carrel S. Characterization of an established human malignant glioma cell line: LN-18. Acta Neuropathol. 1981;53:21–28. doi: 10.1007/BF00697180. [DOI] [PubMed] [Google Scholar]
- 28.Green A.A., Berman M., et al. Craig M.D. A transformation for ordering multispectral data in terms of image quality with implications for noise removal. IEEE Trans. Geosci. Remote Sens. 1988;26:65–74. [Google Scholar]
- 29.Reddy R.K., Bhargava R. Accurate histopathology from low signal-to-noise ratio spectroscopic imaging data. Analyst (Lond.) 2010;135:2818–2825. doi: 10.1039/c0an00350f. [DOI] [PubMed] [Google Scholar]
- 30.Maes F., Collignon A., et al. Suetens P. Multimodality image registration by maximization of mutual information. IEEE Trans. Med. Imaging. 1997;16:187–198. doi: 10.1109/42.563664. [DOI] [PubMed] [Google Scholar]
- 31.Pluim J.P.W., Maintz J.B.A., Viergever M.A. Mutual-information-based registration of medical images: a survey. IEEE Trans. Med. Imaging. 2003;22:986–1004. doi: 10.1109/TMI.2003.815867. [DOI] [PubMed] [Google Scholar]