

Abstract
Myofibroblasts are specialized contractile cells that participate in tissue fibrosis and remodeling, including idiopathic pulmonary fibrosis (IPF). Mechanotransduction, a process by which mechanical stimuli are converted into biochemical signals, regulates myofibroblast differentiation. Relaxin is a peptide hormone that mediates antifibrotic effects through regulation of collagen synthesis and turnover. In this study, we demonstrate enhanced myofibroblast contraction in bleomycin-induced lung fibrosis in mice and in fibroblastic foci of human subjects with IPF, using phosphorylation of the regulatory myosin light chain (MLC20) as a biomarker of in vivo cellular contractility. Compared with wild-type mice, relaxin knockout mice express higher lung levels of phospho-MLC20 and develop more severe bleomycin-induced lung fibrosis. Exogenous relaxin inhibits MLC20 phosphorylation and bleomycin-induced lung fibrosis in both relaxin knockout and wild-type mice. Ex vivo studies of IPF lung myofibroblasts demonstrate decreases in MLC20 phosphorylation and reduced contractility in response to relaxin. Characterization of the signaling pathway reveals that relaxin regulates MLC20 dephosphorylation and lung myofibroblast contraction by inactivating RhoA/Rho-associated protein kinase through a nitric oxide/cGMP/protein kinase G–dependent mechanism. These studies identify a novel antifibrotic role of relaxin involving the inhibition of the contractile phenotype of lung myofibroblasts and suggest that targeting myofibroblast contractility with relaxin-like peptides may be of therapeutic benefit in the treatment of fibrotic lung disease.
Idiopathic pulmonary fibrosis (IPF) is a lethal fibrotic lung disease characterized by excessive deposition of extracellular matrix (ECM) in the lung parenchyma. Myofibroblasts are key effectors of the tissue remodeling process in IPF.1 These cells are specialized contractile cells that possess characteristics of both ECM-producing fibroblasts and α-smooth muscle actin (α-SMA)–expressing smooth muscle cells (SMCs).2 Recent studies3, 4, 5, 6 suggest that acquisition of contractile activity may not simply be a phenotypic marker of myofibroblasts; rather, myofibroblast contractile force generation may provide a feed-forward mechanism for maintaining persistent myofibroblast differentiation in progressive fibrosis through the conversion of mechanical stimuli into biochemical signals, a process known as mechanotransduction.7 Understanding the (dys)regulation of myofibroblast contraction will provide necessary means for the determination of the role of myofibroblast contraction in the regulation of persistent/progressive fibrosis and may potentially lead to effective therapeutic approaches for the treatment of devastating fibrotic diseases.
Recent evidence5 supports the concept that myofibroblast mechanotransduction involves fibrogenic signaling via contractile force-mediated activation of latent transforming growth factor (TGF)-β1 bound to the ECM. In this process, stress fiber–generated contractility is transmitted from the cytoskeleton to the ECM through the transmembrane integrins, primarily integrin αvβ5.5 The force transmission causes a conformational change of the ECM-bound latent TGF-β1 complex, leading to the release (or exposure) of active TGF-β1 that is then able to bind to its cognate receptor(s). Recent studies6 from our laboratory demonstrated that interruption of integrin αvβ5–TGF-β1 interactions by Thy-1, a glycosyl-phosphatidylinositol–linked cell surface protein, blocks fibroblast contraction–induced latent TGF-β1 activation and TGF-β1–dependent lung myofibroblast differentiation. In addition to this extrinsic pathway, myofibroblast contraction may trigger fibrogenic mechanotransduction via an intrinsic pathway that involves the release/activation of intracellular transcription factor(s). Contractile force generation is primarily regulated by Rho/Rho-associated kinase (ROCK) signaling, which regulates actin cytoskeleton dynamics.8, 9, 10, 11 Activation of Rho/ROCK promotes monomeric G-actin polymerization into filamentous actin (F-actin), resulting in nuclear import of myocardin-related transcription factor-A, a serum-responsive factor coactivator.12 In the nucleus, myocardin-related transcription factor-A binds serum-responsive factor and activates fibrogenic gene programs that promote myofibroblast differentiation, collagen synthesis, and myofibroblast survival.13, 14, 15, 16 In addition, cell-derived contractile forces unfold the cryptic sites of ECM protein fibrils, which may potentially trigger autofibrillogenesis and long matrix fibril formation.17 Similarly, fibroblast contraction-induced mechanical deformation renders an extra domain-A fibronectin segment available for specific integrins, a process essential for TGF-β1–induced myofibroblast differentiation.18, 19 In addition to cell-derived contractile forces, externally applied forces (eg, stretching and breathing) and changes in the mechanical properties of the ECM (eg, matrix stiffness) may also activate mechanotransduction events that regulate cellular functions.
Both muscle contraction and nonmuscle cell contraction are regulated by myosin light chain (MLC20) phosphorylation.2 When phosphorylated, MLC20 triggers actin filaments to slide past one another to power actin cytoskeletal contraction.20 Dephosphorylation of MLC20 results in relaxation of the initial and sustained phase of contraction.21, 22 Mechanisms invoked for sustained myofibroblast contraction involve inhibition of MLC phosphatase activity via activation of the small GTPase RhoA and its downstream target ROCK. On activation, ROCK promotes MLC20 phosphorylation by inhibition of MLC phosphatase via phosphorylation of the regulatory myosin phosphatase targeting subunit-1 (MYPT1) and the MLC phosphatase inhibitors PKC-potentiated phosphatase inhibitor of 17 kDa and phosphatase holoenzyme inhibitor-1.23, 24
Relaxin is a 6-kDa peptide hormone that is structurally related to the insulin family of peptides25 and inhibits fibrogenesis in multiple organs, including the lung.26 Continuous infusion of relaxin using s.c. implanted osmotic minipumps reduces alveolar thickening and collagen deposition in bleomycin-treated CBA/J mice and inhibits the accumulation of collagen in the airways of sensitized mice and guinea pigs in response to ovalbumin.27, 28, 29 Relaxin knockout (KO) mice develop age-associated pulmonary fibrosis, which can be partially reversed by supplementation of exogenous relaxin.30 In addition, established fibrosis in aged relaxin KO mice appears to be further exacerbated by exposure to ovalbumin.31 In vitro studies32, 33, 34, 35, 36, 37, 38 have shown that relaxin inhibits collagen production and increases matrix metalloproteinase expression/activity by cardiac, renal, and skin fibroblasts. Exposure of activated hepatic stellate cells to relaxin results in decreased collagen synthesis and deposition; tissue inhibitors of metalloproteinases 1 and 2 are decreased in the cell-conditioned media.39 Based on these findings, the ability to regulate collagen synthesis and turnover may mediate the antifibrotic effect of relaxin. More recent studies40, 41 report that relaxin inhibits TGF-β1–stimulated myofibroblast differentiation, both in cultured renal fibroblasts in vitro and in myocardial infarction–induced cardiac fibrosis in vivo, suggesting that relaxin regulates myofibroblast differentiation in addition to modulating collagen synthesis and turnover.
Relaxin inhibits uterine artery and myometrium contraction in pregnant rats and cervical smooth muscle contraction in women in early pregnancy.42, 43, 44 In vitro studies45, 46, 47 showed that relaxin induces relaxation of myometrial cells, vascular SMCs, and TGF-β1–treated renal fibroblasts. In the present study, we tested the hypothesis that relaxin inhibits myofibroblast contractility and mediates antifibrotic effects in vivo. We show that relaxin induces dephosphorylation of MLC20 and inhibits contraction of lung myofibroblasts isolated from patients with IPF. Overexpression of phosphomimetic MLC20 mutant blocks relaxin inhibition of lung myofibroblast contraction. Furthermore, our studies indicate that relaxin mediates MLC20 dephosphorylation and lung myofibroblast relaxation by induction of RhoA phosphorylation and ROCK inhibition through a nitric oxide (NO)/cGMP/protein kinase G (PKG)–dependent mechanism. In vivo studies show that MLC20 phosphorylation is increased in the fibroblastic foci in both human IPF and bleomycin-treated mouse lung fibrosis. Mice null for relaxin express higher levels of phospho-MLC20 and develop more severe lung fibrosis in response to bleomycin compared with wild-type (WT) mice. Continuous infusion of recombinant relaxin for 7 days (days 14 to 21 after bleomycin injury) inhibits bleomycin-induced MLC20 phosphorylation and attenuates lung fibrosis in both KO and WT mice. Together, this study identifies a novel antifibrotic mechanism of relaxin involving the inhibition of myofibroblast contractility.
Materials and Methods
Antibodies, Plasmids, and Reagents
Anti-phospho-MLC20 antibody for immunohistochemical (IHC) and immunofluorescent staining and anti-green fluorescent protein antibody were obtained from Rockland Immunochemicals (Gilbertsville, PA). Anti-phospho-MLC20 antibody and anti-MLC20 antibody for immunoblot analyses were obtained from Cell Signaling (Danvers, MA). Isoform-specific anti-neuronal NO synthase (nNOS), anti-inducible NO synthase (iNOS), and anti-endothelial NOS (eNOS) antibodies were from Enzo Life Sciences (Plymouth Meeting, PA). Anti-phospho-RhoA (Ser188) was from EMD Chemicals (Gibbstown, NJ). Anti-ROKα/ROCK II antibody was from Millipore (Billerica, MA). Anti-RhoA antibody and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-α-SMA antibody was from Sigma (St. Louis, MO). Anti-phospho-moesin (Thr558) antibody and anti-moesin antibody were from Abcam (Cambridge, MA). All secondary antibodies were from Southern Biotech (Birmingham, AL).
Plasmids encoding an N-terminal enhanced GFP tag and human WT MLC20 or mutated MLC20, in which Thr18 and Ser19 were replaced with aspartic acids (DD-MLC20, a phosphomimetic mutant), were constructed as previously described.48 pEGFP-RhoAV14 plasmid was generated by subcloning a RhoAV14 fragment from pcDNA3RhoAV14 (UMR cDNA Resource Center, Rolla, MO) into pEGFPC1 plasmid (Clontech, Mountain View, CA). pEGFP-RhoAV14A188 was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Recombinant Human Relaxin 2 was obtained from R&D Systems (Minneapolis, MN). Pure Silicone Fluid (30,000cSt) was from Clearco (Bensalem, PA). Cyclic AMP Enzyme Immunoassay kit was obtained from Assay Designs (Ann Arbor, MI). Masson's Trichrome Method for Connective Tissue was obtained from Poly Scientific (Bay Shore, NY). PepTag assay for nonradioactive detection of cAMP-dependent protein kinase was from Promega (Madison, WI). Jasplakinolide was from Fisher Scientific (Pittsburgh, PA). NG-methyl-L-arginine acetate (L-NMMA), 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), S-nitroso-N-acetyl penicillamine (SNAP), curcumin, 8-(4-chlorophenylthio)-guanosine 3′,5′-cyclic monophosphate sodium (8-PCPT-cGMP), and DT-2 were from Sigma. Rhodamine phalloidin was from Invitrogen (Carlsbad, CA). Tissue Protein Extraction reagent and Bicinchoninic Acid Protein Assay kit were from Pierce (Rockford, IL).
Lung Fibroblast Isolation, Culture, Transfection, Sorting, and Treatment
Human lung (myo)fibroblasts were established from tissue samples from patients undergoing lung transplantation for IPF, as approved by the Human Studies Committee of the University of Alabama at Birmingham. Lungs were minced in sterile PBS, and tissue pieces were placed in 100-mm tissue culture dishes containing Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum (FBS), 1% penicillin/streptomycin/glutamine, nonessential amino acids, and sodium pyruvate (supplemented DMEM). Medium was replenished every 3 days. After 14 days, cells growing out of the explants were trypsinized and plated in supplemented DMEM. Lung (myo)fibroblasts were used between passages 6 and 10.
Plasmid transfection was performed using a Nucleofector device (Amaxa, Inc., Cologne, Germany), as previously described.49 Cells expressing EGFP and EGFP-tagged WT MLC20, DD-MLC20, RhoAV14, and RhoAV14A188 were sorted using a FACSTARPlus Cell Sorter (Becton-Dickinson, Mountain View, CA) under sterile conditions.
Lung (myo)fibroblasts were grown in DMEM containing 10% FBS until 80% confluence. Cells were rinsed with DMEM plus 0.1% FBS twice and were incubated in DMEM plus 0.1% FBS containing 1 to 100 nmol/L relaxin in the presence or absence of 100 μmol/L L-NMMA, 20 μmol/L PTIO, 100 μmol/L SNAP, 200 μmol/L 8-PCPT-cGMP, 1 nmol/L to 10 μmol/L forskolin, 30 μmol/L curcumin, or 1 μmol/L DT-2 for 24 hours.
Wrinkle Assay
The wrinkle assay for measurement of fibroblast contractility was performed as described in the previous studies.6, 50, 51 Briefly, approximately 15 μL of silicone monomer was applied onto 18-mm glass coverslips and allowed to spread for 30 minutes. The upper layer of silicone was polymerized by exposure of the coverslip to an open flame for 1.5 seconds. The silicone coverslips were placed into a 12-well plate and were equilibrated with 10 μg/mL collagen type I in serum-free DMEM media, sterilized by UV light exposure, and left overnight in the incubator at 37°C. Cells (104 cells/cm2) were plated onto silicone coverslips in DMEM containing 0.1% FBS in the presence or absence of 100 nmol/L relaxin for 24 hours. Cell contraction on deformable silicone substrates was assessed by the formation of wrinkles seen with a ×20 objective on a Nikon Eclipse TE 300 microscope (Nikon Instruments Inc., Melville, NY). Wrinkling cells were calculated as a percentage of the total cells. Mean values were calculated from 10 random regions, and at least three independent experiments were performed.
Immunofluorescence and Confocal Laser Scanning Microscopy
To observe wrinkles and α-SMA–containing stress fibers, silicone-coated coverslips were fixed in 4% paraformaldehyde in PBS for 20 minutes and permeabilized with 0.5% Triton X-100 in PBS for 10 minutes. Cells were incubated with anti-α-SMA antibody at a concentration recommended by the manufacturer at room temperature for 1 hour. Fluorescein isothiocyanate–conjugated goat anti-mouse IgG1 was used as a secondary antibody. To stain F-actin, cells were incubated with rhodamine-conjugated phalloidin at room temperature for 1 hour in the dark. Nuclei were stained with DAPI. Results were imaged with a Zeiss LSM510 confocal microscope equipped with a digital color camera (AxioCam) and MetaMorph Imaging software (Universal Imaging Corp, Downingtown, PA).
Measurement of NO Production
NO production was evaluated by spectrophotometrical measurement of nitrite and nitrate accumulation in cell culture supernatants using a Nitric Oxide Detection kit (Assay Designs), according to the manufacturer's recommendations. Absorbance was measured at 546 nm using a microplate reader. Results are expressed as picomoles of total nitrite plus nitrate produced per 106 cells.
Measurement of Intracellular cGMP and cAMP Production
Intracellular cGMP/cAMP levels were determined by colorimetric competitive immunoassay using cGMP/cAMP Complete EIA kits (Assay Design). Cells were lysed in 0.1 mol/L HCl to stop endogenous phosphodiesterase activity and to stabilize the released cGMP/cAMP. Standards and samples were loaded into 96-well plates coated with goat anti-rabbit IgG. Plates were incubated with cGMP/cAMP conjugated to alkaline phosphatase, followed by rabbit polyclonal antibodies to cGMP/cAMP. Plates were washed, leaving only bound cGMP/cAMP. pNpp substrate solution was added to generate yellow color when catalyzed by the alkaline phosphatase on the cGMP/cAMP conjugates. Optical density was determined in a plate reader at 450 nm. cGMP/cAMP levels were calculated from a standard curve.
PKG and PKA Activity Assay
PKG activity was assayed using a cGK Assay kit (MBL International, Woburn, MA). Briefly, cell lysates were diluted in kinase buffer and pipetted into the wells precoated with PKG-specific substrates. The amounts of phosphorylated substrates were measured by binding with a horseradish peroxidase–conjugated PKG substrate–specific antibody, which then catalyzed the conversion of the chromogenic substrate tetramethylbenzidine from a colorless solution to a yellow solution. The color is quantified by spectrophotometry at 450 nm.
Protein kinase A (PKA) activity was determined using a PepTag Assay (Promega). This assay uses brightly colored fluorescent peptide substrates, LRRASLG (Kemptide), highly specific for PKA. The phosphorylation of Kemptide by PKA alters the peptide's net charge from 1 to -1. This change in the net charge of the substrate allows the phosphorylated and nonphosphorylated substrates to be rapidly separated on a 0.8% agarose gel. The phosphorylated substrates migrated toward the cathode, whereas the nonphosphorylated substrates migrated toward the anode. Gel was photographed under UV light. PKA activity was assessed by the intensity of phosphorylated substrates that migrated toward the cathode.
Rho Kinase Activity Assay
Rho kinase activity was assessed using the ROCK Activity Assay kit (Cell Biolabs, San Diego, CA). Plates were precoated with substrates corresponding to the C-terminus of MYPT1, which contains a threonine residue that was phosphorylated by ROCK. The specific detector antibody was horseradish peroxidase–conjugated anti-phospho-MYPT1 (AF20). The amount of phosphorylated substrates was measured by binding with horseradish peroxidase–conjugated AF20, which catalyzed the conversion of tetramethylbenzidine to a yellow solution. Results were quantified by a plate reader at 450 nm.
Subcellular Fractionation
Subcellular fractionation was performed as previously described.52 Briefly, cells were lysed in buffer containing 20 mmol/L Tris, pH 7.5; 150 mmol/L NaCl; 1 mmol/L EGTA; 10 mmol/L MgCl2; 25 mmol/L NaF; and 1 mmol/L 4-amidinophenylmethanesulfonyl fluoride. Then, the cells were subjected to ultracentrifugation at 100,000 × g for 60 minutes. The supernatant was saved as the cytosolic fraction. The pellet was sonicated in the previously described buffer containing 1% Triton X-100 and centrifuged at 10,000 × g for 30 minutes. The supernatant was saved as the membrane fraction.
Co-Immunoprecipitation and Immunoblot Analysis
Cells were rinsed three times with ice-cold PBS, pH 7.4, and lysed with ice-cold cell lysis buffer (20 mmol/L Tris, pH 7.5; 150 mmol/L NaCl; 1 mmol/L EDTA; 1 mmol/L EGTA; 1% Triton X-100; 2.5 mmol/L sodium pyrophosphate; 1 mmol/L β-glycerophosphate; 1 mmol/L Na3VO4; 1 μg/mL leupeptin; and 1 mmol/L phenylmethylsulfonyl fluoride) for 5 minutes. After scraping, cell lysates were sonicated on ice three times for 5 seconds each and were centrifuged for 10 minutes at 12,000 × g at 4°C. Co-immunoprecipitation was performed with a ProFound Co-Immunoprecipitation Kit (Pierce, Rockford, IL), as previously described.49
Cell lysates containing 10 to 40 μg total proteins were loaded onto SDS-polyacrylamide gels under reducing conditions. After electrophoresis, proteins were electrophoretically transferred from the gels to nitrocellulose at 100 V for 1.5 hours at 4°C. Membranes were blocked in casein solution (1% casein and 25 mmol/L Na2HPO4, pH 7.1) for 1 hour at room temperature. Primary antibodies were diluted in Tris-buffered saline-tween and casein solution (1:1) at a working concentration recommended by the manufacturers. Membranes were incubated with primary antibodies at room temperature for 1 hour. After extensive washing, membranes were incubated with peroxidase-conjugated secondary antibodies (0.1 μg/mL) diluted in Tris-buffered saline-tween for 1 hour at room temperature. Immunodetection was performed by chemiluminescence.
Animals and Experimental Protocol
Animal use and bleomycin protocols were approved by the Institutional Animal Care and Use Committee, University of Alabama at Birmingham. Pathogen-free female mice, aged 6 to 8 weeks, were used in this study. Relaxin KO mice and WT mice were generated from heterozygous parents (C57B/6J × 129SV background). Bleomycin sulfate (Almirall, Reinbek, Germany) was dissolved in sterile saline solution and intratracheally instilled into mice by a Stepper Repetitive Pipette (Tridak, DYMAX Corporation, Torrington, CT) as a single dose of 0.08 mg in 50 μL of saline solution per animal (5 U/kg body weight). Control mice received 50 μL of saline. For relaxin treatment, osmotic minipumps (model 1007D; Alzet, Cupertino, CA) filled with relaxin at a dosage of 0.5 mg/kg body weight/day over 7 days or an equal volume of PBS was s.c. implanted into mice, 14 days after bleomycin administration.30, 49 Mice were sacrificed at 21 days. Lung tissues were excised and inflated with 4% (mass/volume) paraformaldehyde in PBS for histological and IHC analyses.
To prepare for lung homogenates, fresh lung tissues were transferred to prechilled tubes containing Tissue Protein Extraction reagent and Complete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN) at a proportion of 1 tablet per 10 mL of Tissue Protein Extraction reagent and were homogenized at 4°C. Homogenates were centrifuged at 9000 × g for 10 minutes at 4°C. Supernatants were transferred to clean microcentrifuge tubes. Total protein concentrations in the lung tissue homogenates were determined using a Bicinchoninic Acid Protein Assay kit.
IHC and Histological Characteristics
IHC was performed as previously described.49 Briefly, lung tissue sections (5 μm thick) were immersed in 10 mmol/L sodium citrate buffer (pH 6.0) and heated at 100°C for 10 minutes to unmask antigen. Sections were blocked with 5% normal goat serum and stained with anti-phospho-MLC20 antibody at 2.5 μg/mL. Endogenous peroxidase activity was quenched with 3% H2O2. Staining was developed with biotinylated secondary antibodies, streptavidin-peroxidase, and 3-amino-9-ethylcarbazole chromogen (Vector Laboratories, Burlingame, CA). Masson's trichrome staining for collagen was performed using a kit from Poly Scientific, according to the manufacturer's recommendations. Images were obtained with a Nikon Eclipse TE 300 microscope equipped with a Spot Insight CCD camera and MetaMorph software, version 6.2 r4.
Collagen Content Assay
Levels of collagen in lung tissues were determined by the Sircol collagen assay (Biocolor Ltd, Newtownabbey, UK). Briefly, mouse right lungs were homogenized and collagen was solubilized in 0.5 mol/L acetic acid containing 0.6% pepsin at 4°C overnight. After centrifugation at 15,000 × g for 15 minutes, extracts were incubated with Sirius red dye. Absorbance was determined at 540 nm with a spectrophotometer (Eppendorf, Hauppauge, NY). Collagen content was expressed as micrograms per right lung.53
Statistical Analysis
Statistical differences among treatment conditions were determined using one-way analysis of variance (Newman-Keuls method for multiple comparisons). The analysis was performed with SigmaStat 3.0 software (SPSS Inc., Chicago, IL). P < 0.01 was considered significant.
Results
Phosphorylation of MLC20 Is Prominent in Both the Fibroblastic Foci of Human IPF and Fibrotic Regions of the Murine Lung Injured by Intratracheal Bleomycin
MLC20 phosphorylation regulates both muscle and nonmuscle cell contraction.2 To determine whether increased cell contractility is associated with lung fibrosis in vivo, we performed IHC staining to analyze MLC20 phosphorylation in paraffin-embedded lung tissue sections isolated from patients with IPF and in bleomycin-injured mice. Staining for phospho-MLC20 was prominent in the fibroblastic foci, in which myofibroblasts are predominant, in human IPF lungs and in fibrotic regions of bleomycin-injured mouse lungs (Figure 1, A, C, E, and G). The relatively normal lung areas in IPF patients and saline-treated mice showed no significant alveolar phospho-MLC20 staining (Figure 1, D, F, and H). The arterial smooth muscle, myocardium, and vascular adventitial layer were also positive for phospho-MLC20 (Figure 1, B–D and H), serving as positive controls for staining with this antibody. Tissue sections stained for nonimmune IgG were negative (data not shown). To our knowledge, these data provide the first in vivo evidence that MLC20, a contraction-associated protein, is phosphorylated and activated in fibrotic regions of IPF lung and bleomycin-injured mouse lung.
Figure 1.
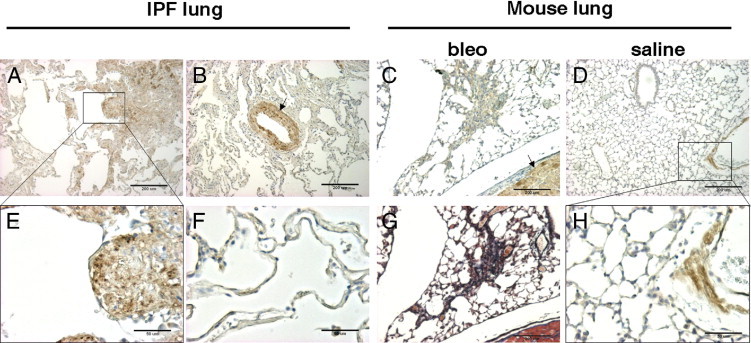
MLC20 phosphorylation is prominent in both the fibroblastic foci in human IPF and in fibrotic regions in bleomycin-induced mouse lung fibrosis. Paraffin-embedded human lung sections from patients with IPF (A, B, E, and F) and mouse lung sections from bleomycin- or saline-treated mice (C, D, G, and H) were stained for phospho-MLC20 (A–F and H) or collagen (G, Masson's trichrome). A: A fibroblastic focus (boxed) and the surrounding area in human IPF. B: An arteriole expressing phospho-MLC20 in SMCs of medial shown as a positive control (arrow). C: A fibrotic region in bleomycin-induced mouse lung fibrosis and adjacent areas; arrow indicates mouse myocardium expressing phospho-MLC20 as a positive control. D: Saline-treated mouse lung, with the boxed area showing a part of the blood vessel wall. E: A magnified view of the fibroblastic focus shown in A. F: A relatively normal lung area in IPF. G: Masson's trichrome staining for the same area shown in C. H: A magnified view of the boxed area shown in D. Scale bars: 200 μm (A, B, and E–G); 50 μm (C, D, and H).
Relaxin Is an Endogenous Inhibitor of MLC20 Phosphorylation and Lung Fibrosis in the Bleomycin Mouse Model
To determine the effect of relaxin on MLC20 phosphorylation in a murine model of lung injury and fibrosis, lungs of relaxin KO and WT mice were injured by intratracheal instillation of the chemotherapeutic agent, bleomycin. Control mice were administered saline. A subgroup of bleomycin-injured mice was given continuous infusions of recombinant relaxin or PBS with s.c. implanted minipumps on days 14 to 21 after bleomycin injury. Lungs were harvested on day 21. Immunoblot analyses showed that saline-treated relaxin KO mice and WT mice expressed low levels of phospho-MLC20 in the lung. Elevated phospho-MLC20 levels were observed in both KO and WT mice injured with bleomycin (Figure 2A). Compared to WT mice, KO mice had a higher level of phospho-MLC20 in response to bleomycin (Figure 2A). The continuous infusion of exogenous relaxin for 7 days inhibited bleomycin-induced MLC20 phosphorylation in both KO and WT mice (Figure 2B). Masson's trichrome staining showed that bleomycin-injured KO mice had more extensive collagen deposition in the lung compared with bleomycin-injured WT mice. Relaxin infusion attenuated collagen deposition in the lungs of both KO and WT mice; these data suggest that relaxin is an endogenous inhibitor of MLC20 phosphorylation and lung fibrosis in vivo.
Figure 2.
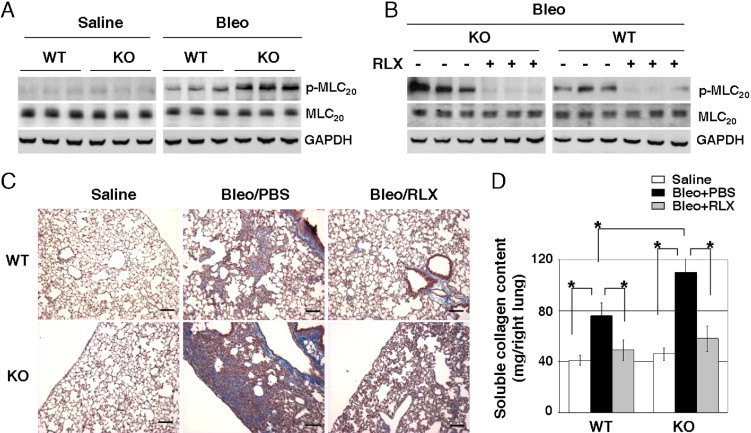
Relaxin (RLX) inhibits MLC20 phosphorylation and lung fibrosis in a bleomycin (Bleo)–induced mouse model. A: Lungs isolated from Bleo- and saline-treated RLX KO and WT mice were homogenized. Levels of phospho-MLC20 (p-MLC20) and total MLC20 in lung homogenates were determined by immunoblot analyses. GAPDH was used as the loading control. B: The RLX KO and WT mice were administered Bleo to induce lung fibrosis. At day 14 after lung injury, mice were given a continuous infusion of exogenous RLX or an equal volume of PBS (vehicle for RLX) through a s.c. implanted minipump for 7 days. Lungs were harvested at day 21. Levels of p-MLC20, total MLC20, and GAPDH were determined by immunoblot analyses. C: Masson's trichrome staining for collagen deposition in the saline and Bleo mouse groups treated with or without continuous infusion of exogenous RLX. Scale bars: 100 μm. D: Sircol-soluble collagen assay for mice treated as described in C (n ≥ 5 mice per group). *P < 0.01 for comparisons, as indicated.
Relaxin Inhibits MLC20 Phosphorylation and Attenuates Contraction of Myofibroblasts Isolated from the Lungs of Patients with IPF
To determine the effect of relaxin on MLC20 phosphorylation and contraction of human lung myofibroblasts, we treated lung (myo)fibroblasts isolated from six patients with IPF with relaxin for 24 hours. Relaxin treatment decreased levels of phospho-MLC20 in all IPF (myo)fibroblasts (Figure 3A). Next, we evaluated cellular contractility using the wrinkle assay,6 in which contractility is determined by the percentage of cells that form wrinkles on flexible silicone substrates (Figure 3B). Unlike the commonly used fibroblast-populated collagen gel assay, which measures averaged contractile strength exerted by a cell population, the wrinkle assay measures contraction on a single-cell basis. By using this approach in combination with immunofluorescent staining techniques, we showed that, among heterogeneous IPF fibroblasts, wrinkle-forming cells were primarily α-SMA–positive myofibroblasts (Figure 3C), whereas most cells that do not form wrinkles were α-SMA negative (Figure 3D); this suggests that the subset of IPF fibroblasts that are contractile is of the myofibroblastic phenotype. The wrinkle assay showed that relaxin treatment decreased the contractile capacity of most wrinkle-forming IPF lung myofibroblasts (Figure 3E), suggesting that relaxin inhibits signaling of the contractile response in human IPF lung myofibroblasts.
Figure 3.
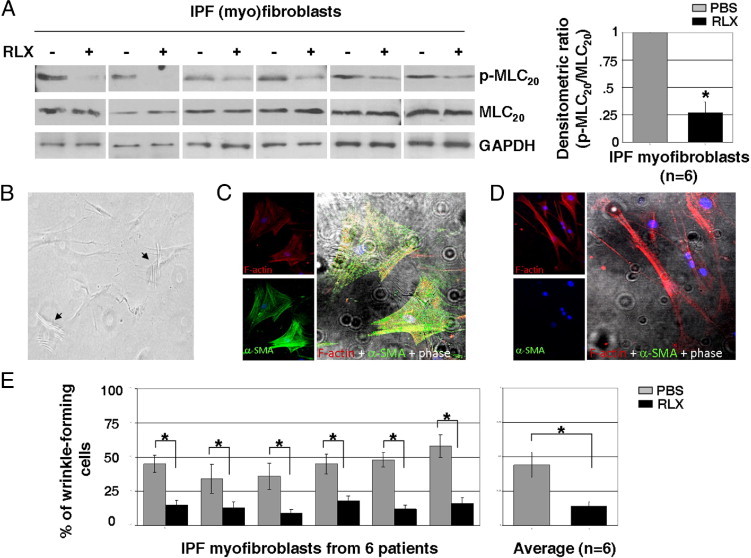
Relaxin (RLX) inhibits MLC20 phosphorylation and contraction of lung myofibroblasts isolated from patients with IPF. A: Lung (myo)fibroblasts isolated from six human subjects with IPF were treated with 100 nmol/L RLX for 24 hours. Levels of p-MLC20 and total MLC20 in cell lysates were determined by immunoblot analyses. GAPDH was used as a loading control. Relative p-MLC20 levels were determined by scanning densitometry of the blots and normalized to total MLC20 levels. The level of p-MLC20 at baseline was set at 1. *P < 0.01 (n = 6). B: (Myo)fibroblasts of IPF, 1 × 104 cells/cm2, were plated on silicone substrates prepared on an 18-mm coverslip and cultured in a 12-well plate for 24 hours. Wrinkles were observed and imaged under a microscope equipped with a digital camera. Arrows indicate wrinkle-forming cells. C and D: IPF (myo)fibroblasts cultured on silicone substrates were fixed and stained for α-SMA with fluorescein isothiocyanate–conjugated anti-α-SMA antibody (green) and F-actin with rhodamine-conjugated phalloidin (red). Phase-contrast and confocal immunofluorescent images were taken and overlaid to show the correlation between α-SMA expression and wrinkle formation. E: IPF (myo)fibroblasts from six patients with IPF were cultured on silicone substrates in the presence of 100 nmol/L RLX or an equal volume of PBS (vehicle control for RLX) for 24 hours. Wrinkle-forming cells were counted and calculated as percentage of total cells. Results are the mean ± SD of three separate experiments, with each experiment performed by random selection of 10 different regions. *P < 0.01 for comparisons, as indicated.
Ectopic Expression of a Phosphomimetic MLC20 Mutant Blocks the Capacity of Relaxin to Inhibit Contractility of Human IPF Myofibroblasts
Next, we determined whether relaxin-induced MLC20 dephosphorylation is essential for its inhibitory effect on IPF myofibroblast contractility. Previous studies48, 54, 55 have shown that a mutant MLC20, in which the phosphorylation target serine and threonine residues are replaced with aspartic acids (DD-MLC20), is phosphomimetic and functions as a constitutively active MLC20 in porcine kidney epithelial cells and cervical cancer cells. In this study, we generated MLC20 mutant plasmids that express the same DD-MLC20 construct as an N-terminal–tagged EGFP fusion protein and transfected this plasmid into IPF (myo)fibroblasts. Immunoblot analyses showed that transfected IPF cells expressed EGFP–DD-MLC20 at the expected molecular mass (50 kDa) (Figure 4A). Immunofluorescent analyses showed that EGFP–DD-MLC20 fusion proteins incorporated into thick stress fibers, whereas EGFP alone did not (Figure 4B), suggesting that DD-MLC20 is structurally integrated into the actomyosin system of myofibroblasts. Ectopic expression of phosphomimetic DD-MLC20 by IPF (myo)fibroblasts abrogated the inhibitory effect of relaxin on lung myofibroblast contraction (Figure 4C). These data suggest that relaxin regulation of myofibroblast contractility targets an intermediary step upstream of MLC20 phosphorylation. Furthermore, overexpression of DD-MLC20 significantly increased IPF myofibroblast contraction compared with nontransfected control cells or cells expressing EGFP alone (Figure 4C). Together, these findings indicate that MLC20 dephosphorylation mediates myofibroblast relaxation and that the effect of relaxin on myofibroblast relaxation is regulated by signaling upstream of MLC20 phosphorylation.
Figure 4.
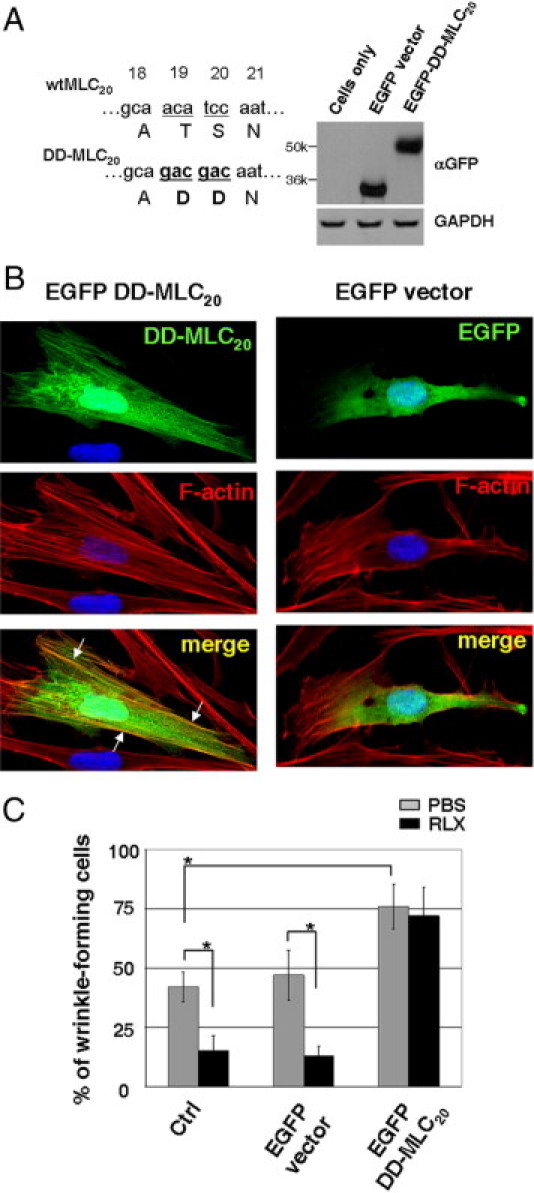
Overexpression of phosphomimetic DD-MLC20 blocks relaxin (RLX)–induced lung myofibroblast relaxation. A: IPF (myo)fibroblasts were transfected with EGFP–DD-MLC20 or EGFP vector alone. Wild-type (wt, underlined letters) and mutated (DD, underlined and bold letters) MLC20 DNA and amino acid sequences are shown. Overexpression of EGFP–DD-MLC20 and EGFP was confirmed by immunoblot analyses. GAPDH was used as a loading control. B: IPF (myo)fibroblasts transfected with EGFP–DD-MLC20 or EGFP vector alone were incubated with rhodamine-conjugated phalloidin to stain F-actin (red). DAPI was used to stain nuclei (blue). Colocalization of DD-MLC20 (green) and F-actin was observed with confocal laser scanning microscopy. Arrows indicate DD-MLC20–containing stress fibers. C: Nontransfected IPF (myo)fibroblasts (Ctrl), EGFP-expressing IPF (myo)fibroblasts, and EGFP–DD-MLC20–expressing IPF (myo)fibroblasts were cultured on silicone substrates in the presence of 100 nmol/L RLX or an equal volume of PBS (vehicle control) for 24 hours. Cell contraction was determined by wrinkle assay. Results are the mean ± SD of three separate experiments, each performed from 10 random regions. *P < 0.01 for comparisons, as indicated.
Relaxin activates the NO/cGMP/PKG signaling pathway in IPF (myo)fibroblasts and regulates multiple cell signaling pathways.25 Of these, the cAMP/PKA- and NO/cGMP/PKG-dependent pathways regulate cell contraction.56, 57, 58, 59, 60 We determined whether cAMP/PKA and NO/cGMP/PKG signals are involved in relaxin-induced IPF lung myofibroblast relaxation. Relaxin treatment increased the level of intracellular cGMP by 2.5-fold, and the NO synthase inhibitors, L-NMMA and PTIO, inhibited relaxin-induced cGMP production; SNAP, an NO donor, promoted cGMP production by IPF (myo)fibroblasts (Figure 5A). In contrast, relaxin (1 nmol/L to 1 μmol/L) did not induce significant changes in cAMP production, whereas forskolin, an adenylyl cyclase activator, induced a dose-dependent increase in cAMP production (Figure 5B). Relaxin promoted a dose-dependent increase in NO synthesis, and L-NMMA and PTIO blocked relaxin-induced NO production in IPF (myo)fibroblasts (Figure 5C). In addition, relaxin elevated PKG activity by 2.0-fold in the same cells (Figure 5D). DT-2, a cell-permeable PKG inhibitor, inhibited relaxin-induced PKG activation; 8-PCPT-cGMP, an analog of the natural cGMP signal molecule, and SNAP (both serving as positive controls) promoted PKG activation (Figure 5D). Relaxin had no effect on PKA activity, whereas forskolin (which increases cAMP production; positive control) induced PKA activation (Figure 5E), corroborating the earlier finding that relaxin does not signal via cAMP production in IPF (myo)fibroblasts.
Figure 5.
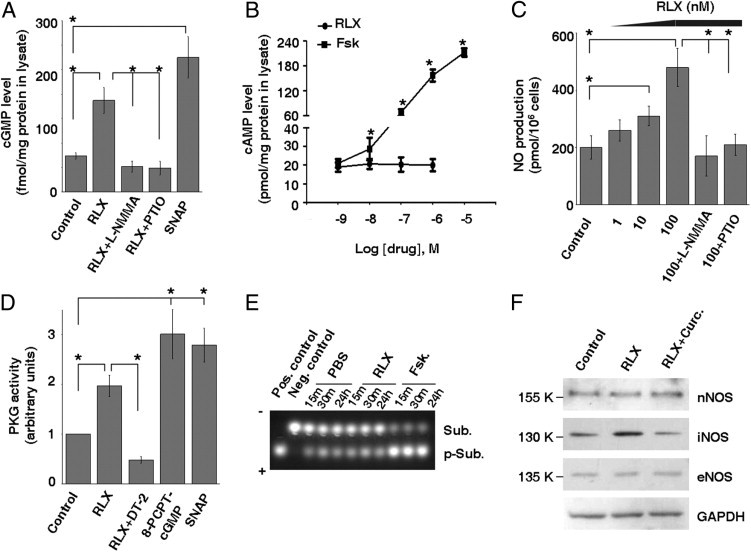
Relaxin (RLX) promotes NO/cGMP/PKG signaling in IPF (myo)fibroblasts. A: Subconfluent IPF (myo)fibroblasts were treated with 100 nmol/L RLX, 100 μmol/L SNAP, and 100 nmol/L RLX in the presence of 100 μmol/L L-NMMA or 20 μmol/L PTIO for 24 hours. Intracellular cGMP levels were determined by colorimetric competitive immunoassay. B: IPF (myo)fibroblasts were treated with 1 nmol/L to 1 μmol/L of RLX or 1 nmol/L to 10 μmol/L of forskolin (Fsk) for 24 hours. Intracellular cAMP levels were determined by colorimetric competitive immunoassay. C: IPF (myo)fibroblasts were treated with 1 to 100 nmol/L of RLX in the presence or absence of 100 μmol/L L-NMMA or 20 μmol/L PTIO for 24 hours. NO production in supernatants was measured by nitrite and nitrate accumulation. D: IPF (myo)fibroblasts were treated with 100 nmol/L RLX in the presence or absence of 1 μmol/L DT-2 or 200 μmol/L 8-PCPT-cGMP or with 100 μmol/L SNAP for 24 hours. PKG activity was measured by colorimetric immunoassay. E: IPF (myo)fibroblasts were treated with PBS, 100 nmol/L RLX, or 1 μmol/L Fsk for various times, as indicated. Cell lysates were incubated with PKA-specific substrates. Phosphorylated substrates (p-Sub) and nonphosphorylated substrates (Sub) were separated on a 0.8% agarose gel and used for evaluating PKA activity. F: IPF (myo)fibroblasts were treated with 100 nmol/L RLX in the presence or absence of 30 μmol/L curcumin (Curc.) for 24 hours. Levels of nNOS, iNOS, and eNOS were determined by immunoblot analyses. Results are the mean ± SD of three separate experiments. *P < 0.01 for comparisons, as indicated.
Relaxin increased inducible NO synthase (iNOS or NOS II) expression in IPF (myo)fibroblasts; neither neuronal NOS (nNOS or NOS I) nor endothelial NOS (eNOS or NOS III) expression was affected by relaxin treatment (Figure 5F). Curcumin, a plant extract known to inhibit iNOS expression by preventing the nuclear import of NF-κB,61 abrogated relaxin-induced iNOS expression. These data indicate that, in IPF (myo)fibroblasts, relaxin promotes NO production by up-regulation of iNOS. Together, our data suggest that relaxin signals via the NO/cGMP/PKG, not the cAMP/PKA, pathway in IPF (myo)fibroblasts.
Blocking NO/cGMP/PKG Signal Transduction Abrogates the Ability of Relaxin to Inhibit MLC20 Phosphorylation and Lung Myofibroblast Contraction
To determine whether the NO/cGMP/PKG pathway mediates relaxin inhibition of IPF lung myofibroblast contraction, we treated IPF (myo)fibroblasts cultured on silicone substrates with or without relaxin in the presence or absence of NO/cGMP/PKG signaling antagonists, L-NMMA, PTIO, curcumin, and DT-2. Blocking NO/cGMP/PKG signal transduction with these antagonists abrogated relaxin-induced lung myofibroblast relaxation, whereas activation of NO/cGMP/PKG signaling with 8-PCPT-cGMP and SNAP promoted IPF lung myofibroblast relaxation (Figure 6A). These data suggest that NO/cGMP/PKG signaling mediates relaxin inhibition of IPF lung myofibroblast contraction.
Figure 6.
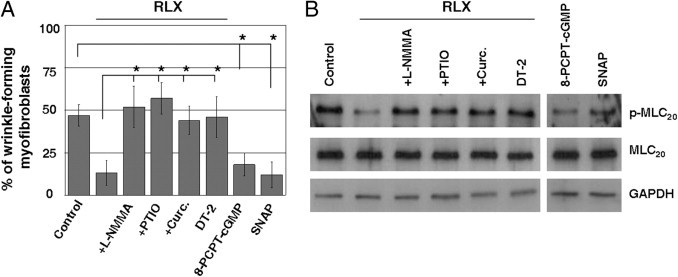
Blocking NO/cGMP/PKG signaling inhibits relaxin (RLX)–induced MLC20 dephosphorylation and lung myofibroblast relaxation. A: IPF (myo)fibroblasts were cultured on silicone substrates and treated with 100 nmol/L RLX, 200 μmol/L 8-PCPT-cGMP, 100 μmol/L SNAP, and 100 nmol/L RLX in the presence of 100 μmol/L L-NMMA, 20 μmol/L PTIO, 30 μmol/L curcumin (Curc.), or 1 μmol/L DT-2 for 24 hours. Cell contraction was measured by the wrinkle assay. Results are the mean ± SD of three separate experiments, each performed from 10 random regions. *P < 0.01 for comparisons, as indicated. B: Subconfluent IPF (myo)fibroblasts were treated as previously described. Levels of p-MLC20 and total MLC20 were determined by immunoblot analyses. GAPDH was used as a loading control.
By demonstrating that the induction of MLC20 dephosphorylation is essential for the inhibitory effect of relaxin on IPF myofibroblast contraction, we next determined whether the NO/cGMP/PKG signaling mediates relaxin-regulated MLC20 dephosphorylation. The augmentation of NO/cGMP/PKG signaling by 8-PCPT-cGMP and SNAP induced MLC20 dephosphorylation, whereas relaxin-induced MLC20 dephosphorylation was blocked by L-NMMA, PTIO, curcumin, and DT-2 (Figure 6B). These data suggest that the NO/cGMP/PKG signaling pathway mediates relaxin regulation of MLC20 dephosphorylation and IPF lung myofibroblast relaxation.
Relaxin-Induced NO/cGMP/PKG Activation Promotes RhoA Phosphorylation at Ser188, Resulting in RhoA/ROCK Inactivation
RhoA/ROCK signaling regulates myofibroblast contraction.2 MLC20 phosphorylation, a downstream target of RhoA/ROCK signaling, is increased in the fibroblastic foci of IPF lungs (Figure 1, A and E), suggesting that lung myofibroblast contraction may be linked to RhoA/ROCK activation. The phosphorylation of RhoA at Ser188 inactivates RhoA, which uncouples the classic GDP/GTP cycle.62, 63 Previous studies52, 64, 65 have shown that PKG promotes RhoA phosphorylation at Ser188, resulting in the inactivation of RhoA. Herein, we show that treatment of IPF (myo)fibroblasts with relaxin and NO/cGMP/PKG agonists (8-PCPT-cGMP and SNAP) promoted RhoA phosphorylation at Ser188 (Figure 7A). NO/cGMP/PKG antagonists (L-NMMA, PTIO, curcumin, and DT-2) blocked relaxin-induced RhoA phosphorylation (Figure 7A). Relaxin decreased the level of membrane-bound RhoA (by cellular fractionation analyses), which was reversed by the NO/cGMP/PKG antagonists, PTIO and DT-2 (Figure 7B), suggesting that relaxin treatment induces RhoA translocation from a membrane-bound active form to a cytosolic inactive form. Immunoprecipitation studies showed that binding of RhoA to ROCK was decreased by relaxin in IPF (myo)fibroblasts (Figure 7C). Finally, relaxin decreased ROCK activity by an enzyme-linked immunosorbent assay that measures MYPT1 phosphorylation (Figure 7D) and by immunoblot analyses of ROCK-specific moesin phosphorylation at Thr558 (Figure 7E). The NO/cGMP/PKG antagonists, PTIO and DT-2, abrogated relaxin-induced ROCK inactivation (Figure 7, D and E). Together, these data suggest that the relaxin-activated NO/cGMP/PKG signaling pathway induces RhoA phosphorylation on Ser188, resulting in RhoA disassociation from ROCK and consequent ROCK inactivation.
Figure 7.
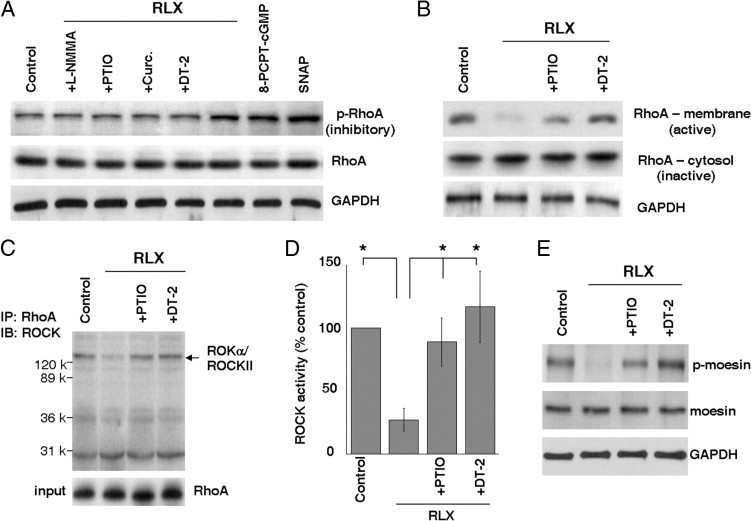
Relaxin (RLX)–induced NO/cGMP/PKG activation promotes RhoA phosphorylation at Ser188, leading to RhoA/ROCK inactivation. A: Subconfluent IPF (myo)fibroblasts were treated with RLX in the presence or absence of NO/cGMP/PKG antagonists or agonists, as indicated. Cells were lysed, and levels of phospho-RhoA at Ser188 (p-RhoA) and total RhoA (RhoA) were determined by immunoblot analyses. GAPDH was used as a loading control. B: Subconfluent IPF (myo)fibroblasts were treated with RLX in the presence or absence of PTIO or DT-2 for 24 hours. Cytosolic and membrane proteins were fractionated. Levels of RhoA from the cytosolic and membrane fractions were determined by immunoblot analyses. GAPDH was used as a loading control. C: IPF (myo)fibroblasts were treated as described in B. Cells were lysed, and cell lysates were immunoprecipitated with anti-RhoA antibody. Immunoprecipitated proteins were blotted with anti-ROKα/ROCK II antibody. A portion of cell lysates was immunoblotted for RhoA levels in cell lysates (input) for loading control. D: An ROCK activity assay was performed by incubation of cell lysates with an ROCK-specific substrate, MYPT1, followed by a colorimetric immunoassay. The level of ROCK activity from cells without any treatments (control) was set at 100%. Results are the mean ± SD of three separate experiments. *P < 0.01 for comparisons. E: IPF (myo)fibroblasts were treated as described in B. Cells were lysed, and levels of phospho-moesin (Thr558) (p-moesin) and total moesin were determined by immunoblot analyses. GAPDH was used as a loading control.
Overexpression of Nonphosphorylable Constitutively Active RhoA (RhoAV14A188) Renders IPF Lung Myofibroblasts Resistant to Relaxin-Induced MLC20 Dephosphorylation and Relaxation
To study the role of RhoA phosphorylation in relaxin regulation of lung myofibroblast relaxation, we constructed an EGFP-tagged, nonphosphorylable, constitutively active RhoA (EGFP-RhoAV14A188) plasmid. The EGFP-RhoAV14A188 plasmid, EGFP-tagged constitutively active RhoA (EGFP-RhoAV14) plasmid, or EGFP plasmid (control) was transfected into IPF (myo)fibroblasts (Figure 8A). As expected, overexpression of constitutively active RhoA increased MLC20 phosphorylation in IPF (myo)fibroblasts, compared with EGFP control (Figure 8B). Cells expressing EGFP-RhoAV14 (constitutively active) and EGFP (control plasmid) responded to relaxin with decreased MLC20 phosphorylation; in contrast, cells expressing EGFP-RhoAV14A188 were resistant to relaxin-induced MLC20 dephosphorylation (Figure 8B). Consistent with the findings on MLC20 phosphorylation, the wrinkle assay showed that overexpression of both constitutively active RhoA plasmids increased IPF myofibroblast contractility (Figure 8C). Relaxin attenuated myofibroblast contraction in both EGFP (control) cells and EGFP-RhoAV14 (constitutively active, phosphorylable) cells. However, EGFP-RhoAV14A188 (constitutively active, nonphosphorylable) cells were resistant to relaxin-induced myofibroblast relaxation. These results demonstrate that RhoA Ser188 phosphorylation mediates relaxin-induced MLC20 dephosphorylation and IPF myofibroblast relaxation.
Figure 8.
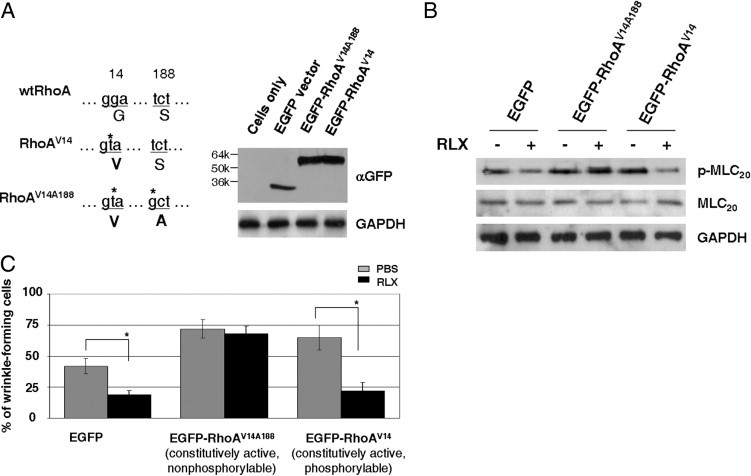
Overexpression of nonphosphorylable constitutively active RhoA (RhoAV14A188) renders IPF (myo)fibroblasts resistant to relaxin (RLX)–induced MLC20 dephosphorylation and relaxation. A: EGFP vector, EGFP-RhoAV14, and EGFP-RhoAV14A188 were transfected into IPF (myo)fibroblasts. Expression levels of EGFP and EGFP fusion proteins were determined by immunnoblot analyses. GAPDH was used as a loading control. B: IPF (myo)fibroblasts expressing EGFP, EGFP-RhoAV14, and EGFP-RhoAV14A188 were treated with or without 100 nmol/L RLX for 24 hours. Levels of p-MLC20 and total MLC20 were determined by immunoblot analyses. GAPDH was used as a loading control. C: Cell contraction was measured using the wrinkle assay. Results are the mean ± SD of three separate experiments, each performed from 10 random regions. *P < 0.01 for comparisons, as indicated.
Discussion
In this study, we elucidated a novel antifibrotic mechanism of relaxin via the inhibition of myofibroblast contractility. This antifibrotic action of relaxin is transduced by increasing intracellular cGMP levels through an iNOS-dependent mechanism and downstream activation of PKG, which phosphorylates RhoA. RhoA phosphorylation inactivates ROCK. ROCK inactivation induces MLC20 dephosphorylation, leading to myofibroblast relaxation (Figure 9).
Figure 9.
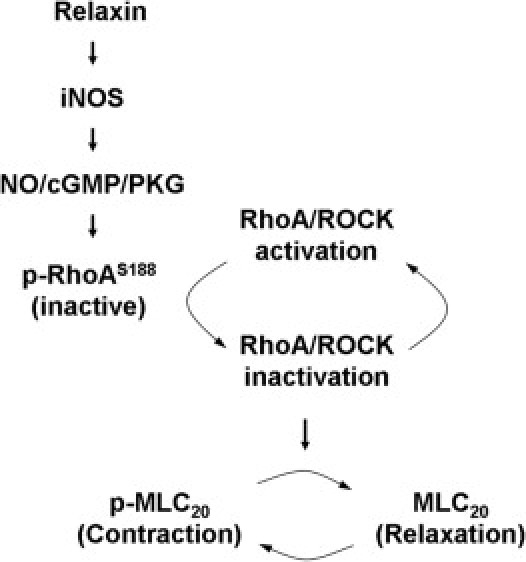
A model for relaxin-mediated cell signaling to inhibit lung myofibroblast contraction. Relaxin promotes iNOS production by lung myofibroblasts, resulting in activation of NO/cGMP/PKG signaling. Elevated PKG activity induces phosphorylation of RhoA at Ser188, which inactivates RhoA and its downstream target, ROCK. Decreased ROCK activity dephosphorylates MLC20 directly and/or through an MLC phosphatase–dependent mechanism,2 leading to lung myofibroblast relaxation.
Rho/ROCK inhibition attenuates lung fibrosis in both bleomycin- and radiation-induced animal models.66, 67 The molecular mechanisms underlying Rho/ROCK inhibition in the regulation of fibrogenesis are not clear. Y-27632, a highly selective ROCK inhibitor, blocks migration of human neutrophils, macrophages, and fibroblasts in response to chemoattractants in vitro, suggesting that prevention of inflammatory cell infiltration and fibroblast migration into injured lung sites may contribute to the antifibrotic effect of Rho/ROCK inhibition.66, 68 In addition to regulating fibroblast migration, the Rho/ROCK signal regulates myofibroblast contraction as well.2, 69 Compared with fibroblasts, fully differentiated myofibroblasts are more hypersynthetic and contractile but less migratory.70, 71 In this study, we demonstrate that relaxin inhibits IPF lung myofibroblast contraction by inactivation of RhoA/ROCK. In a bleomycin-induced mouse model of lung fibrosis, we administered relaxin into mice at day 14 after bleomycin. At this time, injured murine lungs have passed the acute inflammatory phase (0 to 7 days) and the early fibrotic phase (7 to 14 days), during which fibroblast migration occurs, and have entered into the fibrotic phase, during which myofibroblasts are established. This approach limits the inhibitory effect of relaxin-induced RhoA/ROCK inactivaction on fibroblast migration while it preserves the effect on myofibroblast contraction. The results show that inhibition of RhoA/ROCK-induced myofibroblast contraction by relaxin reduced lung fibrosis formation. These data suggest that attenuation of myofibroblast contractility contributes to the antifibrotic effects of Rho/ROCK inhibition.
Myofibroblasts acquire the contractile property similar to SMCs. Both myofibroblast and SMC contractions are determined by phosphorylation (contraction) and dephosphorylation (relaxation) of MLC20.2, 69 Rho/ROCK signaling phosphorylates myosin phosphatase and inactivates myosin phosphatase activity, which maintains the phosphorylated state of MLC20, resulting in sustained myofibroblast contraction.72, 73, 74, 75 In this study, we showed that relaxin induces MLC20 dephosphorylation through inhibition of RhoA/ROCK in human lung myofibroblasts, whereas relaxin deficiency results in higher levels of phospho–MLC20 in the lungs of bleomycin–treated mice, compared with levels in WT mice. These data suggest that relaxin is a naturally occurring antagonist of the contractile myofibroblast phenotype in lung fibrosis.
It has been well established that relaxin inhibits myometrial contractility in response to oxytocin and prostaglandin.76 cAMP/PKA signaling mediates relaxin-regulated uterine contraction through a mechanism involving the coupling of the relaxin receptor, RXFP1, to multiple G proteins.25, 77 A previous study56 showed that prostacyclin-induced human lung fibroblast and bronchial fibroblast relaxation in three-dimensional collagen matrices occurs through a cAMP/PKA-dependent mechanism, suggesting that the cAMP/PKA signal is involved in the regulation of lung fibroblast contraction. In our studies, neither cAMP levels nor PKA activity is altered by relaxin in IPF (myo)fibroblasts, suggesting that it is unlikely that a cAMP/PKA signal plays a role in relaxin-regulated lung myofibroblast relaxation. Consistent with this, other studies32, 78, 79 have shown that relaxin has little effect on cAMP/PKA signal transduction in human uterine fibroblasts, cardiac fibroblasts, and renal myofibroblasts.
Signaling by NO is another important pathway targeted by relaxin.80 NO mediates IL-1β–, tumor necrosis factor-α–, interferon-γ–, and 3-nitrotyrosine–induced lung fibroblast relaxation in three-dimensional collagen matrices.81, 82, 83 In addition, a recent study84 showed that NO protects the lung from ischemia-reperfusion–induced injury. In the current study, we showed that relaxin promotes NO production, resulting in an increase in the intracellular cGMP level and consequent PKG activation in IPF lung myofibroblasts. Relaxin up-regulation of NO production is iNOS dependent because the expression of iNOS increases in response to relaxin, whereas curcumin, an iNOS-specific inhibitor, blocks relaxin-induced iNOS production. On the other hand, nNOS and eNOS expression levels are not affected by relaxin in the lung. Consistent with these findings, relaxin promotes renal myofibroblast differentiation by increasing NO production40; in this study, nNOS, but not iNOS and eNOS, is responsible for relaxin-induced NO production in renal (myo)fibroblasts. These findings suggest that specific NOS isoform(s) are involved in the regulation of NO production by relaxin and may vary depending on the species, organ, and cell types studied.80
The small GTPase, RhoA, cycles between an active GTP-bound form and an inactive GDP-bound form. The GDP/GTP cycle controls RhoA activity through three major groups of regulatory proteins: guanine nucleotide exchange factors, GTPase-activating proteins, and guanine dissociation inhibitors.85 Recent studies63, 86 revealed that Rho protein phosphorylation provides an additional means for the regulation of Rho activity that uncouples the classic GDP/GTP cycle. The phosphorylation of RhoA at Ser188 increases the interactions of RhoA with guanine dissociation inhibitors and enhances the ability of guanine dissociation inhibitors to extract RhoA from membranes, leading to RhoA inactivation.62, 63 In this study, we showed that relaxin-induced NO/cGMP/PKG activation promotes RhoA phosphorylation at Ser188, resulting in RhoA translocation from the plasma membrane to cytosol and disassociation from ROCK that leads to ROCK inactivation. The overexpression of a phosphorylation-resistant RhoA mutant renders IPF lung myofibroblasts refractory to relaxin-induced MLC20 dephosphorylation and relaxation. These results indicate that NO/cGMP/PKG signal-dependent RhoA phosphorylation and ROCK inactivation mediate the inhibitory effect of relaxin on lung myofibroblast contraction.
In this study, we identified a novel antifibrotic mechanism of relaxin involving the inhibition of the contractile phenotype of lung myofibroblasts. Our studies suggest that targeting myofibroblast contraction-induced mechanotransduction may be of therapeutic benefit for the treatment of persistent/progressive fibrotic diseases.
Footnotes
Supported by NIH grant HL097215 and American Heart Association Scientist Development Grant 0835432N (Y.Z.); NIH grants HL067967 and HL107181 (V.J.T.); National Heart Foundation of Australia and National Health & Medical Research Council of Australia R.D. Wright Fellowship (C.S.S.).
References
- 1.Hinz B., Phan S.H., Thannickal V.J., Galli A., Bochaton-Piallat M.L., Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasek J.J., Gabbiani G., Hinz B., Chaponnier C., Brown R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 3.Desmouliere A., Chaponnier C., Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- 4.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 5.Wipff P.J., Rifkin D.B., Meister J.J., Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y., Hagood J.S., Lu B., Merryman W.D., Murphy-Ullrich J.E. Thy-1-integrin alphav beta5 interactions inhibit lung fibroblast contraction-induced latent transforming growth factor-beta1 activation and myofibroblast differentiation. J Biol Chem. 2010;285:22382–22393. doi: 10.1074/jbc.M110.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaalouk D.E., Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunst S.J., Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol. 2008;295:C576–C587. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard T.D., Borisy G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 10.Pak C.W., Flynn K.C., Bamburg J.R. Actin-binding proteins take the reins in growth cones. Nat Rev Neurosci. 2008;9:136–147. doi: 10.1038/nrn2236. [DOI] [PubMed] [Google Scholar]
- 11.Ridley A.J., Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 12.Miralles F., Posern G., Zaromytidou A.I., Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 13.Small E.M., O'Rourke J.R., Moresi V., Sutherland L.B., McAnally J., Gerard R.D., Richardson J.A., Olson E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc Natl Acad Sci U S A. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Small E.M., Thatcher J.E., Sutherland L.B., Kinoshita H., Gerard R.D., Richardson J.A., Dimaio J.M., Sadek H., Kuwahara K., Olson E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morita T., Mayanagi T., Sobue K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J Cell Biol. 2007;179:1027–1042. doi: 10.1083/jcb.200708174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masszi A., Speight P., Charbonney E., Lodyga M., Nakano H., Szaszi K., Kapus A. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol. 2010;188:383–399. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong C., Chrzanowska-Wodnicka M., Brown J., Shaub A., Belkin A.M., Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol. 1998;141:539–551. doi: 10.1083/jcb.141.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz B., Gabbiani G. Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb Haemost. 2003;90:993–1002. doi: 10.1160/TH03-05-0328. [DOI] [PubMed] [Google Scholar]
- 19.Serini G. Loss of E-cadherin tyrosine phosphorylation in human cancers. Mol Med Today. 1999;5:336. doi: 10.1016/s1357-4310(99)01531-2. [DOI] [PubMed] [Google Scholar]
- 20.Maciver S.K. Myosin II function in non-muscle cells. Bioessays. 1996;18:179–182. doi: 10.1002/bies.950180304. [DOI] [PubMed] [Google Scholar]
- 21.Barron J.T., Barany M., Barany K., Storti R.V. Reversible phosphorylation and dephosphorylation of the 20,000-dalton light chain of myosin during the contraction-relaxation-contraction cycle of arterial smooth muscle. J Biol Chem. 1980;255:6238–6244. [PubMed] [Google Scholar]
- 22.Wu X., Haystead T.A., Nakamoto R.K., Somlyo A.V., Somlyo A.P. Acceleration of myosin light chain dephosphorylation and relaxation of smooth muscle by telokin: synergism with cyclic nucleotide-activated kinase. J Biol Chem. 1998;273:11362–11369. doi: 10.1074/jbc.273.18.11362. [DOI] [PubMed] [Google Scholar]
- 23.Pang H., Guo Z., Su W., Xie Z., Eto M., Gong M.C. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2005;289:C352–C360. doi: 10.1152/ajpcell.00111.2005. [DOI] [PubMed] [Google Scholar]
- 24.Pang H., Guo Z., Xie Z., Su W., Gong M.C. Divergent kinase signaling mediates agonist-induced phosphorylation of phosphatase inhibitory proteins PHI-1 and CPI-17 in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;290:C892–C899. doi: 10.1152/ajpcell.00378.2005. [DOI] [PubMed] [Google Scholar]
- 25.Du X.J., Bathgate R.A., Samuel C.S., Dart A.M., Summers R.J. Cardiovascular effects of relaxin: from basic science to clinical therapy. Nat Rev Cardiol. 2010;7:48–58. doi: 10.1038/nrcardio.2009.198. [DOI] [PubMed] [Google Scholar]
- 26.Bennett R.G. Relaxin and its role in the development and treatment of fibrosis. Transl Res. 2009;154:1–6. doi: 10.1016/j.trsl.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Unemori E.N., Pickford L.B., Salles A.L., Piercy C.E., Grove B.H., Erikson M.E., Amento E.P. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest. 1996;98:2739–2745. doi: 10.1172/JCI119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenyon N.J., Ward R.W., Last J.A. Airway fibrosis in a mouse model of airway inflammation. Toxicol Appl Pharmacol. 2003;186:90–100. doi: 10.1016/s0041-008x(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 29.Bani D., Ballati L., Masini E., Bigazzi M., Sacchi T.B. Relaxin counteracts asthma-like reaction induced by inhaled antigen in sensitized guinea pigs. Endocrinology. 1997;138:1909–1915. doi: 10.1210/endo.138.5.5147. [DOI] [PubMed] [Google Scholar]
- 30.Samuel C.S., Zhao C., Bathgate R.A., Bond C.P., Burton M.D., Parry L.J., Summers R.J., Tang M.L., Amento E.P., Tregear G.W. Relaxin deficiency in mice is associated with an age-related progression of pulmonary fibrosis. FASEB J. 2003;17:121–123. doi: 10.1096/fj.02-0449fje. [DOI] [PubMed] [Google Scholar]
- 31.Samuel C.S., Royce S.G., Burton M.D., Zhao C., Tregear G.W., Tang M.L. Relaxin plays an important role in the regulation of airway structure and function. Endocrinology. 2007;148:4259–4266. doi: 10.1210/en.2007-0577. [DOI] [PubMed] [Google Scholar]
- 32.Samuel C.S., Unemori E.N., Mookerjee I., Bathgate R.A., Layfield S.L., Mak J., Tregear G.W., Du X.J. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. doi: 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- 33.Mookerjee I., Unemori E.N., Du X.J., Tregear G.W., Samuel C.S. Relaxin modulates fibroblast function, collagen production, and matrix metalloproteinase-2 expression by cardiac fibroblasts. Ann N Y Acad Sci. 2005;1041:190–193. doi: 10.1196/annals.1282.028. [DOI] [PubMed] [Google Scholar]
- 34.McDonald G.A., Sarkar P., Rennke H., Unemori E., Kalluri R., Sukhatme V.P. Relaxin increases ubiquitin-dependent degradation of fibronectin in vitro and ameliorates renal fibrosis in vivo. Am J Physiol Renal Physiol. 2003;285:F59–F67. doi: 10.1152/ajprenal.00157.2002. [DOI] [PubMed] [Google Scholar]
- 35.Heeg M.H., Koziolek M.J., Vasko R., Schaefer L., Sharma K., Muller G.A., Strutz F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005;68:96–109. doi: 10.1111/j.1523-1755.2005.00384.x. [DOI] [PubMed] [Google Scholar]
- 36.Samuel C.S., Mookerjee I., Masterson R., Tregear G.W., Hewitson T.D. Relaxin regulates collagen overproduction associated with experimental progressive renal fibrosis. Ann N Y Acad Sci. 2005;1041:182–184. doi: 10.1196/annals.1282.026. [DOI] [PubMed] [Google Scholar]
- 37.Unemori E.N., Amento E.P. Relaxin modulates synthesis and secretion of procollagenase and collagen by human dermal fibroblasts. J Biol Chem. 1990;265:10681–10685. [PubMed] [Google Scholar]
- 38.Unemori E.N., Bauer E.A., Amento E.P. Relaxin alone and in conjunction with interferon-gamma decreases collagen synthesis by cultured human scleroderma fibroblasts. J Invest Dermatol. 1992;99:337–342. doi: 10.1111/1523-1747.ep12616665. [DOI] [PubMed] [Google Scholar]
- 39.Williams E.J., Benyon R.C., Trim N., Hadwin R., Grove B.H., Arthur M.J., Unemori E.N., Iredale J.P. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49:577–583. doi: 10.1136/gut.49.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mookerjee I., Hewitson T.D., Halls M.L., Summers R.J., Mathai M.L., Bathgate R.A., Tregear G.W., Samuel C.S. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009;23:1219–1229. doi: 10.1096/fj.08-120857. [DOI] [PubMed] [Google Scholar]
- 41.Samuel C.S., Cendrawan S., Gao X.M., Ming Z., Zhao C., Kiriazis H., Xu Q., Tregear G.W., Bathgate R.A., Du X.J. Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest. 2011;91:675–690. doi: 10.1038/labinvest.2010.198. [DOI] [PubMed] [Google Scholar]
- 42.Longo M., Jain V., Vedernikov Y.P., Garfield R.E., Saade G.R. Effects of recombinant human relaxin on pregnant rat uterine artery and myometrium in vitro. Am J Obstet Gynecol. 2003;188:1468–1474. doi: 10.1067/mob.2003.454. discussion 1474–1476. [DOI] [PubMed] [Google Scholar]
- 43.Norstrom A., Bryman I. Adenosine 3′,5′-monophosphate in relation to inhibition of cervical smooth muscle activity in early pregnant women. Acta Endocrinol (Copenh) 1991;125:122–126. doi: 10.1530/acta.0.1250122. [DOI] [PubMed] [Google Scholar]
- 44.Baccari M.C., Calamai F., Chiappini L., Vannucchi M.G., Bani D. Relaxin restores altered ileal spontaneous contractions in dystrophic (mdx) mice. Ann N Y Acad Sci. 2005;1041:308–310. doi: 10.1196/annals.1282.047. [DOI] [PubMed] [Google Scholar]
- 45.Rao M.R., Sanborn B.M. Relaxin increases calcium efflux from rat myometrial cells in culture. Endocrinology. 1986;119:435–437. doi: 10.1210/endo-119-1-435. [DOI] [PubMed] [Google Scholar]
- 46.Bani D., Failli P., Bello M.G., Thiemermann C., Bani Sacchi T., Bigazzi M., Masini E. Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension. 1998;31:1240–1247. doi: 10.1161/01.hyp.31.6.1240. [DOI] [PubMed] [Google Scholar]
- 47.Masterson R., Hewitson T.D., Kelynack K., Martic M., Parry L., Bathgate R., Darby I., Becker G. Relaxin down-regulates renal fibroblast function and promotes matrix remodelling in vitro. Nephrol Dial Transplant. 2004;19:544–552. doi: 10.1093/ndt/gfg598. [DOI] [PubMed] [Google Scholar]
- 48.Di Ciano-Oliveira C., Lodyga M., Fan L., Szaszi K., Hosoya H., Rotstein O.D., Kapus A. Is myosin light-chain phosphorylation a regulatory signal for the osmotic activation of the Na+-K+-2Cl- cotransporter? Am J Physiol Cell Physiol. 2005;289:C68–C81. doi: 10.1152/ajpcell.00631.2004. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y., Koli K., Hagood J.S., Miao M., Mavalli M., Rifkin D.B., Murphy-Ullrich J.E. Latent transforming growth factor-beta-binding protein-4 regulates transforming growth factor-beta1 bioavailability for activation by fibrogenic lung fibroblasts in response to bleomycin. Am J Pathol. 2009;174:21–33. doi: 10.2353/ajpath.2009.080620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hinz B., Celetta G., Tomasek J.J., Gabbiani G., Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wrobel L.K., Fray T.R., Molloy J.E., Adams J.J., Armitage M.P., Sparrow J.C. Contractility of single human dermal myofibroblasts and fibroblasts. Cell Motil Cytoskeleton. 2002;52:82–90. doi: 10.1002/cm.10034. [DOI] [PubMed] [Google Scholar]
- 52.Sawada N., Itoh H., Miyashita K., Tsujimoto H., Sone M., Yamahara K., Arany Z.P., Hofmann F., Nakao K. Cyclic GMP kinase and RhoA Ser188 phosphorylation integrate pro- and antifibrotic signals in blood vessels. Mol Cell Biol. 2009;29:6018–6032. doi: 10.1128/MCB.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu G., Friggeri A., Yang Y., Milosevic J., Ding Q., Thannickal V.J., Kaminski N., Abraham E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fumoto K., Uchimura T., Iwasaki T., Ueda K., Hosoya H. Phosphorylation of myosin II regulatory light chain is necessary for migration of HeLa cells but not for localization of myosin II at the leading edge. Biochem J. 2003;370:551–556. doi: 10.1042/BJ20021559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwasaki T., Murata-Hori M., Ishitobi S., Hosoya H. Diphosphorylated MRLC is required for organization of stress fibers in interphase cells and the contractile ring in dividing cells. Cell Struct Funct. 2001;26:677–683. doi: 10.1247/csf.26.677. [DOI] [PubMed] [Google Scholar]
- 56.Kamio K., Liu X., Sugiura H., Togo S., Kobayashi T., Kawasaki S., Wang X., Mao L., Ahn Y., Hogaboam C., Toews M.L., Rennard S.I. Prostacyclin analogs inhibit fibroblast contraction of collagen gels through the cAMP-PKA pathway. Am J Respir Cell Mol Biol. 2007;37:113–120. doi: 10.1165/rcmb.2007-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Togo S., Liu X., Wang X., Sugiura H., Kamio K., Kawasaki S., Kobayashi T., Ertl R.F., Ahn Y., Holz O., Magnussen H., Fredriksson K., Skold C.M., Rennard S.I. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-{beta}1-stimulated fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;296:L959–L969. doi: 10.1152/ajplung.00508.2007. [DOI] [PubMed] [Google Scholar]
- 58.Murthy K.S., Zhou H., Grider J.R., Makhlouf G.M. Inhibition of sustained smooth muscle contraction by PKA and PKG preferentially mediated by phosphorylation of RhoA. Am J Physiol Gastrointest Liver Physiol. 2003;284:G1006–G1016. doi: 10.1152/ajpgi.00465.2002. [DOI] [PubMed] [Google Scholar]
- 59.Rivero-Vilches F.J., de Frutos S., Saura M., Rodriguez-Puyol D., Rodriguez-Puyol M. Differential relaxing responses to particulate or soluble guanylyl cyclase activation on endothelial cells: a mechanism dependent on PKG-I alpha activation by NO/cGMP. Am J Physiol Cell Physiol. 2003;285:C891–C898. doi: 10.1152/ajpcell.00590.2002. [DOI] [PubMed] [Google Scholar]
- 60.Perez-Zoghbi J.F., Bai Y., Sanderson M.J. Nitric oxide induces airway smooth muscle cell relaxation by decreasing the frequency of agonist-induced Ca2+ oscillations. J Gen Physiol. 2010;135:247–259. doi: 10.1085/jgp.200910365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quattrone S., Chiappini L., Scapagnini G., Bigazzi B., Bani D. Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod. 2004;10:325–330. doi: 10.1093/molehr/gah040. [DOI] [PubMed] [Google Scholar]
- 62.Ellerbroek S.M., Wennerberg K., Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem. 2003;278:19023–19031. doi: 10.1074/jbc.M213066200. [DOI] [PubMed] [Google Scholar]
- 63.Forget M.A., Desrosiers R.R., Gingras D., Beliveau R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–254. doi: 10.1042/0264-6021:3610243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sauzeau V., Le Jeune H., Cario-Toumaniantz C., Smolenski A., Lohmann S.M., Bertoglio J., Chardin P., Pacaud P., Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2000;275:21722–21729. doi: 10.1074/jbc.M000753200. [DOI] [PubMed] [Google Scholar]
- 65.Rolli-Derkinderen M., Sauzeau V., Boyer L., Lemichez E., Baron C., Henrion D., Loirand G., Pacaud P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ Res. 2005;96:1152–1160. doi: 10.1161/01.RES.0000170084.88780.ea. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu Y., Dobashi K., Iizuka K., Horie T., Suzuki K., Tukagoshi H., Nakazawa T., Nakazato Y., Mori M. Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis. Am J Respir Crit Care Med. 2001;163:210–217. doi: 10.1164/ajrccm.163.1.2001089. [DOI] [PubMed] [Google Scholar]
- 67.Monceau V., Pasinetti N., Schupp C., Pouzoulet F., Opolon P., Vozenin M.C. Modulation of the Rho/ROCK pathway in heart and lung after thorax irradiation reveals targets to improve normal tissue toxicity. Curr Drug Targets. 2010;11:1395–1404. doi: 10.2174/1389450111009011395. [DOI] [PubMed] [Google Scholar]
- 68.Niggli V. Rho-kinase in human neutrophils: a role in signalling for myosin light chain phosphorylation and cell migration. FEBS Lett. 1999;445:69–72. doi: 10.1016/s0014-5793(99)00098-8. [DOI] [PubMed] [Google Scholar]
- 69.Follonier Castella L., Gabbiani G., McCulloch C.A., Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316:2390–2401. doi: 10.1016/j.yexcr.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 70.Ronnov-Jessen L., Petersen O.W. A function for filamentous alpha-smooth muscle actin: retardation of motility in fibroblasts. J Cell Biol. 1996;134:67–80. doi: 10.1083/jcb.134.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hinz B., Dugina V., Ballestrem C., Wehrle-Haller B., Chaponnier C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell. 2003;14:2508–2519. doi: 10.1091/mbc.E02-11-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomasek J.J., Vaughan M.B., Kropp B.P., Gabbiani G., Martin M.D., Haaksma C.J., Hinz B. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen. 2006;14:313–320. doi: 10.1111/j.1743-6109.2006.00126.x. [DOI] [PubMed] [Google Scholar]
- 73.Parizi M., Howard E.W., Tomasek J.J. Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res. 2000;254:210–220. doi: 10.1006/excr.1999.4754. [DOI] [PubMed] [Google Scholar]
- 74.Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 75.Kawano Y., Fukata Y., Oshiro N., Amano M., Nakamura T., Ito M., Matsumura F., Inagaki M., Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–1038. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sherwood O.D. Relaxin's physiological roles and other diverse actions. Endocr Rev. 2004;25:205–234. doi: 10.1210/er.2003-0013. [DOI] [PubMed] [Google Scholar]
- 77.Downing S.J., Hollingsworth M. Action of relaxin on uterine contractions: a review. J Reprod Fertil. 1993;99:275–282. doi: 10.1530/jrf.0.0990275. [DOI] [PubMed] [Google Scholar]
- 78.Palejwala S., Stein D., Wojtczuk A., Weiss G., Goldsmith L.T. Demonstration of a relaxin receptor and relaxin-stimulated tyrosine phosphorylation in human lower uterine segment fibroblasts. Endocrinology. 1998;139:1208–1212. doi: 10.1210/endo.139.3.5772. [DOI] [PubMed] [Google Scholar]
- 79.Samuel C.S., Mookerjee I., Halls M.L., Summers R.J., Chew E., Bathgate R.A., Tregear G.W., Hewitson T.D. Investigations into the inhibitory effects of relaxin on renal myofibroblast differentiation. Ann N Y Acad Sci. 2009;1160:294–299. doi: 10.1111/j.1749-6632.2008.03823.x. [DOI] [PubMed] [Google Scholar]
- 80.Baccari M.C., Bani D. Relaxin and nitric oxide signalling. Curr Protein Pept Sci. 2008;9:638–645. doi: 10.2174/138920308786733921. [DOI] [PubMed] [Google Scholar]
- 81.Zhang H.Y., Gharaee-Kermani M., Phan S.H. Regulation of lung fibroblast alpha-smooth muscle actin expression, contractile phenotype, and apoptosis by IL-1beta. J Immunol. 1997;158:1392–1399. [PubMed] [Google Scholar]
- 82.Zhu Y.K., Liu X.D., Skold M.C., Umino T., Wang H., Romberger D.J., Spurzem J.R., Kohyama T., Wen F.Q., Rennard S.I. Cytokine inhibition of fibroblast-induced gel contraction is mediated by PGE(2) and NO acting through separate parallel pathways. Am J Respir Cell Mol Biol. 2001;25:245–253. doi: 10.1165/ajrcmb.25.2.4383. [DOI] [PubMed] [Google Scholar]
- 83.Sugiura H., Liu X., Ichikawa T., Ichinose M., Rennard S.I. 3-Nitrotyrosine inhibits fibroblast-mediated collagen gel contraction and chemotaxis. Eur Respir J. 2009;34:1452–1460. doi: 10.1183/09031936.00195808. [DOI] [PubMed] [Google Scholar]
- 84.Alexiou K., Matschke K., Westphal A., Stangl K., Dschietzig T. Relaxin is a candidate drug for lung preservation: relaxin-induced protection of rat lungs from ischemia-reperfusion injury. J Heart Lung Transplant. 2010;29:454–460. doi: 10.1016/j.healun.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 85.Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 86.Lang P., Gesbert F., Delespine-Carmagnat M., Stancou R., Pouchelet M., Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]