

Abstract
Particulate matter heavily pollutes the urban atmosphere, and several studies show a link between increased ambient particulate air pollution and exacerbation of pre-existing pulmonary diseases, including asthma. We investigated how diesel exhaust particulates (DEPs) aggravate asthma-like pulmonary inflammation in a mouse model of asthma induced by a house dust extract (HDE) containing cockroach allergens and endotoxin. BALB/c mice were exposed to three pulmonary challenges via hypopharyngeal administration of an HDE collected from the home of an asthmatic child. One hour before each pulmonary challenge, mice were exposed to DEP or PBS. Pulmonary inflammation was assessed by histological features, oxidative stress, respiratory physiological features, inflammatory cell recruitment, and local CXC chemokine production. To prove the role of CXC chemokines in the augmented inflammation, CXC chemokine–specific antibodies were delivered to the lungs before DEP exposure. DEP exacerbated HDE-induced airway inflammation, with increased airway mucus production, oxidative stress, inflammatory cell infiltration, bronchoalveolar lavage concentrations of CXC chemokines, and airway hyperreactivity. Neutralization of airway keratinocyte-derived chemokine and macrophage inflammatory protein-2 significantly improves the respiratory function in addition to decreasing the infiltration of neutrophils and eosinophils. Blocking the chemokines also decreased airway mucus production. These results demonstrate that DEP exacerbates airway inflammation induced by allergen through increased pulmonary expression of the CXC chemokines (keratinocyte-derived chemokine and macrophage inflammatory protein-2).
See related Commentary on page 2678
Asthma is a unique form of chronic respiratory disease characterized by reversible airway obstruction, airway inflammation, and airway hyperresponsiveness (AHR).1 It represents one of the most common chronic inflammatory diseases, affecting an estimated 300 million people worldwide, with an expected significant increase to 400 million people by 2025.2 The sharply increasing prevalence and incidence of asthma causes global concern, both in developing and developed countries.3, 4 In the United States, the prevalence of asthma among children increased from 3.6% in 19805 to 9.6% in 2009.6
To shed light on the cause of recent increases of allergic conditions, such as asthma and allergic rhinitis, several factors have been proposed, including genetic and environmental changes. Although the relative contribution of genetics and the environment in the development of asthma remains to be elucidated, numerous studies have documented the effects of environmental exposures on the risk of pulmonary diseases. As several epidemiological and clinical studies emerged, it has become clear that increased ambient air pollutants,7 including particular matter,8, 9 are correlated with dramatic increases in the risk of respiratory and cardiovascular diseases. Recently, epidemiological research10 demonstrated an association between the degree of traffic exposure and the lung function of asthmatic patients.
Among common air pollutants, suspended particulate pollutants and ambient particulate matter (PM) are the most serious pollutants that have been consistently correlated with adverse health effects.4, 11, 12 Diesel exhaust particulates (DEPs) from the diesel-powered motor vehicles constitute the largest single source of PM (≤90%) in the atmosphere of cities.4 Consequently, DEP is widely used for studying the effects of PM.11 DEP is a complex mixture of solid and liquid PM, including elemental carbon, polycyclic aromatic hydrocarbons, acid aerosols, volatile organic compounds, and gases (ie, carbon dioxide and nitrogen dioxide).13 The effects of air pollutants on the development and exacerbation of asthma have been demonstrated in animal and human studies.11 Although the effects are clearly demonstrable, the mechanisms responsible for air pollution–induced asthma exacerbations have yet to be elucidated.
Substantial evidence, including the oxidant properties of PM, implicates reactive oxygen species (ROS) in DEP-induced pulmonary inflammation.11, 14 Highly elevated oxidative stress will induce pro-inflammatory cytokines and chemokines,14, 15, 16 in addition to its direct effects on airway smooth muscle and mucin secretion.17 Chemokines may be particularly important in the regulation of inflammatory responses induced by DEP.15, 16 DEP enhances CXCL8 (IL-8)18 and chemokine ligand 2 (monocyte chemoattractant protein-1) expression in human cell lines. The CXC chemokines [keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2)] are considered important neutrophil chemoattractants released in the lung in many animal models of airway inflammation, induced by allergens and exposure to air pollutants.13, 19
To investigate the biological mechanisms responsible for the exacerbation of pulmonary inflammation and AHR in a mouse model of asthma, we determined whether asthma-like inflammatory responses in mice are exacerbated by the combination of the air pollutant DEP and allergen challenge. Because CXC chemokines help drive the asthmatic response in the absence of air pollution, we specifically examined if these mediators were the mechanism of DEP aggravation of asthma. Our data demonstrate, for the first time to our knowledge, that KC and MIP-2, two CXC chemokines, orchestrate DEP-induced exacerbation of airway inflammation and AHR in vivo.
Materials and Methods
Mice
Female BALB/c mice (aged 8 weeks) were obtained from Jackson Laboratory (Bar Harbor, ME) and maintained under standard laboratory conditions. The mice were housed in a temperature-controlled room (22°C) with a 12-hour dark-light cycle, with food and water allowed ad libitum. All experiments were performed in accordance with the NIH guidelines and approved by the Boston University or University of Michigan Committee on the Use and Care of Animals.
Sensitization Procedures
The household dust used for all sensitizations and airway challenges was collected from a house in Detroit, MI, and then extracted as previously reported.20, 21 Each mouse received 50 μL of house dust extract (HDE) that contains 1.9 U of Bla g1, 31 ng of Bla g2, and 0.91 ng of endotoxin.
We used our previously described model of asthma to induce the response with the significant modification of eliminating the use of an adjuvant.22 The pulmonary-only exposure protocol has strongly induced asthma-like pulmonary inflammation.22 Briefly, mice were sensitized with three hypopharyngeal instillations of HDE on days 0, 14, and 21.22 AHR was measured 2 hours after the last challenge on day 21, and mice were then sacrificed to collect samples.
DEP Challenge
DEPs were obtained from the National Institute of Standards and Technology (Standard Reference Material 2975, Gaithersburg, MD) and prepared in PBS with 0.05% Tween-80, a nontoxic and nonionic surfactant, following established procedures.15, 23 The suspension was sonicated for 2 minutes (Misonix, Newtown, CT) immediately before use. One hour before each HDE challenge, mice received a total of 0.05 mg of DEP suspension by hypopharyngeal instillation on days 0, 14, and 21. Control mice received 50 μL of PBS with 0.05% Tween-80. The DEP suspension contains 107 ng/mL of endotoxin (5.4 ng per mouse), and the pH is 6.8.
Antibody Treatment
Groups of BALB/c mice were sensitized and challenged as previously described. One hour before the third challenge of DEP on day 21, mice were given a combination of 10 μg of rat anti-mouse KC monoclonal antibody and 4 μg of rat anti-mouse MIP-2 monoclonal antibody (anti-KC/MIP-2 Ab; R&D Systems, Inc., Minneapolis, MN) by hypopharyngeal challenge. For controls, mice received 14 μg of rat IgG (Jackson Immuno Research Laboratories) via the same route.
Measurement of Respiratory Function
AHR was measured 2 hours after the final allergen challenge by two separate methods. Whole-body plethysmography (WBP; Buxco, Troy, NY) was used to measure AHR in response to increasing doses of aerosolized acetyl β-methylcholine (Methacholine; Sigma, St Louis, MO) in unrestrained and conscious mice, as previously reported.20, 22
Changes of airway resistance were measured by a forced oscillation technique24 on a Flexivent instrument (Scireq Scientific Respiratory Equipment, Montreal, QC, Canada). Mice were anesthetized with an i.p. injection of 1:5 diluted pentobarbital (Nembutal, 0.016 mL/g body weight; Ovation Pharmaceutical, Deerfield, IL). The paralytic was pancuronium (Sigma-Aldrich, St Louis) at 0.5 μg/body weight. When adequate surgical sedation was reached, as determined by a firm squeeze of the foot pad, a tracheotomy was performed, followed by insertion of an 18-gauge polyethylene cannula into the distal trachea. The mouse was then placed on the Flexivent mechanical ventilator (Scireq Scientific Respiratory Equipment) and ventilated at 190 breaths/minute with positive-end expiratory pressure set at 3 cm H2O. The measurement of airway resistance in response to increasing doses of aerosolized methacholine was obtained through periodic computer-generated “snapshot 150” forced-maneuver interruptions in ventilation. Data are then presented as resistance change from baseline (in cm H2O/mL per second).
Sample Collection and Analyses
Blood and BALF
Blood and bronchoalveolar lavage fluid (BALF) were collected and processed as previously reported.25
Preparation of Lung Homogenates and MPO Assay
After the BALF was collected, the right lung was removed and immediately processed, as previously reported.26 The myeloperoxidase (MPO) activity was assayed as described by Schneider and Issekutz,27 with minor modifications. The supernatant from sonicated and centrifuged lung homogenates was diluted with 10 mmol/L citrate buffer, pH 5.0, on a 96-well plate. An equal volume (75 μL) of substrate solution containing 3,3′,5,5′-tetramethylbenzidine, 120 mmol/L resorcinol (an eosinophil peroxidase inhibitor), and 0.007% H2O2 in deionized H2O was added to the sample and incubated in the dark for 2 minutes. Ice-cold 4N H2SO4 (150 μL) was added to stop the reaction, and the absorbance at 450 nm was measured. MPO data are expressed as ΔOD, reflecting the difference in absorbance between the sample and the blank wells.
Histopathological Analysis
Immediately after collecting the BALF, the left lung from each mouse was removed, fixed in 70% ethanol, and processed for routine histological analysis in paraffin. Tissue sections were stained with H&E or PAS and then examined under light microscopy. Digital images were taken of each of the lungs from each animal. The entire histological section on the glass slide was selected, and areas were quantified using NIH ImageJ software (http://rsbweb.nih.gov/ij, last accessed December 9, 2010), as previously described.22
Measuring 8-Isoprostane Concentrations in BALF
The concentration of 8-isoprostane in BALF was measured by a commercially available specific enzyme immunoassay kit (Cayman Chemical Co, Ann Arbor, MI).
Cytokine and Chemokine Analysis
All chemokine, cytokine, and IgE measurements were performed simultaneously to reduce errors due to interassay variation. The concentrations of all chemokines and cytokines were measured by enzyme-linked immunosorbent assay using matched antibody pairs (R&D Systems, Inc.), as previously described.20
Microarray Data Analysis
The microarray analysis was performed as previously reported.22 Briefly, using freeware, TM4-MeV 4.6.2 (http://www.tm4.org/mev.html, last accessed December 9, 2010), the heat map and the hierarchal clustering map were generated after Z-score normalization was performed.
Statistical Analyses
Mean ± SEM was used for summary statistics in all figures. Differences between all treatment groups were compared by unpaired Student's t-test or one-way analysis of variance with Tukey's posttest using GraphPad Prism version 5.0 software (GraphPad Software, San Diego, CA). Statistical significance was achieved when P ≤ 0.05 at the 95% CI.
Results
DEP Exposure Exacerbates Cockroach Allergen–Induced Asthma-Like Pulmonary Inflammation
Three pulmonary exposures to an HDE containing high levels of cockroach allergens induce substantial asthma-like pulmonary inflammation. This inflammation is characterized by infiltrates of lymphocytes, eosinophils, and neutrophils in the peribronchial and perivascular space (Figure 1A). Mice exposed to DEP have the same inflammatory pattern; however, macrophages containing phagocytosed material are clearly evident as intracellular anthracotic pigment (Figure 1B). Airway mucus production in the model was determined by PAS staining. DEP/asthma mice (Figure 1D) showed a marked increase of mucus staining compared with PBS/asthma mice (Figure 1C). The increased mucus production was confirmed by computer-aided morphometric quantification of the PAS staining (Figure 1E).
Figure 1.
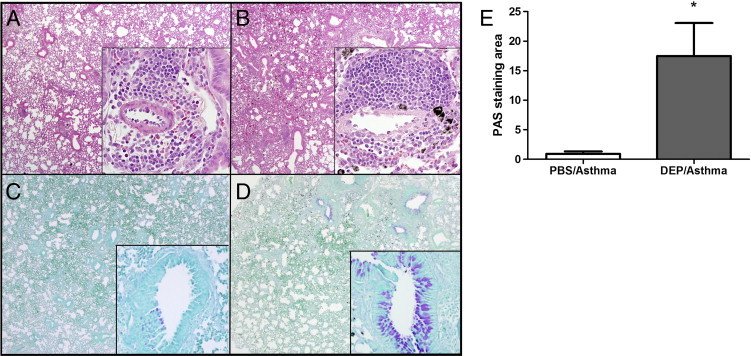
Representative pulmonary histological features 4 hours after the last challenge of HDE in the presence of DEPs. PBS/asthma were HDE-challenged mice in the presence of PBS in place of DEP. H&E-stained sections from PBS/asthma mice (A) show interstitial inflammation with peribronchial infiltration of inflammatory cells (inset). In the H&E sections from DEP/asthma mice, the anthracotic pigment is clearly visible (B). PAS-stained lung from PBS/asthma mice shows limited mucus in the airway (C). PAS sections from DEP/asthma mice show abundant mucus production (D). Computer-assisted quantification of the PAS staining area shows a significant increase (E). Each value is the mean ± SEM for six to eight mice per group from two separate experiments. *P ≤ 0.05 when compared with the PBS/asthma group.
Increased Pulmonary Oxidative Stress
Cellular oxidative stress is considered a primary factor in the biological effects of air pollutants.11 An imbalance in oxidative stress and antioxidant defenses induces a wide spectrum of pro-inflammatory mediators.14 Since first discovered in 1990,28 8-isoprostane (8-iso prostaglandin F2α) has been considered a reliable marker to assess the in vivo status of oxidative stress.29 To investigate potential exacerbation of asthma-like pulmonary inflammation by DEP, isoprostane levels in BALF were measured 4 hours after the last allergen challenge. Isoprostane concentrations in BALF were significantly greater in DEP/asthma compared with asthma alone (Figure 2). As a control, mice were exposed only to DEP without allergens. This group of mice did not have elevated isoprostane levels, indicating that the combination of stimulation with the allergen in the presence of the pollutants induces the highest level of oxidative stress.
Figure 2.
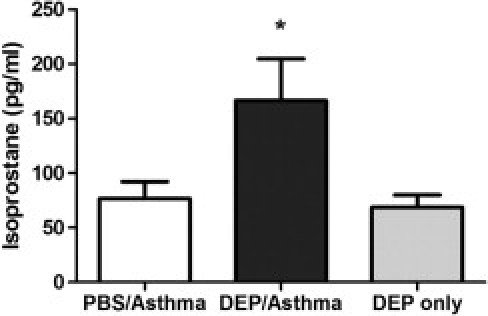
Isoprostane concentration in BALF. The DEP/asthma group received the combination of DEP and HDE, the PBS/asthma group received PBS and HDE, and the DEP-only group just received the DEP challenge (no HDE). Asthma challenge in the presence of DEP significantly increased isoprostane levels in BALF. Each value is the mean ± SEM for six to eight mice per group from two separate experiments. *P ≤ 0.05 when compared with the PBS-challenged group.
DEP Enhances AHR
DEP-induced ROS have exacerbated asthma by sensitizing airway smooth muscle to acetylcholine-induced contraction.17 When delivered concomitantly or after allergen challenge, DEP exacerbated AHR in addition to pulmonary inflammation.30, 31 AHR was measured by WBP and confirmed with a direct invasive method, given the controversy of WBP.24, 32 Bronchopulmonary hyperresponsiveness to methacholine (50 mg/mL) challenge was measured 2 hours after the last challenge and was significantly increased in DEP/asthma mice compared with PBS/asthma mice (Figure 3A). The DEP-only group showed no significant changes in enhanced pause (data not shown). Forced oscillation experiments compared the airway resistance of the DEP/asthma with the PBS/asthma group, which verified the WBP studies by demonstrating a significant increase in airway resistance in the DEP/asthma mice (Figure 3B).
Figure 3.
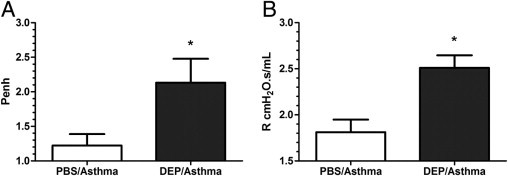
DEP significantly exacerbates asthma AHR. Enhanced pause (Penh; A) and resistance (R; B) values in response to methacholine (50 and 25 mg/mL, respectively) were obtained 2 hours after the last challenge via the WBP (A) or forced oscillation (B) techniques. Data are the mean ± SEM (n = 8 per group for A, from two separate experiments, and n = 4 per group for B). *P ≤ 0.05 when compared with the PBS/asthma group.
Exacerbation of Pulmonary Neutrophil Infiltration
The early response to asthma includes recruitment of neutrophils to the pulmonary airspace.20, 33 We examined whether DEP would increase the inflammatory response in the early phase by assessing parameters 4 hours after the allergen challenge. DEP/asthma mice had significantly more neutrophils within the BALF compared with PBS/asthma mice (Figure 4A). Pulmonary MPO, which measures neutrophils and monocytes sequestered in the interstitium, was also significantly increased by exposure to DEP plus the allergen challenge. The numbers of other inflammatory cells in the BALF, including eosinophils, lymphocytes, and macrophages, were not increased by the DEP and allergen challenge (data not shown).
Figure 4.
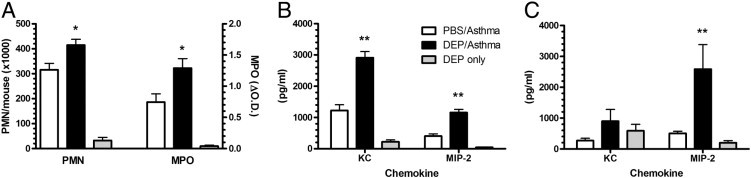
Increased pulmonary neutrophils and CXC chemokines in the DEP/asthma group 4 hours after allergen challenge. Asthma challenge after DEP exposure significantly increases pulmonary neutrophil recruitment and MPO activity in the lungs (A). The CXC chemokines KC and MIP-2 were also significantly increased in the BALF (B) and within the lung homogenate (C). The data are expressed as the mean ± SEM for six to eight mice per group from two separate experiments. *P ≤ 0.05, **P ≤ 0.01 when compared with the PBS/asthma group.
To investigate the potential role of CXC chemokines in the enhanced neutrophil recruitment induced by DEP/asthma, we measured the concentrations of KC and MIP-2 in the BALF and lung tissue during the early response. The BALF levels of both chemokines were significantly increased (Figure 4B) in the DEP/asthma mice. In addition, MIP-2 levels were elevated in the lung homogenate in the DEP/asthma group (Figure 4C). We also analyzed the BALF levels of CC chemokines, including regulated on activation normal T-cell expressed and secreted and MIP-1α, as previous reports demonstrated.19, 34 and found no significant differences between the two groups (data not shown). DEP exposure in asthma mice also had systemic effects, because the plasma levels of MIP-2 and total IgE were increased compared with PBS/asthma or DEP only (data not shown).
CXC Chemokines Are Responsible for Enhanced Pulmonary Neutrophil Recruitment in Airway Inflammation and AHR by DEP
Particulate matter–induced oxidative stress has augmented CXC chemokines (ie, IL-8, KC, and MIP-2) in cystic fibrosis airway cells35 and airway epithelial cells.16, 36 As previously shown, in this model, KC and MIP-2 were significantly increased locally (BALF and lung homogenates) in the DEP/asthma group. We hypothesized that DEP/asthma may promote the development and exacerbation of allergic pulmonary inflammation via up-regulation of CXC chemokines.
To rigorously test this hypothesis, mice were treated with a combination of neutralizing antibodies to KC and MIP-2 at 1 hour before DEP challenge on day 21 by the oropharyngeal route. At 1 hour after the DEP exposure, mice were challenged with HDE-containing cockroach allergens; at 2 hours after the HDE challenge, respiratory parameters were measured via WBP. After WBP, mice were sacrificed, which was 4 hours after the allergen challenge. For this study, mice challenged with PBS/asthma or DEP only were not included because KC and MIP-2 were substantially elevated only in the DEP/asthma group.
All respiratory parameters, the number of inflammatory cells in airways, and the inflammatory mediator data set from the IgG-treated and anti-CXC chemokine–treated mice were converted to a heat map to perform a comprehensive unbiased analysis, as previously reported.22 The raw values that were subjected to Z-score transformation include all respiratory parameters (ie, respiratory rate, tidal volume, minute ventilation, time inspiration, time expiration, peak inspiratory flow, peak expiratory flow, and enhanced pause); differential counting of BALF (ie, number of total cells, eosinophils, lymphocytes, macrophages, and neutrophils); neutrophil MPO; eosinophil-specific peroxidase; inflammatory mediators in BALF, plasma, and lung homogenates; and quantified PAS staining. Once Z-score normalization was completed, a heat map was generated from the averaged Z-score by using the TM4-MeV program and further simplified by unbiased hierarchal clustering based on sample groups and parameters.
The final analysis of the clustered heat map demonstrated the pattern that anti-KC/MIP-2 antibody–treated mice shared more common features with healthy naïve mice (see Supplemental Figure S1 at http://ajp.amjpathol.org), and airway obstruction and pulmonary inflammatory cell infiltrations induced by DEP were significantly improved by anti-KC/MIP-2 Ab treatment. To further demonstrate the improvement of respiratory function by anti-CXC chemokine Ab treatment, we analyzed and compared the minute ventilation and time expiration in control IgG-treated, anti-CXC antibody–treated, and naïve mice. As shown in Figure 5, A and B, anti-KC/MIP-2 Ab treatment significantly improved minute ventilation and time expiration. Among the common features of respiratory parameters, minute ventilation and time expiration are correlated with improved airway obstruction.22
Figure 5.
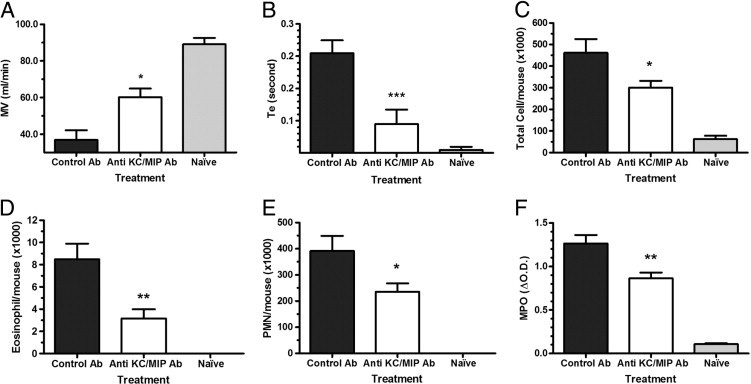
Anti-KC and MIP-2 antibody treatment significantly improved lung functions [minute ventilation (MV; A) and time of expiration (Te; B)] and pulmonary inflammatory cell recruitment (C–F). Mice received a combination of anti-mouse KC and MIP-2 monoclonal antibodies or control rat IgG before DEP exposure and allergen challenge. Lung function (MV and Te) changes in response to nebulized methacholine (50 mg/mL) were measured 2 hours after the final allergen challenge via WBP. Cells in BALF recovered after 4 hours were quantified. Antibody treatment significantly improved MV (A), Te (B), total BALF cells (C), eosinophils (D), and neutrophils (E). Lung MPO was also reduced significantly (F). The data are expressed as the mean ± SEM for eight mice per group from two separate experiments. *P ≤ 0.05, **P ≤ 0.01 when compared with the IgG-treated group.
Analysis of the clustered heat map also suggested that pulmonary infiltration of inflammatory cells was significantly improved by anti-CXC antibody treatment. CXC chemokines are major neutrophil chemoattractants, and it is anticipated that antibody inhibition of their biological activity would decrease pulmonary inflammatory cell recruitment, particularly neutrophils. This was examined by quantifying the cells recovered from the BALF obtained 4 hours after the last allergen challenge. Neutralization of CXC chemokines resulted in fewer cells in the BALF (Figure 5C). CXC chemokines will increase the chemotaxis of eosinophils,37, 38 and our studies show that antibody blockade of the CXC chemokines reduced the number of eosinophils in the BALF (Figure 5D). Given the prominent role of KC and MIP-2 as neutrophil chemoattractants, we anticipated that the number of neutrophils recruited to the lung in response to the DEP and HDE challenge would be significantly decreased. As expected, the number of neutrophils recovered from the BALF was significantly reduced in the mice treated with anti-KC/MIP-2 Ab (Figure 5E). Along with the reduction of neutrophils in the airway, there was a marked reduction in lung MPO activity, which reflects neutrophil sequestration within the lung (Figure 5F). There was no significant reduction in the numbers of lymphocytes or macrophages, the expected result because these cells do not respond to CXC chemokines (data not shown). These data demonstrate that neutralization of the CXC chemokines in the airway reduced the number of inflammatory cells in the lung tissue and airways.
In our previous report,22 we demonstrated that excess production of mucus in airways is correlated with the airway obstruction, and a decrease in mucus production significantly improved air exchange in dexamethasone-treated mice. With improved respiratory function and fewer pulmonary neutrophils and eosinophils after neutralization of KC and MIP-2, we then examined how the antibody treatment affects airway mucin expression via PAS staining. The anti-CXC antibody–treated mice (Figure 6B) had significantly less production of airway mucin compared with the control antibody–treated mice (Figure 6A), with levels similar to the amounts quantified in Figure 1E. The reduction in the levels of mucin expression with antibody inhibition of CXC chemokines was further demonstrated by computer-assisted morphometric quantification of the PAS staining (Figure 6C).
Figure 6.
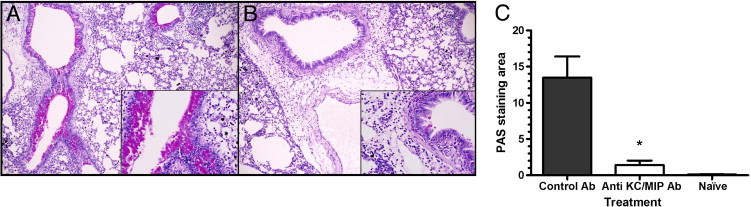
CXC chemokine inhibition reduces airway mucus expression. PAS-stained lungs from control IgG-treated mice show abundant mucus production (A). Anti-KC and MIP-2 antibody-treated mice show significantly reduced mucus in the airway (B). Computer-assisted quantification of PAS staining showed a significant reduction of PAS staining with anti-KC/MIP-2 treatment (C). Each value is the mean ± SEM for eight mice per group from two separate experiments. *P ≤ 0.01 when compared with the control IgG group.
Discussion
In this study, we investigated the role of CXC chemokines KC and MIP-2 in a mouse model of asthma to define the mechanisms and mediators responsible for the exacerbation of pulmonary inflammation induced by co-exposure of the air pollutant, DEP, and HDE. The current study defined that DEP augmented the pulmonary expression of CXC chemokines, followed by an increase in the number of neutrophils in the airways. Neutralization of KC and MIP-2 chemokines significantly attenuated the pulmonary inflammation, including reduced inflammatory cells, mucin production, and AHR.
A significant association between exposure to air pollutants, including PM, and pre-existing pulmonary diseases, including asthma, has been demonstrated by many epidemiological39, 40and clinical10, 41 studies. While pinpointing the exact mechanisms underlying the pathophysiological characteristics, the evidence for the role of DEP as an adjuvant in promoting sensitization and allergic diseases has been demonstrated in human42, 43, 44 and animal31, 45, 46, 47 studies. When DEP is administered concurrently with allergen, the expression of type 2 helper T-cell cytokines is increased, with decreased type 1 helper T-cell cytokines.30 Furthermore, when mice were exposed to ultrafine particles before allergen challenge, significant increases in inflammatory cells, including neutrophils and eosinophils, airway mucus production, and AHR, were induced.31, 48 Although DEP exposure was significantly delayed compared with our study, these results are consistent with the current study.
Although the mechanisms that promote sensitization and allergic diseases by air pollutants are yet to be fully elucidated, a growing body of studies suggests potential epigenetic modifications, including DNA methylation and histone modification induced by exposure to air pollution, play some role in the development of allergy and asthma.49, 50 A recent study by Liu et al51 demonstrated that augmented IgE expression in combined exposure to DEP and allergen is potentially induced via DNA hypermethylation of interferon-γ promoter and DNA hypomethylation of IL-4 promoter. Viera et al52 suggest that expanding the investigation in the epigenetic changes could provide potential insight into the mechanism of promoting the development of allergic inflammation, including asthma, by DEP exposure.
The pulmonary expressions of the CXC chemokines KC and MIP-2 have contributed to neutrophil-mediated acute lung injury.53 Selective recruitment of neutrophils into the airways during early allergic inflammation suggests that neutrophil-specific chemoattractants are produced and released throughout pulmonary inflammation. Airway accumulation of neutrophils and production of inflammatory mediators, including cytokines and chemokines, are characteristics of an inhaled ozone-induced early inflammatory response.54 A strong correlation has been established between neutrophilic inflammation of the airways and asthma severity, as well as the presence of activated neutrophils.55, 56 Our studies are in agreement with previous reports, in which neutralization of KC and MIP-2 in the mouse model of asthma, induced by allergen challenge, ovalbumin,57 or HDE,25 showed a significant reduction of the number of airway neutrophils.
Considering the fact that DEPs used in this experiment include a low level of lipopolysaccharide (LPS; 5.4 ng per mouse), an increased number of neutrophils infiltrated in the lung might result from the presence of the LPS in the DEPs. However, the number of neutrophils in the lung and the expression of CXC chemokines in the DEP–only mice [that received three DEP challenges (ie, no allergen)] remained close to the baseline, suggesting that the low level of LPS in the DEPs is not the contributing factor to the increased pulmonary recruitment of neutrophils. Although the role of endotoxin in the onset of asthma is complicated because of the difference and complexity in the model, including allergen used, timing of endotoxin exposure, and dose,58, 59 animal and human studies60, 61 have shown that endotoxin co-exposure is associated with the exacerbation of pre-existing pulmonary disease, including asthma. Recent human studies demonstrated that endotoxin co-exposure with cockroach allergen62 or air pollutant60 synergistically affects pulmonary inflammation and wheezing in children. In our recent study63 using BALB/c mice, the administration of endotoxin before allergen challenge significantly reduced AHR, mucus production, and pulmonary eosinophilia. A transitory state of LPS hyporesponsiveness64 has recently been identified and postulated as a protective mechanism from excessive inflammatory responses via reduced expression of tumor necrosis factor-α and CC chemokine, including eotaxin, while maintaining pulmonary neutrophil recruitment to fight against bacterial challenge.26, 65
In addition to the anticipated reduction of the number of neutrophils infiltrated into airway and lungs, the number of eosinophils in BALF was also decreased by neutralization of the CXC chemokines. The decreased pulmonary influx of eosinophils may be the direct result of reduced chemokine levels or an indirect result. In a study66 using human lung epithelial cells, neutrophils stimulated eosinophils to move toward the complement fragment 5a. In another ex vivo system using isolated neutrophils and eosinophils, IL-8 (CXCL8) stimulated neutrophils to significantly enhance the trans-basement membrane migration of eosinophils.38 The same authors67 also reported that the percentage of eosinophils in the sputum of patients with severe persistent asthma strongly correlated with the number of neutrophils and concluded that increased airway accumulation of neutrophils may be a major contributing factor in the development of pulmonary eosinophilia. Our data are in agreement with these results, although they do not fully address the controversy about whether eosinophils are chemotactic to IL-8.37, 68, 69 Mice lack the exact homolog of human IL-8, and KC and MIP-2 are considered to be the mouse homologs of IL-8.70
The recruitment of inflammatory cells, such as neutrophils, and the decline of lung function by oxidative injury from exposure to air pollutants have been shown in clinical71, 72 and animal73 studies. Increased ROS production by macrophages, eosinophils, and neutrophils in patients with asthma induces elevated hydrogen peroxide in their breath74 and superoxide dismutase in their lung cells.75 Furthermore, eosinophil peroxidase76 and MPO77 activities in the BALF of patients with asthma were significantly increased. Although the mechanisms of oxidant toxicity in the pre-existing pulmonary diseases remain to be elucidated, reactions between ROS and surrounding biological compounds, such as proteins, lipids, and DNA, induce various pathological consequences. Based on this finding, enhancing the antioxidant defense systems of the airway would be a therapeutic strategy,14, 78 especially for populations at risk with increased DEP exposure.9
It is also possible that the oxidant enhancement of asthma-like pulmonary inflammation was not the result of a direct toxic effect but rather the result of augmentation of chemokine production. Previous work has demonstrated that ROS will increase the production of CXC chemokines, including IL-879 and the murine CXC chemokines.80 The proposed sequence of events would involve HDE and DEP augmenting ROS production, which, in turn, would increase CXC chemokine levels and drive the inflammatory process. Blocking the elevated CXC chemokines resulted in significant improvements of several parameters of pulmonary inflammation, including reduced neutrophil and eosinophil numbers, improved respiratory function, and decreased airway mucin production.
Footnotes
Supported by a grant from the NIH (R01 ES013528).
CME Disclosure: None of the authors disclosed any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.08.008.
Supplementary data
Supplemental Figure S1.
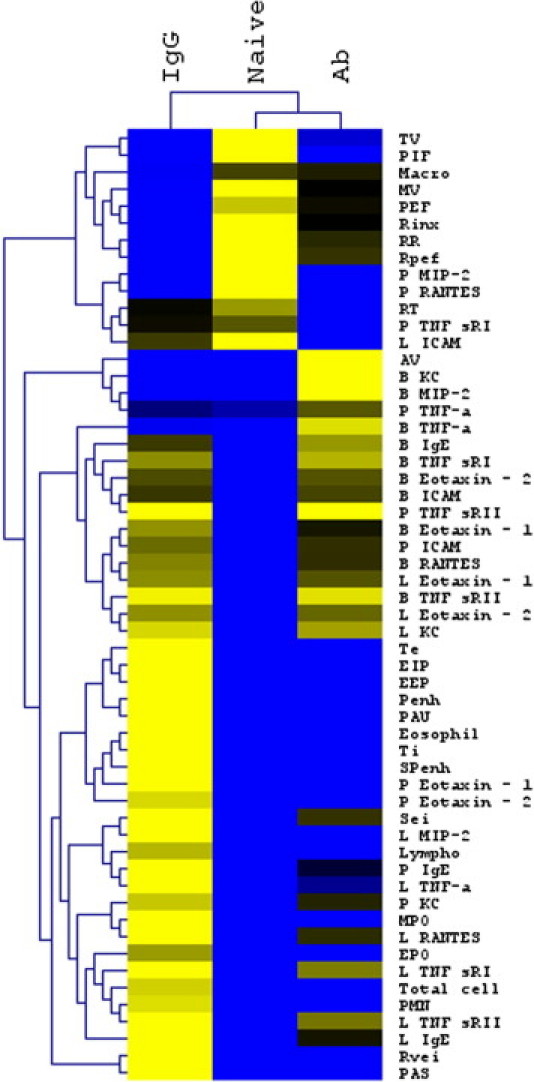
Heat map analysis and hierarchal clustering of the entire data set. Inflammatory cells in the BALF, entire respiratory parameters, and inflammatory mediators in BALF (B), plasma (P), and lung homogenates (L) were subject to Z-score normalization. Normalized Z-scores were averaged and converted to a heat map. The heat map data were subject to hierarchal clustering by sample group (naïve, IgG-treated, and anti-CXC antibody–treated mice) and by parameter to demonstrate the pattern in each group's response. The hierarchal clustering demonstrated that anti-KC and MIP-2 antibody-treated mice shared many features with the naïve mice (n = 8 for naïve mice, and n =12 for IgG-treated and CXC antibody-treated group from three separate experiments).
References
- 1.Busse W.W., Lemanske R.F., Jr Asthma. N Engl J Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Masoli M., Fabian D., Holt S., Beasley R. Global Initiative for Asthma (GINA) Program: The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 3.Eder W., Ege M.J., von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 4.D'Amato G., Liccardi G., D'Amato M., Holgate S. Environmental risk factors and allergic bronchial asthma. Clin Exp Allergy. 2005;35:1113–1124. doi: 10.1111/j.1365-2222.2005.02328.x. [DOI] [PubMed] [Google Scholar]
- 5.Akinbami L.J., Schoendorf K.C. Trends in childhood asthma: prevalence, health care utilization, and mortality. Pediatrics. 2002;110(Pt 1):315–322. doi: 10.1542/peds.110.2.315. [DOI] [PubMed] [Google Scholar]
- 6.Akinbami L.J., Moorman J.E., Liu X. Asthma prevalence, health care use, and mortality: United States, 2005–2009. Natl Health Stat Report. 2011;(32):1–14. [PubMed] [Google Scholar]
- 7.Ciencewicki J., Trivedi S., Kleeberger S.R. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol. 2008;122:456. doi: 10.1016/j.jaci.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pope C.A., 3rd, Burnett R.T., Thurston G.D., Thun M.J., Calle E.E., Krewski D., Godleski J.J. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- 9.Riedl M., Diaz-Sanchez D. Biology of diesel exhaust effects on respiratory function. J Allergy Clin Immunol. 2005;115:221. doi: 10.1016/j.jaci.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 10.Balmes J.R., Earnest G., Katz P.P., Yelin E.H., Eisner M.D., Chen H., Trupin L., Lurmann F., Blanc P.D. Exposure to traffic: lung function and health status in adults with asthma. J Allergy Clin Immunol. 2009;123:626. doi: 10.1016/j.jaci.2008.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riedl M.A. The effect of air pollution on asthma and allergy. Curr Allergy Asthma Rep. 2008;8:139–146. doi: 10.1007/s11882-008-0024-8. [DOI] [PubMed] [Google Scholar]
- 12.Saxon A., Diaz-Sanchez D. Air pollution and allergy: you are what you breathe. Nat Immunol. 2005;6:223. doi: 10.1038/ni0305-223. [DOI] [PubMed] [Google Scholar]
- 13.Pandya R.J., Solomon G., Kinner A., Balmes J.R. Diesel exhaust and asthma: hypotheses and molecular mechanisms of action. Environ Health Perspect. 2002;110(Suppl 1):103–112. doi: 10.1289/ehp.02110s1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dozor A.J. The role of oxidative stress in the pathogenesis and treatment of asthma. Ann N Y Acad Sci. 2010;1203:133–137. doi: 10.1111/j.1749-6632.2010.05562.x. [DOI] [PubMed] [Google Scholar]
- 15.Seagrave J. Mechanisms and implications of air pollution particle associations with chemokines. Toxicol Appl Pharmacol. 2008;232:469–477. doi: 10.1016/j.taap.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ovrevik J., Lag M., Holme J.A., Schwarze P.E., Refsnes M. Cytokine and chemokine expression patterns in lung epithelial cells exposed to components characteristic of particulate air pollution. Toxicology. 2009;259:46–53. doi: 10.1016/j.tox.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 17.Bowler R.P. Oxidative stress in the pathogenesis of asthma. Curr Allergy Asthma Rep. 2004;4:116–122. doi: 10.1007/s11882-004-0056-7. [DOI] [PubMed] [Google Scholar]
- 18.Bömmel H., Haake M., Luft P., Horejs-Hoeck J., Hein H., Bartels J., Schauer C., Pöschl U., Kracht M., Duschl A. The diesel exhaust component pyrene induces expression of IL-8 but not of eotaxin. Int Immunopharmacol. 2003;3:1371–1379. doi: 10.1016/S1567-5769(03)00135-8. [DOI] [PubMed] [Google Scholar]
- 19.Takano H., Yanagisawa R., Ichinose T., Sadakane K., Yoshino S., Yoshikawa T., Morita M. Diesel exhaust particles enhance lung injury related to bacterial endotoxin through expression of proinflammatory cytokines, chemokines, and intercellular adhesion molecule-1. Am J Respir Crit Care Med. 2002;165:1329–1335. doi: 10.1164/rccm.2108122. [DOI] [PubMed] [Google Scholar]
- 20.Kim J., Merry A.C., Nemzek J.A., Bolgos G.L., Siddiqui J., Remick D.G. Eotaxin represents the principal eosinophil chemoattractant in a novel murine asthma model induced by house dust containing cockroach allergens. J Immunol. 2001;167:2808–2815. doi: 10.4049/jimmunol.167.5.2808. [DOI] [PubMed] [Google Scholar]
- 21.McKinley L., Kim J., Bolgos G.L., Siddiqui J., Remick D.G. Reproducibility of a novel model of murine asthma-like pulmonary inflammation. Clin Exp Immunol. 2004;136:224–231. doi: 10.1111/j.1365-2249.2004.02461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaickus L.J., Bouchard J., Kim J., Natarajan S., Remick D.G. Assessing pulmonary pathology by detailed examination of respiratory function. Am J Pathol. 2010;177:1861–1869. doi: 10.2353/ajpath.2010.100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohtani T., Nakagawa S., Kurosawa M., Mizuashi M., Ozawa M., Aiba S. Cellular basis of the role of diesel exhaust particles in inducing Th2-dominant response. J Immunol. 2005;174:2412–2419. doi: 10.4049/jimmunol.174.4.2412. [DOI] [PubMed] [Google Scholar]
- 24.Vanoirbeek J.A.J., Rinaldi M., De Vooght V., Haenen S., Bobic S., Gayan-Ramirez G., Hoet P.H., Verbeken E., Decramer M., Nemery B., Janssens W. Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol. 2010;42:96–104. doi: 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 25.McKinley L., Kim J., Bolgos G.L., Siddiqui J., Remick D.G. CXC chemokines modulate IgE secretion and pulmonary inflammation in a model of allergic asthma. Cytokine. 2005;32:178–185. doi: 10.1016/j.cyto.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Natarajan S., Kim J.Y., Remick D.G. Acute pulmonary lipopolysaccharide tolerance decreases TNF-alpha without reducing neutrophil recruitment. J Immunol. 2008;181:8402–8408. doi: 10.4049/jimmunol.181.12.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider T., Issekutz A.C. Quantitation of eosinophil and neutrophil infiltration into rat lung by specific assays for eosinophil peroxidase and myeloperoxidase: application in a Brown Norway rat model of allergic pulmonary inflammation. J Immunol Methods. 1996;198:1–14. doi: 10.1016/0022-1759(96)00143-3. [DOI] [PubMed] [Google Scholar]
- 28.Morrow J.D., Hill K.E., Burk R.F., Nammour T.M., Badr K.F., Roberts L.J., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montuschi P., Barnes P.J., Roberts L.J. Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 30.Hao M., Comier S., Wang M., Lee J.J., Nel A. Diesel exhaust particles exert acute effects on airway inflammation and function in murine allergen provocation models. J Allergy Clin Immunol. 2003;112:905. doi: 10.1016/j.jaci.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Alessandrini F., Schulz H., Takenaka S., Lentner B., Karg E., Behrendt H., Jakob T. Effects of ultrafine carbon particle inhalation on allergic inflammation of the lung. J Allergy Clin Immunol. 2006;117:824–830. doi: 10.1016/j.jaci.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 32.Finkelman F.D. Use of unrestrained, single-chamber barometric plethysmography to evaluate sensitivity to cholinergic stimulation in mouse models of allergic airway disease. J Allergy Clin Immunol. 2008;121:334–335. doi: 10.1016/j.jaci.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen B., Dahl R., Karlstrom R., Peterson C.G., Venge P. Eosinophil and neutrophil activity in asthma in a one-year trial with inhaled budesonide: the impact of smoking. Am J Respir Crit Care Med. 1996;153:1519–1529. doi: 10.1164/ajrccm.153.5.8630596. [DOI] [PubMed] [Google Scholar]
- 34.Diaz-Sanchez D., Jyrala M., Ng D., Nel A., Saxon A. In vivo nasal challenge with diesel exhaust particles enhances expression of the CC chemokines rantes, MIP-1alpha, and MCP-3 in humans. Clin Immunol. 2000;97:140–145. doi: 10.1006/clim.2000.4921. [DOI] [PubMed] [Google Scholar]
- 35.Bartling T.R., Drumm M.L. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. Am J Respir Cell Mol Biol. 2009;40:58–65. doi: 10.1165/rcmb.2007-0464OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomita K., Barnes P.J., Adcock I.M. The effect of oxidative stress on histone acetylation and IL-8 release. Biochem Biophys Res Commun. 2003;301:572–577. doi: 10.1016/s0006-291x(02)03029-2. [DOI] [PubMed] [Google Scholar]
- 37.Bochenska-Marciniak M., Kupczyk M., Gorski P., Kuna P. The effect of recombinant interleukin-8 on eosinophils' and neutrophils' migration in vivo and in vitro. Allergy. 2003;58:795–801. doi: 10.1034/j.1398-9995.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- 38.Kikuchi I., Kikuchi S., Kobayashi T., Hagiwara K., Sakamoto Y., Kanazawa M., Nagata M. Eosinophil trans-basement membrane migration induced by interleukin-8 and neutrophils. Am J Respir Cell Mol Biol. 2006;34:760–765. doi: 10.1165/rcmb.2005-0303OC. [DOI] [PubMed] [Google Scholar]
- 39.D'Amato G., Liccardi G., D'Amato M., Cazzola M. Outdoor air pollution, climatic changes and allergic bronchial asthma. Eur Respir J. 2002;20:763–776. doi: 10.1183/09031936.02.00401402. [DOI] [PubMed] [Google Scholar]
- 40.Peden D.B. The epidemiology and genetics of asthma risk associated with air pollution. J Allergy Clin Immunol. 2005;115:213–219. doi: 10.1016/j.jaci.2004.12.003. quiz 220. [DOI] [PubMed] [Google Scholar]
- 41.Downs S.H., Schindler C., Liu L.J., Keidel D., Bayer-Oglesby L., Brutsche M.H., Gerbase M.W., Keller R., Kunzli N., Leuenberger P., Probst-Hensch N.M., Tschopp J.M., Zellweger J.P., Rochat T., Schwartz J., Ackermann-Liebrich U., Team S. Reduced exposure to PM10 and attenuated age-related decline in lung function. N Engl J Med. 2007;357:2338–2347. doi: 10.1056/NEJMoa073625. [DOI] [PubMed] [Google Scholar]
- 42.Diaz-Sanchez D., Garcia M.P., Wang M., Jyrala M., Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol. 1999;104:1183–1188. doi: 10.1016/s0091-6749(99)70011-4. [DOI] [PubMed] [Google Scholar]
- 43.Takenaka H., Zhang K., Diazsanchez D., Tsien A., Saxon A. Enhanced human IgE production results from exposure to the aromatic-hydrocarbons from diesel exhaust: direct effects on B-cell IgE production. J Allergy Clin Immunol. 1995;95:103–115. doi: 10.1016/s0091-6749(95)70158-3. [DOI] [PubMed] [Google Scholar]
- 44.Nel A.E., Diaz-Sanchez D., Ng D., Hiura T., Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. 1998;102:539–554. doi: 10.1016/s0091-6749(98)70269-6. [DOI] [PubMed] [Google Scholar]
- 45.Li N., Harkema J.R., Lewandowski R.P., Wang M.Y., Bramble L.A., Gookin G.R., Ning Z., Kleinman M.T., Sioutas C., Nel A.E. Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol. 2010;299:L374–L383. doi: 10.1152/ajplung.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alberg T., Hansen J., Lovik M. Allergic adjuvant activity of air pollutants in an intranasal mouse model. Allergy. 2007;62(Suppl 83):170. [Google Scholar]
- 47.Rogerieux F., Abou C.O., Remy F., Robidel F., Lacroix G. A pollen guinea pig model to study adjuvant effects of air pollutants. Allergy. 2008;63:1530. [Google Scholar]
- 48.Miyabara Y., Ichinose T., Takano H., Lim H.B., Sagai M. Effects of diesel exhaust on allergic airway inflammation in mice. J Allergy Clin Immunol. 1998;102:805–812. doi: 10.1016/s0091-6749(98)70021-1. [DOI] [PubMed] [Google Scholar]
- 49.Kuriakose J.S., Miller R.L. Environmental epigenetics and allergic diseases: recent advances. Clin Exp Allergy. 2010;40:1602–1610. doi: 10.1111/j.1365-2222.2010.03599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barnes P.J. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:693–696. doi: 10.1513/pats.200907-071DP. [DOI] [PubMed] [Google Scholar]
- 51.Liu J., Ballaney M., Al-alem U., Quan C., Jin X., Perera F., Chen L.-C., Miller R.L. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci. 2008;102:76–81. doi: 10.1093/toxsci/kfm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viera L., Chen K., Nel A., Lloret M.G. The impact of air pollutants as an adjuvant for allergic sensitization and asthma. Curr Allergy Asthma Rep. 2009;9:327–333. doi: 10.1007/s11882-009-0046-x. [DOI] [PubMed] [Google Scholar]
- 53.Lomas-Neira J.L., Chung C.S., Wesche D.E., Perl M., Ayala A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J Leukoc Biol. 2005;77:846–853. doi: 10.1189/jlb.1004617. [DOI] [PubMed] [Google Scholar]
- 54.Yoon H.K., Cho H.Y., Kleeberger S.R. Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ Health Perspect. 2007;115:1557–1563. doi: 10.1289/ehp.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jatakanon A., Uasuf C., Maziak W., Lim S., Chung K.F., Barnes P.J. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. 1999;160:1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 56.Macdowell A.L., Peters S.P. Neutrophils in asthma. Curr Allergy Asthma Rep. 2007;7:464–468. doi: 10.1007/s11882-007-0071-6. [DOI] [PubMed] [Google Scholar]
- 57.Knott P.G., Gater P.R., Dunford P.J., Fuentes M.E., Bertrand C.P. Rapid up-regulation of CXC chemokines in the airways after Ag-specific CD4+ T cell activation. J Immunol. 2001;166:1233–1240. doi: 10.4049/jimmunol.166.2.1233. [DOI] [PubMed] [Google Scholar]
- 58.Delfino R.J. Do endotoxin and air pollution have a synergistic relationship to asthma onset or exacerbation? Am J Respir Crit Care Med. 2009;180:1037–1038. doi: 10.1164/rccm.200908-1285ED. [DOI] [PubMed] [Google Scholar]
- 59.Zhu Z., Oh S.Y., Zheng T., Kim Y.K. Immunomodulating effects of endotoxin in mouse models of allergic asthma. Clin Exp Allergy. 2010;40:536–546. doi: 10.1111/j.1365-2222.2010.03477.x. [DOI] [PubMed] [Google Scholar]
- 60.Ryan P.H., Bernstein D.I., Lockey J., Reponen T., Levin L., Grinshpun S., Villareal M., Hershey G.K.K., Burkle J., LeMasters G. Exposure to traffic-related particles and endotoxin during infancy is associated with wheezing at age 3 years. Am J Respir Crit Care Med. 2009;180:1068–1075. doi: 10.1164/rccm.200808-1307OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yanagisawa R., Takano H., Inoue K., Ichinose T., Sadakane K., Yoshino S., Yamaki K., Kumagai Y., Uchiyama K., Yoshikawa T., Morita M. Enhancement of acute lung injury related to bacterial endotoxin by components of diesel exhaust particles. Thorax. 2003;58:605–612. doi: 10.1136/thorax.58.7.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kulhankova K., George C.L.S., Kline J.N., Snyder J.M., Darling M., Field E.H., Thorne P.S. Early-life co-administration of cockroach allergen and endotoxin augments pulmonary and systemic responses. Clin Exp Allergy. 2009;39:1069–1079. doi: 10.1111/j.1365-2222.2009.03254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Natarajan S., Kim J., Bouchard J., Cruikshank W., Remick D.G. Pulmonary endotoxin tolerance protects against cockroach allergen-induced asthma-like inflammation in a mouse model. Int Arch Allergy Appl Immunol. 2011 doi: 10.1159/000330896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Medvedev A.E., Sabroe I., Hasday J.D., Vogel S.N. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133–150. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]
- 65.Natarajan S., Kim J., Remick D.G. Chronic pulmonary LPS tolerance induces selective immunosuppression while maintaining the neutrophilic response. Shock. 2010;33:162–169. doi: 10.1097/SHK.0b013e3181aa9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zuurbier A.E.M., Liu L., Mul F.P.J., Verhoeven A.J., Knol E.F., Roos D. Neutrophils enhance eosinophil migration across monolayers of lung epithelial cells. Clin Exp Allergy. 2001;31:444–452. doi: 10.1046/j.1365-2222.2001.01073.x. [DOI] [PubMed] [Google Scholar]
- 67.Kikuchi S., Nagata M., Kikuchi I., Hagiwara K., Kanazawa M. Association between neutrophilic and eosinophilic inflammation in patients with severe persistent asthma. Int Arch Allergy Immunol. 2005;137(Suppl 1):7–11. doi: 10.1159/000085425. [DOI] [PubMed] [Google Scholar]
- 68.Petering H., Gotze O., Kimmig D., Smolarski R., Kapp A., Elsner J. The biologic role of interleukin-8: functional analysis and expression of CXCR1 and CXCR2 on human eosinophils. Blood. 1999;93:694–702. [PubMed] [Google Scholar]
- 69.Ulfman L.H., Joosten D.P.H., van der Linden J.A.M., Lammers J.W.J., Zwaginga J.J., Koenderman L. IL-8 induces a transient arrest of rolling eosinophils on human endothelial cells. J Immunol. 2001;166:588–595. doi: 10.4049/jimmunol.166.1.588. [DOI] [PubMed] [Google Scholar]
- 70.Rubio N., Sanz-Rodriguez F. Induction of the CXCL1 (KC) chemokine in mouse astrocytes by infection with the murine encephalomyelitis virus of Theiler. Virology. 2007;358:98–108. doi: 10.1016/j.virol.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 71.Vagaggini B., Carnevali S., Macchioni P., Taccola M., Fornai E., Bacci E., Bartoli M.L., Cianchetti S., Dente F.L., Di Franco A., Giannini D., Paggiaro P.L. Airway inflammatory response to ozone in subjects with different asthma severity. Eur Respir J. 1999;13:274–280. doi: 10.1034/j.1399-3003.1999.13b09.x. [DOI] [PubMed] [Google Scholar]
- 72.Louis R., Lau L.C., Bron A.O., Roldaan A.C., Radermecker M., Djukanovic R. The relationship between airways inflammation and asthma severity. Am J Respir Crit Care Med. 2000;161:9–16. doi: 10.1164/ajrccm.161.1.9802048. [DOI] [PubMed] [Google Scholar]
- 73.Kleeberger S.R., Levitt R.C., Zhang L.Y., Longphre M., Harkema J., Jedlicka A., Eleff S.M., DiSilvestre D., Holroyd K.J. Linkage analysis of susceptibility to ozone-induced lung inflammation in inbred mice. Nat Genet. 1997;17:475–478. doi: 10.1038/ng1297-475. [DOI] [PubMed] [Google Scholar]
- 74.Emelyanov A., Fedoseev G., Abulimity A., Rudinski K., Fedoulov A., Karabanov A., Barnes P.J. Elevated concentrations of exhaled hydrogen peroxide in asthmatic patients. Chest. 2001;120:1136–1139. doi: 10.1378/chest.120.4.1136. [DOI] [PubMed] [Google Scholar]
- 75.Smith L.J., Shamsuddin M., Sporn P.H., Denenberg M., Anderson J. Reduced superoxide dismutase in lung cells of patients with asthma. Free Radic Biol Med. 1997;22:1301–1307. doi: 10.1016/s0891-5849(96)00550-3. [DOI] [PubMed] [Google Scholar]
- 76.Aldridge R.E., Chan T., van Dalen C.J., Senthilmohan R., Winn M., Venge P., Town G.I., Kettle A.J. Eosinophil peroxidase produces hypobromous acid in the airways of stable asthmatics. Free Radic Biol Med. 2002;33:847–856. doi: 10.1016/s0891-5849(02)00976-0. [DOI] [PubMed] [Google Scholar]
- 77.Monteseirin J., Bonilla I., Camacho J., Conde J., Sobrino F. Elevated secretion of myeloperoxidase by neutrophils from asthmatic patients: the effect of immunotherapy. J Allergy Clin Immunol. 2001;107:623–626. doi: 10.1067/mai.2001.113566. [DOI] [PubMed] [Google Scholar]
- 78.Bjelakovic G., Nikolova D., Gluud L.L., Simonetti R.G., Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2008;(2) doi: 10.1002/14651858.CD007176. CD007176. [DOI] [PubMed] [Google Scholar]
- 79.DeForge L.E., Fantone J.C., Kenney J.S., Remick D.G. Oxygen radical scavengers selectively inhibit interleukin 8 production in human whole blood. J Clin Investigation. 1992;90:2123–2129. doi: 10.1172/JCI116097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim Y.J., Kim D., Lee Y., Choi S.Y., Park J., Lee S.Y., Park J.W., Kwon H.J. Effects of nanoparticulate saponin-platinum conjugates on 2,4-dinitrofluorobenzene-induced macrophage inflammatory protein-2 gene expression via reactive oxygen species production in RAW 264.7 cells. BMB Rep. 2009;42:304–309. doi: 10.5483/bmbrep.2009.42.5.304. [DOI] [PubMed] [Google Scholar]