

Abstract
The risk of diabetic retinopathy is associated with the presence of both oxidative stress and toxic eicosanoids. Whether oxidative stress actually causes diabetic retinopathy via the generation of toxic eicosanoids, however, remains unknown. The aim of the present study was to determine whether tyrosine nitration of prostacyclin synthase (PGIS) contributes to retinal cell death in vitro and in vivo. Exposure of human retinal pericytes to heavily oxidized and glycated LDL (HOG-LDL), but not native forms of LDL (N-LDL), for 24 hours significantly increased pericyte apoptosis, accompanied by increased tyrosine nitration of PGIS and decreased PGIS activity. Inhibition of the thromboxane receptor or cyclooxygenase-2 dramatically attenuated HOG-LDL–induced apoptosis without restoring PGIS activity. Administration of superoxide dismutase (to scavenge superoxide anions) or l-NG-nitroarginine methyl ester (l-NAME, a nonselective nitric oxide synthase inhibitor) restored PGIS activity and attenuated pericyte apoptosis. In Akita mouse retinas, diabetes increased intraretinal levels of oxidized LDL and glycated LDL, induced PGIS nitration, enhanced apoptotic cell death, and impaired blood–retinal barrier function. Chronic administration of tempol, a superoxide scavenger, reduced intraretinal oxidized LDL and glycated LDL levels, PGIS nitration, and retina cell apoptosis, thereby preserving the integrity of blood–retinal barriers. In conclusion, oxidized LDL-mediated PGIS nitration and associated thromboxane receptor stimulation might be important in the initiation and progression of diabetic retinopathy.
Loss of pericytes from retinal microvessels is an important event in the early stage of diabetic retinopathy, which is associated with increased pericyte apoptosis. Although the mechanisms responsible for pericyte apoptosis remain unclear, several studies have shown associations between dyslipidemia and the severity of diabetic retinopathy.1, 2 Specifically, elevated levels of triglycerides, low density lipoprotein (LDL), and apolipoprotein B, a principal lipoprotein component of LDL, have been associated with diabetic retinopathy.1 However, these associations are relatively weak, leading us to hypothesize that the key contribution of lipoproteins to the propagation of diabetic retinopathy is not through changes in their plasma levels but, rather, through extravasation via a disrupted blood–retinal barrier and subsequent modification by glycation and oxidation. In support of this model, we have demonstrated that oxidized LDL is present in the retina in diabetic humans in proportion to the severity of diabetic retinopathy,3 and in cell culture studies we have shown that oxidized LDL has injurious effects on retinal pericytes.4, 5
Oxidative stress, defined as increased production of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) or decreased antioxidant defenses, has been implicated in the development of diabetic retinopathy. This proposed relationship is supported by the findings that oxidative stress is increased in the retinas of diabetic animals, and antioxidants inhibit the development of retinopathy in diabetic animals.6 Many proteins are likely affected by increased oxidative stress. We have found7, 8 that prostacyclin synthase (PGIS), an enzyme that releases vasoprotective prostacyclin (PGI2), is particularly susceptible to tyrosine nitration by RNS, including peroxynitrite (ONOO−). In cultured endothelial cells, hyperglycemia increases PGIS nitration and decreases its activity.9, 10 Tyrosine nitration of PGIS and associated thromboxane receptor activation are important contributors to the initiation and progression of vascular complications in diabetes.11 Although there is evidence that the risk of diabetic retinopathy is associated with the presence of both oxidative stress and toxic eicosanoids,6, 12 the role of ONOO− and nitration of PGIS in the pathogenesis of diabetic retinopathy remains to be determined. In this study, we investigated the mechanisms by which LDL oxidation and glycation contribute to pericyte loss in diabetes. Here we demonstrate that oxidized and glycated LDL induces human pericyte apoptosis, at least in part, by inducing tyrosine nitration of PGIS and inhibiting its activity.
Materials and Methods
Materials
The following antibodies were used: antibodies against inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) (BD Bioscience, San Jose, CA), antibodies against tumor necrosis factor (TNF)–α (Abcam, Cambridge, MA), antibodies against 3-nitrotyrosine (poly- and monoclonal) and rabbit antibody to HOCl-oxidized LDL (Upstate Biotechnologies, Lake Placid, NY), anti-human glycated LDL antibody (Exocell. Philadelphia, PA), antibodies against cleaved caspase-3 and poly (ADP-ribose) polymerase (PARP) (Cell Signaling Technology, Danvers, MA). Monoclonal antibodies against PGIS and enzyme-linked immunosorbent assay (ELISA) kits to measure 6-keto-PGF1α were purchased from Cayman Chemicals (Ann Arbor, MI). l-NG-Nitroarginine methyl ester (l-NAME) and tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) were from Sigma (Atlanta, GA). Other chemicals and organic solvents of the highest grade were purchased from local suppliers. Native LDL (N-LDL) from healthy human donors and in vitro–heavily oxidized and glycated LDL (HOG-LDL) were prepared as described previously.4 Briefly, human LDL was isolated by sequential ultracentrifugation of pooled plasma. N-LDL and glycated LDL were prepared by incubating LDL with and without freshly prepared 50 mmol/L glucose for 72 hours at 37°C under antioxidant conditions (1 mmol/L N,N-bis[2-(bis[carboxymethyl]-amino)ethyl] glycine (DTPA) and 270 μmol/L EDTA, with sustained nitrogen gas). HOG-LDL was prepared from glycated LDL by oxidizing in the presence of 10 μmol/L CuCl2 at 37°C for 24 hours, followed by dialysis at 4°C for 24 hours. Protein in LDL preparations was determined by BCA protein assay (Pierce, Rockford, IL). LDL preparations were further characterized by measuring fluorescence at 360 nm ex/430 nm em (Gilford Fluorimeter IV, Oberlin, OH), performing agarose gel electrophoresis (Paragon LIPO Gel, Beckman, Fullerton, CA), and measuring absorbance at 234 nm using a Beckman DU 650 spectrophotometer. LDL preparations were stored in the dark under nitrogen at 4°C in the presence of 270 μmol/L EDTA, and were used within 6 weeks.4, 13 The study protocol was approved by the Institutional Review Board of the University of Oklahoma Health Sciences Center.
Animals
Akita and wild-type (WT) C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, MI). All mice were housed in temperature-controlled cages under a 12-hour light–dark cycle and given free access to water and normal rodent chow. At 5 weeks of age, WT and Akita mice were randomly assigned to receive tempol (1 mmol/L, in drinking water) or vehicle for 8 weeks. The animal protocol was reviewed and approved by the University of Oklahoma Institutional Animal Care and Use Committee.
Cell Culture
Human retinal capillary pericytes were purchased from Lonza Walkersville (Walkersville, MD). Characterization of the cells had been performed by the company: CD146/CD105 > 90% positive, CD31/CD34 > 95% negative. The human retinal pericytes were cultured in EBM-2 basal serum-free culture medium supplemented with EGM-2-MV singleQuots growth supplement (Clonetics, Walkersville, MD), which contained 5% fetal bovine serum, 0.1% human epidermal growth factor (hEGF), 0.04% hydrocortisone, 0.1% vascular endothelial growth factor (VEGF), 0.4% human fibroblast growth factor–β (hFGF-β), 0.1% R3-insulin-like growth factor–1 (IGF-1), 0.1% ascorbic acid, and 0.1% GA-1000. After reaching 85% confluence, the cells were maintained in low-serum (0.5%) medium for 24 hours to promote quiescence, and then subjected to the indicated experimental conditions. Pericytes in passages 4 to 10 were used for the experiments. Each condition was evaluated in duplicate, with at least three experiments per series.
Immunoprecipitation and Western Blotting
Cells and retinas were homogenized in lysis buffer (1% Triton X-100, 150 mmol/L NaCl, 10 mmol/L Tris, pH 7.4, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.2 mmol/L phenylmethylsulfonyl fluoride, 0.2 mmol/L sodium orthovanadate, and 0.5% Nonidet P-40), and protein contents were measured by using the Bradford assay. Total cell lysates were immunoprecipitated with the indicated antibodies. Immunoprecipitates or whole-cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and probed with specific antibodies, as described previously.14, 15, 16 The intensities (density × area) of individual bands were measured by densitometry (model GS-700, Imaging Densitometer, Bio-Rad). The background was subtracted from the calculated area.
Measurement of Intracellular ROS Generation
Intracellular ROS generation was measured by the dichlorodihydrofluorescein (DCF) method using carboxy-H2DCFDA, a cell-permeable fluorescent indicator for ROS, as previously described.17 Briefly, cells were gently washed with PBS and incubated for 20 minutes with carboxy-H2DCFDA (2 μmol/L) in phenol red-free medium at 37°C. The medium was discarded and cells were washed with PBS. Fluorescence was measured using a fluorescence microplate reader with an excitation wavelength of 485 nm and an emission wavelength of 538 nm.
Determination of NO Production in Pericytes
NO production was evaluated by measuring the accumulation of nitrites, stable oxidative end products of NO metabolism, in the conditioned medium using the Greiss reagent kit (Molecular Probe, Carlsbad, CA), following the manufacturer's instructions. Briefly, nitrates were converted to nitrites for analysis by first incubating the samples with nitrate reductase (100 mU/mL) and reduced nicotinamide adenine dinucleotide phosphate (NADPH 20 μg/mL). Then, 150 μl of sample was added to 20 μL of premixed Greiss reagent [containing sulfanilamide and N-(1-naphtyl) ethylendiamine] and 130 μL of deionized water in a 96-well microplate, and incubated at room temperature for 30 minutes. The optical density was measured with a Victor microplate reader at a wavelength of 546 nm. Nitrite concentrations in the supernatants were calculated by reference to a standard curve.
Immunohistochemistry and TUNEL Staining
Immunostaining for oxidized LDL, glycated LDL, iNOS and COX-2 was performed as previously described.8 Briefly, sections were incubated with anti-oxidized LDL (1:400), anti-glycated LDL (1:400), anti-iNOS (1:50), or anti-COX-2 (1:50) antibody overnight at 4°C, and then incubated with secondary antibodies for 30 minutes at room temperature. 3,3′-Diaminobenzidine (DAB) was used to visualize immunoreactive proteins. Samples were double stained for 3-nitrotyrosine and PGIS using a kit from Vector Laboratories, following the protocol provided by the company. Quantitative analysis for immunohistochemistry was performed with a personal computer. Sequence images were taken using a Retiga 1300 Color-Cooled Camera and Q capture software, and the staining areas were measured using a Bioquant analysis system (Nashville, TN). The percentage of the total area with positive color for each section was recorded.
Cell death was measured in retina sections and cultured human retinal pericytes by the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining method using a cell death detection assay kit (Roche Applied Science, Indianapolis, IN), as described by manufacturer. Cells were counterstained with DAPI to stain nuclei. TUNEL-positive nuclei were counted, and the total number of nuclei per unit area of the retina was estimated by counting the number of DAPI-stained nuclei under UV illumination. The number of TUNEL positive cell in 15 fields was averaged, and the data were calculated as number of TUNEL-positive nuclei per total number of nuclei, expressed as a percentage.18
Assay of PGIS Activity
PGIS activity was assayed by measuring the stable metabolite of PGI2, 6-keto-PGF1α, after incubating cells with the PGIS substrate, PGH2 (10−5 mol/L for 3 minutes), as described previously.8, 10, 19, 20, 21 Briefly, after incubation with PGH2, the reaction was stopped by acidification with 1 N HCl to pH 3.5. Incubation media were extracted with ethyl acetate and, after centrifugation, the organic phase was evaporated to dryness under nitrogen. Samples were then resuspended in 100 μl of PBS. The amounts of 6-keto-PGF1α and PGE2 were subsequently determined with an ELISA kit (Cayman Chemicals) according to the instructions provided by the supplier.
Statistical Analysis
Data are presented as means ± standard errors of the mean (SEMs). Significance was determined by one-way analysis of variance corrected by Bonferroni's method. P < 0.05 was considered statistically significant.
Results
HOG-LDL Increases NO Production, iNOS Expression, and 3-Nitrotyosine Modification of Proteins in Cultured Human Retinal Pericytes
Oxidative stress has been implicated in oxidized LDL-induced apoptosis22 and diabetic retinopathy. Peroxynitrite (ONOO−), formed through the reaction of superoxide with NO,9 is a potent oxidant that initiates both nitrosative and oxidative reactions that affect cellular proteins, lipids, and DNA.9 In diabetic retinas, inflammatory cytokines, oxidized LDL, and hypoxia may induce iNOS,23 which produces a large amount of NO. The over-production of NO not only provides an essential precursor for ONOO− generation, but also contributes to pericyte loss in diabetic retinopathy.24 Therefore, we determined whether heavily oxidized and glycated LDL (HOG-LDL) induces iNOS expression and oxidative stress in cultured human retinal capillary pericytes. As shown in Figure 1, iNOS expression (Figure 1A) and NO production (Figure 1B) were very low in pericytes treated with vehicle or N-LDL. As expected, exposure of pericytes to HOG-LDL significantly induced iNOS expression (Figure 1A) and increased l-NAME–inhibitable NO production (Figure 1B). Concomitantly, exposure to HOG-LDL, but not N-LDL, for 24 hours markedly increased ROS generation in retinal pericytes (Figure 1C). In line with the observed increase in NO and ROS production, the levels of 3-nitroTyrosine–positive proteins, a “footprint” of ONOO− in cultured cells,25 were elevated in retinal pericytes exposed to HOG-LDL (Figure 1, D and E). In contrast, exposure of pericytes to N-LDL had only a minor effect on 3-nitrotyrosine–positive protein levels (Figure 1, D and E).
Figure 1.
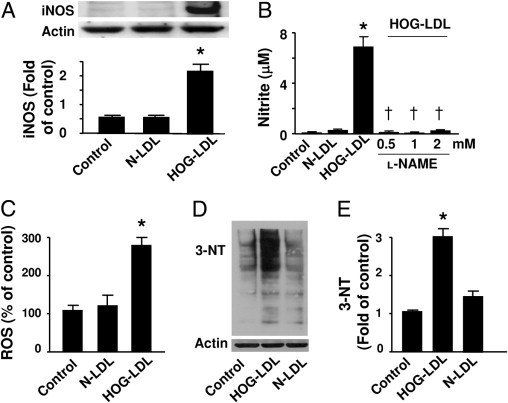
HOG-LDL induces iNOS and increases superoxide and NO production. A: Human retinal pericytes were incubated with HOG-LDL (100 μg/mL) and N-LDL for 24 hours. Cell lysates were analyzed by Western blotting using an antibody against iNOS. The blot shown is a representative of blots from three different experiments (*P < 0.05 vs. control or N-LDL). B: NOS activity was determined by measuring nitrite levels (*P < 0.05 vs. control or N-LDL; †P < 0.05 vs. HOG-LDL; n = 4). C: HOG-LDL increased ROS release. Confluent pericytes were exposed to HOG-LDL (100 μg/mL) for 3 hours, and ROS was measured by detecting DCF fluorescence (*P < 0.01 vs. control or N-LDL; n = 3). D and E: 3-Nitrotyrosine–modified proteins were detected by Western analysis using a specific antibody and quantified relative to control (*P < 0.05 vs. control or N-LDL).
HOG-LDL Induces Tyrosine Nitration of PGIS and Decreases Its Activity
To determine whether HOG-LDL induces PGIS nitration in pericytes, whole-cell lysates were first immunoprecipitated with an antibody against PGIS, and PGIS immunoprecipitates were analyzed by Western blotting using an antibody against 3-nitrotyrosine. As depicted in Figure 2A, exposure of human retinal pericytes to HOG-LDL, but not N-LDL, markedly increased the levels of nitrated PGIS. Additional studies conducted to determine whether the increase in PGIS nitration inhibited PGIS activity revealed that HOG-LDL significantly inhibited PGIS activity in retinal pericytes, as measured by the conversion of exogenous PGH2 to 6-keto-PGF1α (Figure 2B). Because the production of PGI2 is dependent on cyclooxygenases (COX-1 and COX-2) and induction of cyclooxygenase is likely to be required for thromboxane receptor activation, we determined whether HOG-LDL induces COX-2 expression. Incubation of human retinal pericytes with HOG-LDL significantly induced COX-2 expression, whereas N-LDL did not affect COX-2 expression (Figure 2C).
Figure 2.
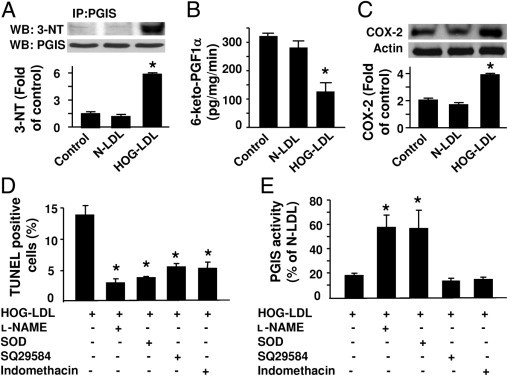
SOD or l-NAME prevents HOG-LDL–induced inactivation of PGIS and apoptosis in pericytes. A: Human retinal pericytes were exposed to HOG-LDL (100 μg/mL), N-LDL, or vehicle for 24 hours. PGIS was immunoprecipitated (IP) using a specific antibody, and PGIS and 3-nitrotyrosine (3-NT) in immunoprecipitates were detected by Western blotting (WB). PGIS tyrosine nitration was quantified by densitometric analysis (*P < 0.05 vs. control or N-LDL, n = 3). B: PGIS activity was assessed by analyzing 6-keto-PGFα1, a metabolite of PGI2, using an enzyme-linked immunoassay (*P < 0.05 vs. control or N-LDL; n = 5). C: Expression of cyclooxygenase-2 (COX-2) was evaluated by Western blotting (*P < 0.05 vs. control or N-LDL; n = 3). D: Pericytes were pretreated with PEG-SOD (300 U/mL), l-NAME (0.5 mmol/L), SQ29548 (10−5 mol/L), or indomethacin (10 μmol/L) for 30 minutes, followed by incubation with HOG-LDL (100 μg/mL) or N-LDL for 24 hours. Pericyte apoptosis was determined by TUNEL staining. The number of TUNEL-positive cells is indicated in the bar graph (*P < 0.05 vs. HOG-LDL; n = 4). E: PGIS activity was assessed by analyzing the PGI2 metabolite PGFα1 using an enzyme-linked immunoassay (*P < 0.05 vs. HOG-LDL; n = 4).
Activation of the Thromboxane Receptor Enhances Pericyte Apoptosis
Our earlier studies9 demonstrated that inactivation of PGIS can trigger endothelial cell apoptosis by consequent overstimulation of the thromboxane receptor. We next determined whether HOG-LDL–inactivated PGIS triggers pericyte apoptosis through thromboxane receptor stimulation. As shown in Figure 2D, the fraction of apoptotic cells was dramatically increased in pericytes exposed to HOG-LDL for 24 hours (14.7% ± 3.4% vs. 3.5% ± 1.5% in controls; P < 0.05), an increase that was accompanied by a significant reduction in PGIS activity (Figure 2E). Pretreatment of human retinal pericytes with either PEG-SOD (300 U/mL) or l-NAME (0.5 mmol/L) restored PGIS activity and attenuated HOG-LDL–induced pericyte apoptosis (Figure 2, D and E). These results suggest that ONOO− is responsible for HOG-LDL–induced PGIS inhibition and apoptosis in human retinal pericytes. Notably, the effect of HOG-LDL on apoptosis was also antagonized by SQ29548, a selective thromboxane receptor antagonist, and indomethacin, a nonselective cyclooxygenase inhibitor. However, neither SQ29548 nor indomethacin restored PGIS activity (Figure 2, D and E) in HOG-LDL–treated pericytes, suggesting that the associated activation of the thromboxane receptor might contribute to HOG-LDL–induced pericyte apoptosis.
Diabetes Enhances Oxidized LDL and Glycated LDL in the Diabetic Retina
The Akita mouse is a well-established animal model of diabetic retinopathy. At 13 weeks of age, Akita mice exhibited significantly lower body weights (Figure 3A) and elevated blood glucose levels (Figure 3B) as compared with WT control mice. In agreement with previous reports, tempol, a potent antioxidant, did not normalize body weight or blood glucose (Figure 3A and B) in these mice.26 Our previous results suggested that, in diabetic retinas, increased vascular permeability results in extravasation and accumulation of LDL in the perivascular space, where it undergoes additional glycation and extensive oxidation, and enhances oxidative stress and retinal vascular injury.3, 4, 5 We therefore used immunohistochemistry to detect the protein levels of oxidized LDL and glycated LDL in diabetic retinas. In contrast to the faint staining observed in WT mouse retinas, oxidized LDL staining in Akita mouse retinas was markedly increased (Figure 4A). Importantly, tempol treatment reduced the intensity of oxidized LDL immunostaining in diabetic mice (Figure 4A). Similar to the change in oxidized LDL, glycated LDL immunostaining was very weak in WT mouse retinas, but was dramatically enhanced in Akita mouse retinas. The intensity of glycated LDL immunostaining in Akita mouse retinas was attenuated by tempol administration (Figure 4A).
Figure 3.
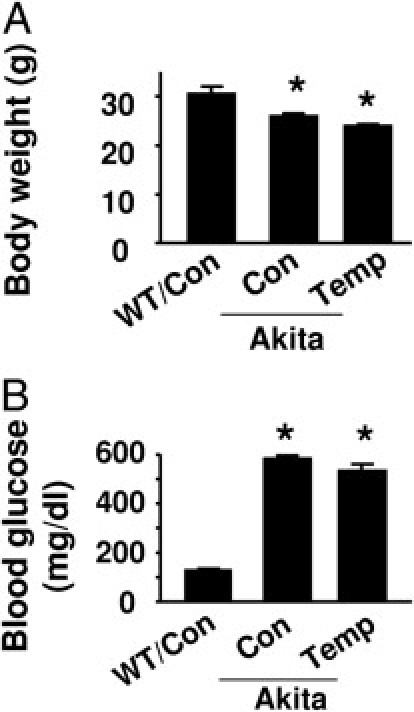
The effects of tempol on body weight and blood glucose in WT and Akita mice. A: Average body weight (*P < 0.05 vs. WT control [Con]; n = 4). B: Blood glucose was determined in tail blood using the Reli-On Ultima Blood Glucose Monitoring System (*P < 0.05 vs. WT Con; n = 4).
Figure 4.
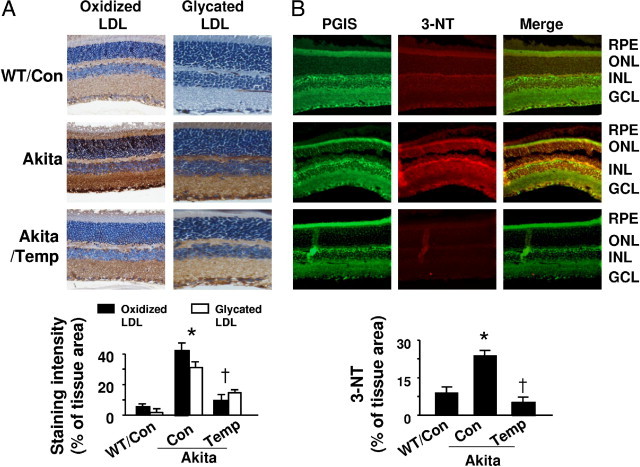
Diabetes increases oxidized LDL, glycated LDL, and PGIS nitration in Akita mouse retinas. A: Upper panel: immunostaining for oxidized LDL and glycated LDL. Retinal sections from WT and Akita mice, treated with or without tempol (Temp), were stained with antibody against oxidized LDL or glycated LDL, and the nuclei were counterstained with hematoxylin. Lower panel: Quantitative analysis of immunohistochemical staining for oxidized LDL and glycated LDL [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4]. B: Upper panel: representative immunostaining for PGIS (green) and 3-nitrotyrosine (3-NT; red) in retinas obtained from WT and Akita mice treated with or without tempol. Yellow in merged images represents overlapping red and green signals, indicating partial colocalization of 3-nitrotyrosine and PGIS staining. Lower panel: Quantification of immunostaining for 3-NT [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4].
Diabetes Induces Nitration of PGIS in the Retina
To investigate whether tyrosine nitration of PGIS occurred and whether PGIS nitration mediated retinal cell death in vivo, we determined the colocalization of PGIS and 3-nitrotyrosine using a double-labeling immunofluorescence technique. PGIS expression was found mainly in retinal pigment epithelial, outer plexiform, and ganglion cell layers. As expected, there was no significant difference in the expression of PGIS (green) among groups (Figure 4B). In contrast, staining for 3-nitrotyrosine–positive proteins (red) was significantly elevated in Akita mouse retinas compared with those of control mice (Figure 4B). Areas of colocalization, indicated by yellow in merged images, suggest tyrosine nitration of PGIS in the diabetic retinas, specifically in the outer plexiform, retinal pigment epithelial, and ganglion cell layers, where pericytes are found in capillaries. Notably, the fluorescence intensity was attenuated by administration of tempol (Figure 4B), supporting the conclusion that ROS-inhibitable 3-nitrotyrosine formed in vivo.
Diabetes Increases iNOS and COX-2 Expression in the Diabetic Retina
We next determined whether iNOS induction in the diabetic retina contributed to PGIS nitration. Compared to the weak staining for iNOS in WT mouse retinas, the much stronger staining in Akita mouse retina indicated a dramatic increase in iNOS expression in diabetes (Figure 5A). In addition, tempol treatment attenuated iNOS immunostaining in diabetic mice (Figure 5A). Enhanced iNOS expression in diabetes and attenuation by tempol administration were confirmed by Western analysis (Figure 5, A and B). Immunohistochemistry and Western blotting also demonstrated an increase in COX-2 expression in diabetic retina, which was diminished by tempol treatment (Figure 5, C and D).
Figure 5.
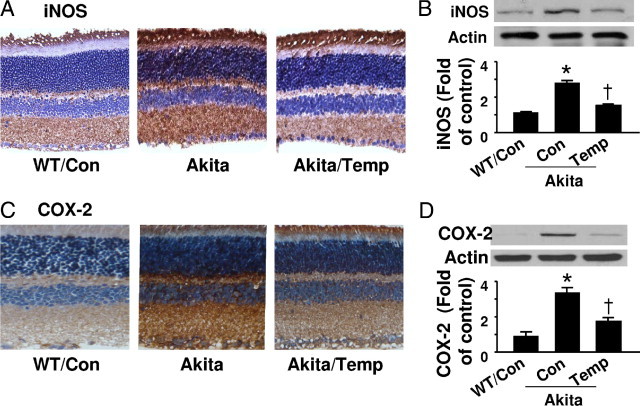
Expression of iNOS and COX-2 in Akita mouse retinas. A: Representative images of iNOS immunostaining. Retinal sections for WT, Akita, and tempol-treated Akita mice were stained with an iNOS specific antibody and the nuclei were counterstained with hematoxylin. B: Protein levels of iNOS in retinal homogenates were analyzed by Western blotting and quantified by densitometry [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4]. C: Retinal sections were stained with an anti-COX-2 antibody and counterstained with hematoxylin. D: COX-2 expression in retinal homogenates were analyzed by Western blotting and quantified by densitometry [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4].
Tempol Prevents Hyperglycemia-Induced Retinal Cell Apoptosis in Akita Mice
Whole-mounted retinas from WT and diabetic Akita mice were processed for TUNEL staining; TUNEL-positive nuclei were identified by the presence of a dark red reaction product. As shown in Figure 6A, TUNEL-positive nuclei were dramatically increased in Akita mouse retinas. Apoptotic cells were found in all layers of the retina, including the retinal pigment epithelial cell layer and ganglion cell layer (Figure 6, A and B). Importantly, tempol treatment prevented this enhancement of apoptotic cell death (Figure 5, A and B). Consistent with these findings, the levels of cleaved caspase-3 and PARP were increased in Akita retinas, and the increase was prevented by tempol treatment (Figure 6, C and D).
Figure 6.
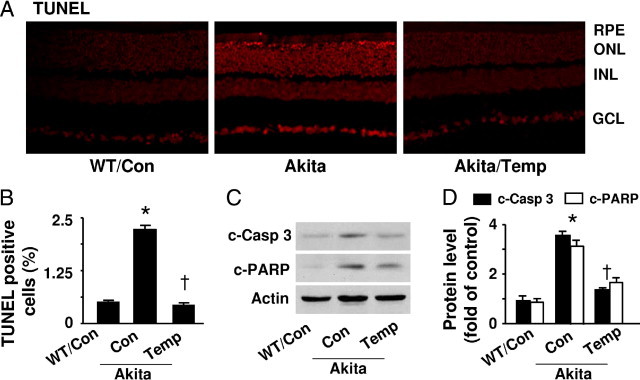
Diabetes induces apoptosis in Akita mouse retinas. A: Representative images of TUNEL staining. Apoptotic cells in retinas from WT, Akita, and tempol-treated mice were labeled by TUNEL staining, and nuclei were counterstained with DAPI. B: The number of TUNEL-positive cells is indicated in the bar graph [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4 in each group]. C: Western analysis of cleaved (C) caspase-3 (c-Casp 3) and PARP in retinal homogenates. D: The protein levels of c-Casp 3 and c-PARP were quantified by densitometry [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4].
Chronic Administration of Tempol Protects Retinal Tight Junctions
To assess the effects of tempol on the blood–retinal barrier, we measured extravascular albumin content in WT and Akita mouse retinas after removing blood from the vascular system thorough perfusion. Western analysis showed that the albumin content was higher in Akita mouse retinas (Figure 7A and B), and the increase was attenuated by tempol treatment (Figure 7, A and B). We also determined whether tempol regulates tight junctions in diabetic retinal vessels using Western blotting. Levels of the tight junction protein occludin-1 were decreased in Akita retinas compared to those of WT control mice (Figure 7, C and D). Tempol dramatically protected the diabetic blood–retinal barrier, as indicated by reduced extravascular albumin content, and restored occludin-1 levels compared with those in untreated controls (Figure 7, C and D).
Figure 7.
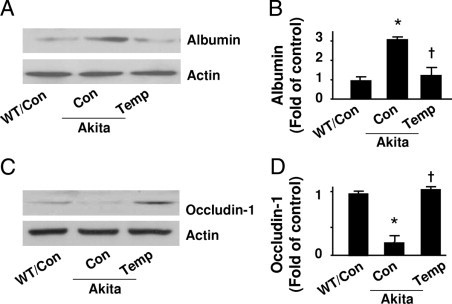
Tempol protects retinal tight junctions in Akita mice. A and B: Albumin in retinas from WT and Akita mice, treated with or without tempol (Temp), were determined by Western blot analysis and quantified by densitometry [*P < 0.05 vs. WT control (Con); †P < 0.05 vs. Akita Con; n = 4]. C and D: Occludin-1 levels were determined by Western blotting and quantified by densitometry (*P < 0.05 vs. WT Con; †P < 0.05 vs. Akita Con; n = 4).
Discussion
In this study, we provide the first demonstration that, by promoting tyrosine nitration of PGIS and consequent thromboxane receptor stimulation, oxidation and glycation of LDL contributes to retinal pericyte apoptosis, the most specific vascular histopathology associated with diabetic retinopathy. In cultured human retinal pericytes, HOG-LDL significantly increased pericyte apoptosis, which was accompanied by increased tyrosine nitration of PGIS and decreased PGIS activity. SOD and l-NAME restored PGIS activity and attenuated HOG-LDL-enhanced apoptosis. Blockade of the thromboxane receptor (SQ29548) or inhibition of cyclooxygenase (indomethacin) also diminished apoptosis, but failed to restore PGIS activity. Diabetes enhanced accumulation of oxidized LDL and glycated LDL in Akita mouse retinas, which led to PGIS nitration and apoptosis. All these effects were attenuated by chronic administration of tempol. Collectively, these studies suggest that PGIS nitration and inactivation play a role in diabetic retinopathy.
In diabetic retinas, inflammatory cytokines, oxidized LDL, and hypoxia may induce iNOS,23 which produces a large amount of NO. NO can exert cytotoxic effects directly or via ONOO− formation in retinal tissues. Several lines of evidence implicate the overproduction of NO in the development of diabetic retinopathy.27 For instance, immunohistochemical analyses have demonstrated enhanced iNOS expression in the retinas of diabetic patients,23 and both genetic knockout and pharmacological inhibition of iNOS reduce apoptosis in the inner nuclear layer of the avascular retina in ischemic proliferative retinopathy.28 In addition, NO may act as an angiogenic mediator by stimulating production of VEGF, which has been strongly implicated in the development of proliferative retinopathy.29, 30 In agreement with these observations, we found that iNOS expression was higher in Akita mouse retinas, and HOG-LDL induced an increase in iNOS expression and NO production in human retinal pericytes.
NO can interact with superoxide to generate the highly reactive ONOO−, inducing nitrosative and oxidative damage to the vascular system. In addition to increasing iNOS expression and NO production, HOG-LDL enhanced the generation of superoxide anions. Major support for the idea that HOG-LDL elevated ONOO− formation is provided by the observation that HOG-LDL increased the formation of 3-nitrotyrosine and elevated the levels of immunodetectable nitrated PGIS, which is reported to be particularly susceptible to tyrosine nitration.7 PGIS is a heme-thiolate protein. The presence of metal ions in PGIS allows an alternative and energetically preferred pathway that will lead to an oxo-species and nitrogen dioxide. ONOO− anion forms a ferryl species with PGIS that causes oxidation of an active site-located tyrosine (430) to a phenoxyradical. The addition of the remaining nitrogen dioxide (.NO2) radical yields 3-nitrotyrosine. In terms of reaction velocities, heme complexes exhibit second-order reaction constants around 106−107M−1S−1 and therefore compete favorably with the protonation/excitation pathway. Thus, PGIS can be inhibited by ONOO− in an extremely low IC50 value.31 In line with such studies was the ability of other heme-thiolate enzymes to protect PGI2 synthase from inactivation and nitration by ONOO−. Consistent with these findings, the present study has demonstrated that oxidative stress increased the levels of nitrated PGIS while inhibiting its activity in human retinal pericytes and diabetic retinas. Chronic administration of tempol, a superoxide scavenger, decreased the levels of intraretinal oxidized LDL and glycated LDL, reduced 3-nitrotyrosine formation, iNOS induction, PGIS nitration, and apoptotic cell death, and protected the blood–retinal barrier. These data imply that oxidized LDL-mediated, ROS-dependent cell death is involved in the initiation and progression of diabetic retinopathy.
Oxidized LDL-mediated inflammation and vascular damage are critical in the pathogenesis of atherosclerosis as well as diabetic complications,32, 33, 34, 35, 36 including diabetic retinopathy, as witnessed by our own studies. Diabetes may amplify LDL oxidation and exacerbate its complications by increasing glycation of plasma LDL37 and rendering the lipoprotein more susceptible to oxidation.38 The healthy retina is protected from extravasation of LDL by the inner blood–retinal barrier; but once this is compromised, extravasated, glycated LDL is sequestered through advanced glycation product cross-linking, and becomes oxidized, aggregated, and cytotoxic. Hyperglycemia may directly enhance oxidative stress,39 promoting further LDL oxidation. Our results demonstrate that oxidized LDL and glycated LDL are widely distributed in Akita mouse retinas. The different patterns of oxidized LDL and glycated LDL between WT and Akita mice suggest that the protein component of LDL may become dissociated from the lipid component during modification and fragmentation; such an effect has been reported in atherosclerotic regions of the thoracic aorta.40 The presence of oxidized LDL and glycated LDL in the retinas was associated with the presence of TUNEL-positive cells. Akita mouse retinas exhibited an increase in the number of TUNEL-positive cells in all layers of the retina, including retinal pigment epithelial cells and the ganglion cell layer. These findings are consistent with a role for oxidized LDL in mediating apoptotic cell death in the retina. Our findings strongly support the view that extravasation and accumulation of LDL in the perivascular space can enhance oxidative stress and retinal vascular injury.
In summary, diabetes-enhanced intraretinal oxidized LDL and glycated LDL and PGIS nitration are associated with retinal cell apoptosis. Exposure of retinal pericytes to HOG-LDL increases the production of ONOO− and tyrosine nitration of PGIS. Associated activation of thromboxane receptors increases retinal pericyte apoptosis. Thus, oxidant stress and nitration of PGIS may contribute to the development of diabetic retinopathy.
Footnotes
Supported by NIH grants (HL079584, HL080499, HL074399, HL089920, HL096032, and HL105157 to M.H.Z.). Additional support was provided by the NIH, National Center for Research Resources grants 1P20RR024215-01 to Z.X. and 3P20RR024215-03S1 to T.J.L., a Scientist Development Grant from the American Heart Association to Z.X., and by separate grants from Oklahoma Center for Advancement of Science and Technology to Z.X. and T.J.L. M.H.Z. is a recipient of an Established Investigator Award of the American Heart Association.
Contributor Information
Timothy J. Lyons, Email: timothy-lyons@ouhsc.edu.
Zhonglin Xie, Email: zxie@ouhsc.edu.
References
- 1.Lyons T.J., Jenkins A.J., Zheng D., Lackland D.T., McGee D., Garvey W.T., Klein R.L. Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Invest Ophthalmol Vis Sci. 2004;45:910–918. doi: 10.1167/iovs.02-0648. [DOI] [PubMed] [Google Scholar]
- 2.Klein R., Sharrett A.R., Klein B.E., Moss S.E., Folsom A.R., Wong T.Y., Brancati F.L., Hubbard L.D., Couper D. The association of atherosclerosis, vascular risk factors, and retinopathy in adults with diabetes: the atherosclerosis risk in communities study. Ophthalmology. 2002;109:1225–1234. doi: 10.1016/s0161-6420(02)01074-6. [DOI] [PubMed] [Google Scholar]
- 3.Wu M., Chen Y., Wilson K., Chirindel A., Ihnat M.A., Yu Y., Boulton M.E., Szweda L.I., MA J.X., Lyons T.J. Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:2679–2685. doi: 10.1167/iovs.07-1440. [DOI] [PubMed] [Google Scholar]
- 4.Song W., Barth J.L., Lu K., Yu Y., Huang Y., Gittinger C.K., Argraves W.S., Lyons T.J. Effects of modified low-density lipoproteins on human retinal pericyte survival. Ann NY Acad Sci. 2005;1043:390–395. doi: 10.1196/annals.1333.045. [DOI] [PubMed] [Google Scholar]
- 5.Song W., Barth J.L., Yu Y., Lu K., Dashti A., Huang Y., Gittinger C.K., Argraves W.S., Lyons T.J. Effects of oxidized and glycated LDL on gene expression in human retinal capillary pericytes. Invest Ophthalmol Vis Sci. 2005;46:2974–2982. doi: 10.1167/iovs.04-1505. [DOI] [PubMed] [Google Scholar]
- 6.Zheng Z., Chen H., Wang H., Ke B., Zheng B., Li Q., Li P., Su L., Gu Q., Xu X. Improvement of retinal vascular injury in diabetic rats by statins is associated with the inhibition of mitochondrial ROS pathway mediated by PGC-1{alpha} Diabetes. 2010;59:2315–2325. doi: 10.2337/db10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou M., Jendral M., Ullrich V. Prostaglandin endoperoxide-dependent vasospasm in bovine coronary arteries after nitration of prostacyclin synthase. Br J Pharmacol. 1999;126:1283–1292. doi: 10.1038/sj.bjp.0702434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He C., Choi H.C., Xie Z. Enhanced tyrosine nitration of prostacyclin synthase is associated with increased inflammation in atherosclerotic carotid arteries from type 2 diabetic patients. Am J Pathol. 2010;176:2542–2549. doi: 10.2353/ajpath.2010.090783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou M.H., Shi C., Cohen R.A. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 10.Xie Z., Zhang J., Wu J., Viollet B., Zou M.H. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57:3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Zou M.H., Cohen R., Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11:89–97. doi: 10.1080/10623320490482619. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Correa J.A., Arrebola M.M., Munoz-Marin J., Mno A., Guerrero A., Arranz I., De La Cuesta F.S., De La Cruz J.P. Gender differences in the effect of aspirin on retinal ischemia, prostanoid synthesis and nitric oxide production in experimental type 1-like diabetes. Vascul Pharmacol. 2007;47:83–89. doi: 10.1016/j.vph.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Diffley J.M., Wu M., Sohn M., Song W., Hammad S.M., Lyons T.J. Apoptosis induction by oxidized glycated LDL in human retinal capillary pericytes is independent of activation of MAPK signaling pathways. Mol Vis. 2009;15:135–145. [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Z., Singh M., Siwik D.A., Joyner W.L., Singh K. Osteopontin inhibits interleukin-1beta-stimulated increases in matrix metalloproteinase activity in adult rat cardiac fibroblasts: role of protein kinase C-zeta. J Biol Chem. 2003;278:48546–48552. doi: 10.1074/jbc.M302727200. [DOI] [PubMed] [Google Scholar]
- 15.Xie Z., Dong Y., Scholz R., Neumann D., Zou M.H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Z., Dong Y., Zhang J., Scholz R., Neumann D., Zou M.H. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol. 2009;29:3582–3596. doi: 10.1128/MCB.01417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M., Dong Y., Xu J., Xie Z., Wu Y., Song P., Guzman M., Wu J., Zou M.H. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Z., Lau K., Eby B., Lozano P., He C., Pennington B., Li H., Rathi S., Dong Y., Tian R., Kem D., Zou M.H. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou M.H., Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
- 20.Pasquet J.P., Zou M.H., Ullrich V. Peroxynitrite inhibition of nitric oxide synthases. Biochimie. 1996;78:785–791. doi: 10.1016/s0300-9084(97)82537-0. [DOI] [PubMed] [Google Scholar]
- 21.Zou M.H., Klein T., Pasquet J.P., Ullrich V. Interleukin 1beta decreases prostacyclin synthase activity in rat mesangial cells via endogenous peroxynitrite formation. Biochem J. 1998;336:507–512. doi: 10.1042/bj3360507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renier G., Mamputu J.C., Desfaits A.C., Serri O. Monocyte adhesion in diabetic angiopathy: effects of free-radical scavenging. J Diabetes Complications. 2003;17:20–29. doi: 10.1016/s1056-8727(02)00271-4. [DOI] [PubMed] [Google Scholar]
- 23.bu El-Asrar A.M., Desmet S., Meersschaert A., Dralands L., Missotten L., Geboes K. Expression of the inducible isoform of nitric oxide synthase in the retinas of human subjects with diabetes mellitus. Am J Ophthalmol. 2001;132 doi: 10.1016/s0002-9394(01)01127-8. 551-516. [DOI] [PubMed] [Google Scholar]
- 24.Miller A.G., Smith D.G., Bhat M., Nagaraj R.H. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol Chem. 2006;281:11864–11871. doi: 10.1074/jbc.M513813200. [DOI] [PubMed] [Google Scholar]
- 25.Obrosova I.G., Pacher P., Szabo C., Zsengeller Z., Hirooka H., Stevens M.J., Yorek M.A. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barber A.J., Antonetti D.A., Kern T.S., Reiter C.E., Soans R.S., Krady J.K., Levison S.W., Gardner T.W., Bronson S.K. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 27.Becquet F., Courtois Y., Goureau O. Nitric oxide in the eye: multifaceted roles and diverse outcomes. Surv Ophthalmol. 1997;42:71–82. doi: 10.1016/s0039-6257(97)84043-x. [DOI] [PubMed] [Google Scholar]
- 28.Hartnett M.E., Stratton R.D., Browne R.W., Rosner B.A., Lanham R.J., Armstrong D. Serum markers of oxidative stress and severity of diabetic retinopathy. Diabetes Care. 2000;23:234–240. doi: 10.2337/diacare.23.2.234. [DOI] [PubMed] [Google Scholar]
- 29.Sennlaub F., Courtois Y., Goureau O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest. 2001;107:717–725. doi: 10.1172/JCI10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiello L.P., Wong J.S. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int Suppl. 2000;77:S113–S119. doi: 10.1046/j.1523-1755.2000.07718.x. [DOI] [PubMed] [Google Scholar]
- 31.Mehl M., Daiber A., Herold S., Shoun H., Ullrich V. Peroxynitrite reaction with heme proteins. Nitric Oxide. 1999;3:142–152. doi: 10.1006/niox.1999.0217. [DOI] [PubMed] [Google Scholar]
- 32.Bloomgarden Z.T. Diabetic retinopathy and neuropathy. Diabetes Care. 2005;28:963–970. doi: 10.2337/diacare.28.4.963. [DOI] [PubMed] [Google Scholar]
- 33.Jenkins A.J., Rowley K.G., Lyons T.J., Best J.D., Hill M.A., Klein R.L. Lipoproteins and diabetic microvascular complications. Curr Pharm Des. 2004;10:3395–3418. doi: 10.2174/1381612043383188. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins A.J., Best J.D., Klein R.L., Lyons T.J. 'Lipoproteins, glycoxidation and diabetic angiopathy'. Diabetes Metab Res Rev. 2004;20:349–368. doi: 10.1002/dmrr.491. [DOI] [PubMed] [Google Scholar]
- 35.Santini S.A., Marra G., Giardina B., Cotroneo P., Mordente A., Martorana G.E., Manto A., Ghirlanda G. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes. 1997;46:1853–1858. doi: 10.2337/diab.46.11.1853. [DOI] [PubMed] [Google Scholar]
- 36.Graier W.F., Kostner G.M. Glycated low-density lipoprotein and atherogenesis: the missing link between diabetes mellitus and hypercholesterolaemia? Eur J Clin Invest. 1997;27:457–459. doi: 10.1046/j.1365-2362.1997.1470696.x. [DOI] [PubMed] [Google Scholar]
- 37.Lyons T.J., Baynes J.W., Patrick J.S., Colwell J.A., Lopes-Virella M.F. Glycosylation of low density lipoprotein in patients with type 1 (insulin-dependent) diabetes: correlations with other parameters of glycaemic control. Diabetologia. 1986;29:685–689. doi: 10.1007/BF00870276. [DOI] [PubMed] [Google Scholar]
- 38.Ravandi A., Kuksis A., Shaikh N.A. Glucosylated glycerophosphoethanolamines are the major LDL glycation products and increase LDL susceptibility to oxidation: evidence of their presence in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:467–477. doi: 10.1161/01.atv.20.2.467. [DOI] [PubMed] [Google Scholar]
- 39.Pennathur S., Heinecke J.W. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid Redox Signal. 2007;9:955–969. doi: 10.1089/ars.2007.1595. [DOI] [PubMed] [Google Scholar]
- 40.Kruth H.S., Shekhonin B. Evidence for loss of apo B from LDL in human atherosclerotic lesions: extracellular cholesteryl ester lipid particles lacking apo B. Atherosclerosis. 1994;105:227–234. doi: 10.1016/0021-9150(94)90053-1. [DOI] [PubMed] [Google Scholar]