

Abstract
The transglutaminase-mediated, covalent cross-linking of proteins is an essential step in tissue remodeling after injury. This process provides tissues with extra rigidity and resistance against proteolytic degradation. Plasma coagulation factor XIII (FXIII) is a transglutaminase that promotes cross-linking of the extracellular matrix (ECM) components fibrin and fibronectin to form a provisional matrix in response to tissue damage. However, the functional requirement for this FXIII-mediated cross-linked provisional matrix in adult tissue remodeling remains to be defined. Although it has been proposed that the formation FXIII-mediated fibrin-fibronectin provisional matrix is a critical step for ECM remodeling, we show in an FXIII subunit A–deficient murine model of acute liver injury that the lack of FXIII subunit A did not interfere with collagen reconstruction and resolution after liver injury. Furthermore, FXIIIA deficiency caused significantly increased hepatocyte apoptosis and a delay in hepatocyte regeneration after injury, which were accompanied by a significantly high induction of p53 expression. These findings suggest novel functions of FXIII that the FXIII-mediated covalently cross-linked matrix could promote survival signals for hepatocytes in adult tissue remodeling.
Covalent cross-linking between proteins, including in the provisional matrix, is catalyzed by transglutaminases (TGs). This is an important process for tissue remodeling as it generates extra rigidity and a resistance against proteolytic degradation.1 Plasma coagulation factor Xlll (FXlll), one of the TG family members, circulates as a heterotetramer composed of two catalytic A subunits (FXIII A) and two noncatalytic B subunits (FXIII B) (A2B2), which are activated by thrombin-mediated cleavage. FXlll is a Ca2+-dependent TG and catalyzes the formation of ε-(γ-glutamyl) lysyl cross-links between specific protein pairs. The cross-linking of fibrin molecules by FXIII to form γ-chain dimerization is the final step of blood coagulation.2, 3, 4 FXIII also acts on fibronectin-fibronectin and fibrin-fibronectin cross-linking.5, 6 Thus, the action of FXIII in forming covalent cross-linkages clearly plays a vital role in establishing the mechanical stability of the clot, which is critical for homeostatic functions.2, 4 Growing evidence demonstrates that the source of FXIII is not only from plasma: Megakaryocytes/platelets and monocytes/macrophages also contain cellular FXIII subunit A in their cytoplasm.7, 8
Acute liver injury mediated by the hepatitis virus or hepatotoxins causes massive hepatocyte apoptosis and/or fatal liver damage, and eventually liver transplantation is required.9, 10 Identification of hepatic survival factors is critical for successful therapeutic intervention in liver failure. After acute injury, the liver undergoes a wound-healing response to restore tissue architecture and maintain homeostasis.11 It requires both a well-orchestrated proliferation of cells and the reconstruction of the extracellular matrix (ECM). A central paradigm of ECMs in the process of wound healing, including after liver injury, is that the “provisional matrix” formation between plasma fibrin(ogen) and plasma type fibronectin during an initial stage stabilizes wound areas and provides a road map for cell migration, and such a provisional matrix acts as a nidus for collagen fibrillogenesis.12 The components of provisional matrix, plasma fibrinogen (∼340 kDa as [Aα, Bβ, γ]2 chains) and plasma-type fibronectin (∼440 kDa as a dimer), are produced by hepatocytes in the liver.5, 6, 12
FXIII is of clinical importance because patients with FXIII deficiency have moderate to severe hemorrhagic diathesis, and impaired wound healing is frequently associated with inherited FXIII deficiency.7, 13 However, the molecular mechanisms underlying impaired wound healing in the absence of FXIII are still largely unknown. Furthermore, very little is known about FXIII dependence during the response to acute tissue damage. The pertinent questions in relation to FXIII are as follows: i) Are exudated fibrin and plasma fibronectin in the initial stage of adult tissue damage largely covalently cross-linked by FXlll? ii) What is the requirement for FXIII-mediated cross-linked provisional matrix during tissue remodeling? iii) Does plasma fibronectin and fibrin deposition without covalent cross-linking sufficiently function as an initial matrix, which is then followed successfully by collagen fibrillogenesis?
To determine the functional role of FXIII during adult tissue remodeling, we used, in the present study, a transgenic mouse strain that lacks FXIII subunit A (FXlllA-null) as an in vivo model system, which completely abolishes FXIII transglutaminase activity.14 Although FXIII mediates covalent cross-linking in several ECM proteins,4 we focused here on the plasma fibronectin–fibrin(ogen) provisional matrix and its roles in response to tissue damage. This study provides evidence demonstrating that FXIII-mediated covalent cross-linking of fibronectin and fibrin is not essential for collagen fibrillogenesis after acute liver injury; we suggest that FXIII mediates covalently cross-linked matrix survival signals for hepatocytes.
Materials and Methods
Mice and Induction of Acute Liver Injury by Carbon Tetrachloride
The generation of FXIIIA-null mice has been described elsewhere.14 Control [FXIIIA(+/+)] and FXIIIA(−/−) young adult mice at 6 to 8 weeks of age from the same litters with the CBA/129 mixed background were used for the analysis. All mice were maintained and bred at the Cleveland Clinic's Biological Research Unit (BRU) in accordance with institutional guidelines and National Institutes of Health standards. This study was approved by the Institutional Animal Care and Use Committee. Mice were regularly monitored and had free access to standard mouse chow and water. Mice received humane care according to National Institutes of Health (NIH) recommendations outlined in the “Guide for the Care and Use of Laboratory Animals.” Acute liver injury was induced intraperitoneously by a single-dose administration of carbon tetrachloride [CCl4; 1.0 ml/kg body weight as a 50% (v/v) solution in olive oil (Fluka)] in sex-matched mice as described previously.15
Antibodies, Cytokines, and Reagents
The following antibodies were used for the analyses: rabbit polyclonal antibody (pAb) against mouse fibronectin (Chemicon, Temecula, CA); goat pAb against fibrinogen (Nordic Immunological Laboratories, Tiburg, The Netherlands); mouse monoclonal antibody (mAb) against α-smooth muscle actin (αSMA: clone 1A4, Sigma, St. Louis, MO); rabbit mAb against Ki-67 (clone SP6, Lab Vision, Fremont, CA); rabbit pAb against cleaved caspase 3 (Cell Signaling, Danvers, MA); rat mAb against F4/80 (clone A3-1, Serotec, Oxford, UK); mouse mAb against phospho-extracellular signal-regulated kinase (Erk)1/2 (pT202/pY204) and rabbit pAb against total Erk (Cell Signaling); rabbit pAbs against phospoho-Akt (pT308), phospho-Akt(pS473) and total Akt (Cell Signaling); rabbit pAbs against p53 and p21 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and mouse mAb against heat shock cognate protein 70 (Hsc70, Santa Cruz Biotechnology, Inc.). Rabbit pAbs against type III and type I collagens were a kind gift from Dr. Takako Sasaki (Department of Experimental Medicine I, University of Erlangen-Nuernberg, Erlangen, Germany). Cy3-conjugated donkey anti-rabbit IgG, and peroxidase-conjugated donkey anti-mouse, anti-rabbit, and anti-goat IgG were from Jackson ImmunoRes Laboratories Inc. (West Grove, PA). Alexa Fluor488-conjugated donkey anti-goat IgG, and DAPI were from Molecular Probes (Eugene, OR).
Histological Analysis, Immunohistochemistry, and Immunofluorescence
Histological analysis, and immunohistochemistry and immunofluorescence studies were performed as described previously.15, 16 To quantify positive areas in response to acute liver injury, images were captured with the same gain, offset, magnitude and exposure time. Then a minimum of four different images from each mouse were randomly selected and the intensities were quantified using Image-Pro Plus software version 6.1 (Media Cybernetics, Bethesda, MA).
One- and Two-Dimensional SDS-PAGE and Western Blot Analysis
Liver samples (50 mg) were homogenized in 500 μl deoxycholate (DOC) buffer (2% deoxycholate and 0.02 mol/L Tris-HCl, pH 8.8). The DOC-insoluble materials were isolated by centrifugation and solubilized with urea buffer (8 mol/L urea, 2% SDS, 0.16 mol/L Tris-HCl, pH 6.8). The 2% DOC-soluble and -insoluble fractions are defined as immaturely and maturely assembled matrices, respectively, as described.17 One- and two-dimensional nonreducing/reducing SDS-PAGE analyses were then performed as described by others.18, 19 For two-dimensional SDS-PAGE, the first dimension was performed using a 4% to 20% gradient gel (Novex, Invitrogen, Carlsbad, CA) under nonreducing conditions. Lanes containing separated nonreduced samples were cut out and soaked with 60 mmol/L Tris, pH 6.8, containing 2% SDS and 10% β-mercaptoethanol for 30 minutes at 60°C. The gel slice was put on top of the stacking gel and the second dimension was performed using a 3.3% stacking gel and a 6% separating gel. Western-blot analyses were performed as described.20 In some immunoblotting analyses, samples were transferred onto Immobilon-FL PDVF membrane (Millipore Corp., Temecula, CA) and probed with primary and IRDye 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Bioscience, Lincoln, NE). Immunoreactive bands were detected using the Odyssey Infrared Imaging System (LI-COR Bioscience) as described previously.15
Real-Time PCR
Real-time PCR was performed as described previously.21 The following primers were used: TG2 forward, 5′-TGTCAAGTTCATCAAGAGTGTGC-3′; TG2 reverse, 5′-CAGGAACTTGGGGTTCATATCC-3′; 18S rRNA forward, 5′-GGCGACGACCCATTCG-3′; 18S rRNA reverse, 5′-ACCCGTGGTCACCATGGTA-3′. All samples were analyzed in triplicate as a minimum. After the reactions, the specificity of amplifications in each sample was confirmed by dissociation analysis showing that each sample gave a single melting peak. The relative mRNA levels were normalized to the level of 18S rRNA.
Data Presentation and Statistical Analysis
All experiments were performed in triplicate as a minimum on separate occasions, and the data shown were chosen as representative of results consistently observed. Results are presented as the mean ± SD. Differences between groups were analyzed using the two-sided Student's t-test on raw data. A P value of <0.05 was considered significant.
Results
FXlllA Deficiency Alters Cross-Linking Activities Between Plasma Fibronectin and/or Fibrin in Response to Acute Liver Injury
Because FXIII stabilizes clots by covalent cross-linking and FXIIIA deficiency is often associated with hemorrhagic diathesis, histopathological analysis of the liver was initially performed in FXIIIA-null mice. Intact FXIIIA-null livers from young adult mice at 8 weeks of age revealed normal morphology and showed no inflammation and/or fibrosis (see Supplemental Figure S1A at http://ajp.amjpathol.org). Identical plasma levels of fibronectin and fibrinogen, including α-, β-, and γ-chains in the control and the FXIIIA-null mice were confirmed by Western blot analysis (see Supplemental Figure S1B at http://ajp.amjpathol.org).
To address the roles of FXIII in the initial stage of adult tissue remodeling, acute liver injury was induced with a well-established model of a single treatment with the liver-damaging agent CCl4 (1 mL/kg body weight).11, 22 Because the initial step in ECM remodeling in response to tissue damage is to form a provisional matrix composed of fibronectin and fibrin, it was first examined whether the deposition and tissue distribution patterns of such exudated plasma proteins were altered in the FXlllA-null liver after acute injury. The depositions of fibronectin and fibrin did not show marked differences between control and FXIIIA-null liver up to day 3 after injury (Figure 1A). In contrast, their colocalization was significantly reduced in the mutant liver at 6 and 24 hours after injury by double immunofluorescence staining, as evidenced by less yellowish fluorescence in merged figures (Figure 1, A and B), suggesting an active role of FXIIIA in stabilizing clots by cross-linking.
Figure 1.
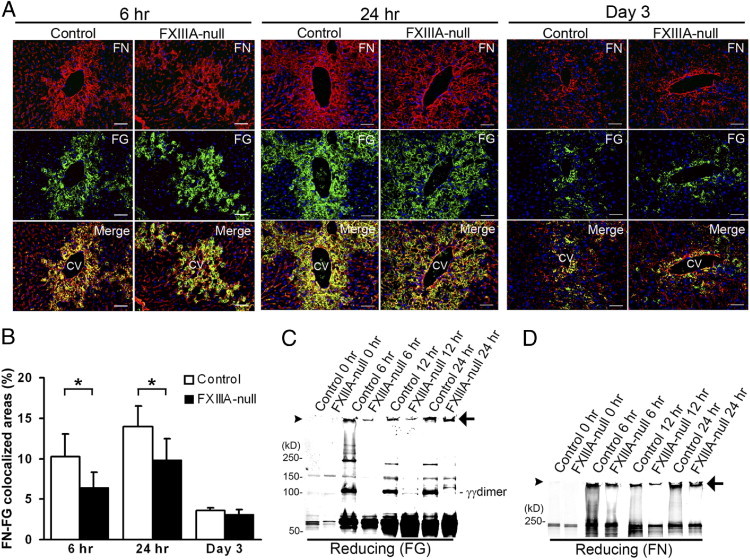
FXIIIA deficiency results in decreased cross-linking activities of clots in the early stages after liver injury. A: Time-course of fibronectin and fibrin deposition in control and FXIIIA-null livers at 6 and 24 hours and day 3 after acute liver injury induced by carbon tetrachloride. Double immunofluorescence staining for fibronectin (FN, in red) and fibrin(ogen) (FG, in green), and the merged images. Scale bar = 50 μm. CV, central vein. B: Quantification of colocalized areas between fibronectin and fibrinogen (yellowish fluorescence in each merged figure shown in A; see Materials and Methods) (n = 5 for each group). *P < 0.05. C and D: Western blot analysis of fibrin (FG) (C) and fibronectin (FN) (D) after one-dimensional SDS-PAGE of DOC-insoluble liver samples under reducing conditions. The detection of both fibronectin and fibrin signals was performed in the same membrane using IRDye 800CW- or IRDye 680-conjugated secondary antibodies and the Odyssey Infrared Imaging System (see Materials and Methods). Arrowheads point to top of gels. The positions of a molecular mass marker (kDa) are indicated. Arrows indicate nonreducible high-molecular-weight complexes. The intensity of nonreducible high-molecular-weight bands was measured by densitometry, and the intensity of the control liver at each time point was set to 1. The relative intensities in mutant liver are: fibrinogen, 0.2 at 6 hours, 0.3 at 12 hours, and 0.8 at 24 hours; fibronectin, 0.4 at 6 hours, 0.4 at 12 hours, and 0.6 at 24 hours.
Next, to determine whether the absence of FXIIIA affected the formation of covalently cross-linked products at the molecular level, one-dimensional Western analysis was performed focusing on early stages (up to 24 hours after injury) under reducing conditions using deoxycholate (DOC)–insoluble liver-tissue fractions (defined as maturely assembled matrices17). The quantity of nonreducible high-molecular-weight complexes with fibrin and fibronectin was lower in the mutant livers compared with controls, particularly at the earlier stage (Figure 1, C and D, arrows: The relative intensity in the mutant liver at 6 hours was 0.2 for fibrinogen and 0.4 for fibronectin). These results suggest the participation of FXIII-mediated, nonreducible, covalent cross-linkages in the early stages after injury. No γγ-dimer chain cross-linking of fibrin was detectable in the mutant liver up to 24 hours after injury (Figure 1C14), confirming the absence of FXIII activity in those mutant mice.
To determine more precisely the contribution of plasma fibronectin and fibrinogen to FXIII-mediated covalent cross-links in the early stages after injury, two-dimensional nonreducing/reducing SDS-PAGE and immunoblotting were performed. In the control liver, the formation of high-molecular-weight complexes with fibrin, fibronectin, and of fibrin γγ-dimer were apparent up to 24 hours after injury (Figure 2, A and B). Importantly, nonreducible cross-linked high-molecular-weight fibrin-fibronectin complexes were evident at the top of the gel in the control liver from 6 hours after injury, as shown by yellow fluorescence in the merged figure (Figure 2, A and B). In addition, nonreducible cross-linked high-molecular-weight fibrin-fibrin and fibronectin-fibronectin complexes were also detected, as evidenced by green and red fluorescence, respectively (Figure 2B). In contrast, in FXIIIA-null livers, those high-molecular-weight fibrin-fibronectin complexes were completely absent at 6 hours after injury (Figure 2B). Such complexes gradually started to form from 12 hours; however, the amounts were still lower than in controls (Figure 2A), suggesting the participation of other transglutaminases from this time point, such as locally produced tissue transglutaminase (also termed transglutaminase type 2 [TG2]) produced by hepatocytes and/or endothelial cells.23, 24, 25 Indeed, real-time PCR analysis revealed that the expression level of TG2 mRNA was upregulated in both control and mutant liver after injury, and the mutant liver showed significantly upregulated expression level at 72 hours after injury (Figure 3). Interestingly, the molecule which migrated to the top of the gel in the first dimension and migrated as a fibronectin monomer (∼220 kDa) in the second dimension was identified as one of the major populations in both control and mutant liver products (Figure 2, A and B, asterisks).19, 26, 27 There was little infiltration of monocytes/macrophages, which contain the cellular FXIIIA type, in both control and mutant liver as shown by F4/80 immunostaining up to day 3 after injury (data not shown). These results indicate that FXlll mediates the covalent cross-linking that forms the provisional matrix (fibrin-fibronectin) as well as fibrin-fibrin and fibronectin-fibronectin complexes in the early stages of acute liver injury.
Figure 2.
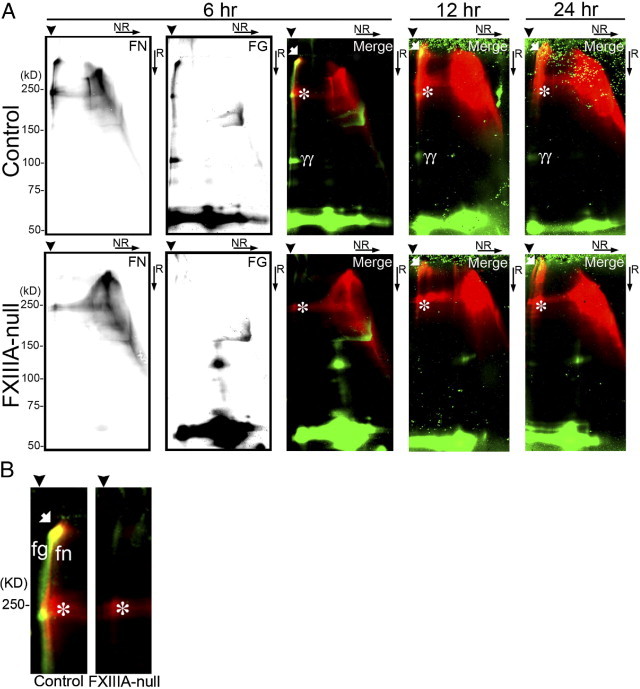
FXIIIA deficiency decreases the formation of non-reducible covalently cross-linked high-molecular-weight complex after injury. A: Western blot analysis of fibronectin (FN) and fibrin (FG) after two-dimensional (2D: NR, non-reducing; R, reducing) SDS-PAGE of DOC-insoluble liver samples from control and FXlllA-null mice at 6, 12, and 24 hours after injury. The detection of both fibronectin and fibrin signals was performed in the same membrane using IRDye 800CW- or IRDye 680-conjugated secondary antibodies and the Odyssey Infrared Imaging System (see Materials and Methods). At 6 hours after injury, the fibronectin and fibrin signal is shown separately in each black and white image, and their merged picture is shown as a color image (fibronectin and fibrin signals are in red and green, respectively). B: Enlarged merged images of the control and FXIIIA-null blot shown in A at 6 hours after injury (high-molecular-weight areas). Note that the non-reducible covalently cross-linked high-molecular-weight complexes between fibronectin and fibrin are detected only in control liver, as evidenced by the yellow spot in the merged figure (white arrow). Arrowheads point to the top of gels in the first dimension. The positions of a molecular mass marker (kDa) are indicated. White arrows, non-reducible covalently cross-linked high-molecular-weight complexes; asterisks, high-molecular-weight disulfide-bonded fibronectin complexes; γγ, fibrin γγ-dimer. fn, covalently cross-linked high-molecular-weight fibronectin complexes; fg, covalently cross-linked high-molecular-weight fibrin complexes.
Figure 3.
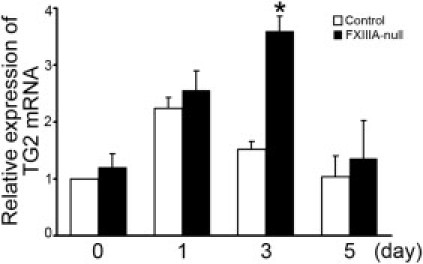
Lack of XIIIA increases the expression level of TG2 mRNA by real-time PCR analysis. The mRNA level of intact control liver (day 0) was set to 1 (control). The mRNA levels determined at each time point are shown relative to control. Data are mean ± SD (n = 3 for each group). *P < 0.05.
Lack of FXIIIA Does Not Affect the Activation of Hepatic Stellate Cells or Collagen Fibrillogenesis After Acute Liver Injury
Activation and transdifferentiation of hepatic stellate cells (Ito cells or fat-storing cells) into myofibroblasts is the central event in mediating collagen production and remodeling in response to liver injury.11 Therefore it was next determined whether the delay in provisional matrix formation in FXIIIA-null liver altered the activation of hepatic stellate cells, which thereby affected collagen organization after injury. The induction level of αSMA, a marker for activated hepatic stellate cells, was identical in control and mutant liver over time (Figure 4A). Furthermore, total ECM deposition by trichrome staining, and the deposition and assembly of collagen type III and type I fibrils did not show any obvious differences between control and mutant liver both at day 3 and 4 (Figure 4, B and C). In addition, those newly organized collagen networks were nearly completely cleared from both control and mutant liver by day 7 (data not shown). These results indicate that FXlllA deficiency does not interfere with hepatic stellate cell activation or normal reconstruction and resolution of collagen organization after acute liver injury.
Figure 4.
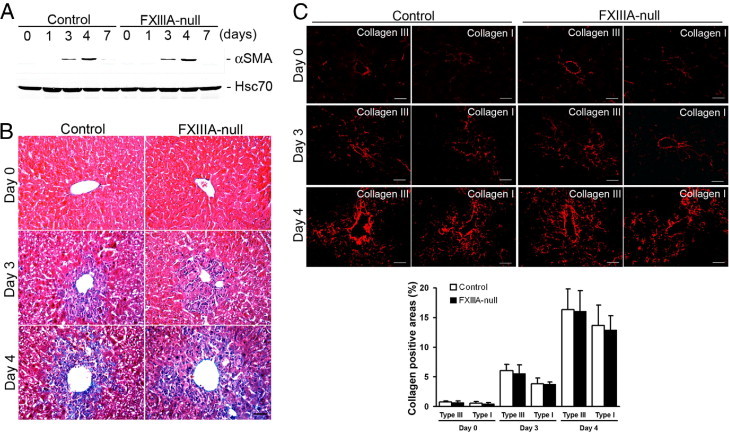
Lack of XIIIA does not affect activation of hepatic stellate cells or collagen fibrillogenesis after injury. A: Western blot of αSMA in the control and FXlll-null livers at 0, 1, 3, 4 and 7 days after injury. The intensity of the bands was measured by densitometry, and each αSMA intensity was normalized to Hsc70, then the intensity of the control at day 3 was set to 1.0. Relative intensities are 1.1 at day 3 in FXIII-null, and 2.8 and 3.0 at day 5 in control and FXIII-null, respectively. B: Masson trichrome staining at day 0 (untreated), 3 and 4 to examine total ECM deposition (blue color). Scale bar = 50 μm. C: Upper panels: Deposition and assembly of type III and type I collagen (in red) at day 0 (untreated), 3 and 4 by immunofluorescence staining. Scale bar = 50 μm. Lower panel: Quantification of areas in assembled collagen fibrils (n = 5 for each group). Note that the collagen positive areas show similar levels between control and mutant liver at day 0 and at days 3 and 4 after injury.
FXIIIA Deficiency Results in Significantly Increased Hepatocyte Apoptosis and Delayed Hepatocyte Proliferation After Injury
Because FXIIIA deficiency is often associated with impaired wound healing,13, 14 we examined whether the lack of FXIIIA affects hepatocyte apoptosis and/or proliferation in response to acute liver damage. There was a significantly increased number of cleaved caspase 3 (a marker for apoptosis)–positive cells in the FXIIIA-null liver at 24 hours after injury (P < 0.05), whereas no significant differences were noted at 3 days after injury (Figure 5A). No obvious extensive hemorrhage was observed in either FXIIIA-null or control liver throughout the periods (data not shown).
Figure 5.
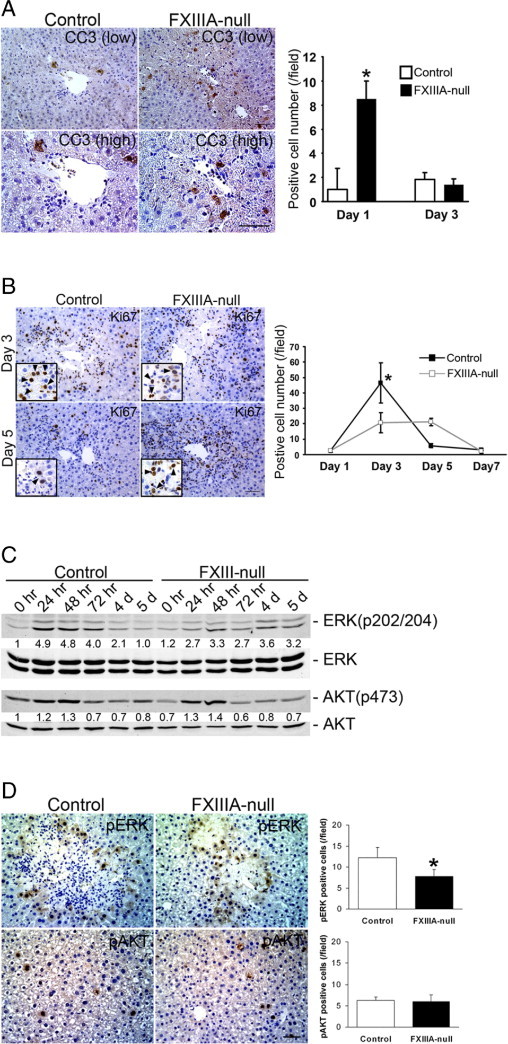
FXIIIA deficiency results in significantly increased hepatocyte apoptosis and delay in hepatocyte proliferation after injury. A: Analysis of cell death. Left panels: Immunostaining for cleaved caspase 3 (CC3) at 24 hours after injury at low (upper panels) and high (lower panels) magnifications. Scale bars: 50 μm (upper panel); 25 μm (lower panel). Right panel: Analysis of cleaved caspase 3–positive cells. Data are mean ± SD [cells/field; field = 0.036 mm2 (n = 4 for each group)]. Note that the number of positive cells in mutant liver is significantly higher at 1 day after treatment. *P < 0.05. B: Analysis of hepatocyte proliferation. Left panels: Immunostaining for Ki-67 at 3 and 5 days after injury. Inset in each box shows Ki-67–positive hepatocytes at higher magnification [hepatocyte nuclei (arrowheads) are ∼two- to threefold larger than those of nonparenchymal cells such as hepatic stellate cells, endothelial cells, and lymphocytes]. Scale bar = 50 μm. Right panel: Analysis of Ki-67–positive cells. Data are mean ± SD [cells/field; field = 0.14 mm2 (n = 4 for each group)]. Note that the number of positive hepatocytes in control liver is significantly higher at 3 days after injury. *P < 0.05. C: Western blot analysis of phospho-Erk [ERK(p202/204)], total Erk, phospho-Akt [AKT(p473)], and total Akt in control and FXIII-null liver. Pooled samples from three mouse livers from each strain were used for the analysis. Intensity of bands was measured by densitometry. Each pErk and pAkt intensity was normalized to total Erk and total Akt, respectively, and then the intensity at 0 hours in the control liver was set to 1. Each intensity is shown relative to control value. D: Left panels: Immunostaining for pErk and pAkt at 72 hours after injury. Scale bar = 50 μm. Right panels: Analysis of pErk- and pAkt-positive cells. Data are mean ± SD [cells/field; field = 0.14 mm2 (n = 3 for each group)]. Note that the number of pErk-positive cells in the mutant liver is significantly lower at 72 hours after injury, whereas the number of pAkt-positive cells does not show significant differences between them. *P < 0.05.
Hepatocyte regeneration is a multistep process. In response to injury, intracellular signaling pathways that involve Ras/extracellular signal–regulated kinase (Erk) and PI3K/Akt pathways are activated, which thereby provide clues to the initiating signals.28 Since the mutant liver showed more extensive hepatocyte damage with increased apoptosis in response to acute injury, it was next examined whether such a phenotype altered the proliferation activity of the hepatocytes. Indeed, the proliferative response in the mutant liver was delayed: The number of Ki-67 (a marker for cell proliferation)-positive hepatocytes in the mutant liver was significantly lower at day 3 compared to controls (Figure 5B, P < 0.05). Interestingly, although proliferation activity in the control liver considerably decreased from day 3 to day 5, no such decrease was observed in the mutant liver (Figure 5B). Therefore the mechanism underlying the altered proliferation response of mutant hepatocytes to injury was further investigated. Considerable phosphorylation of Erk1/2 (∼fivefold induction compared to untreated controls) was induced in the control liver in response to injury with the induction peak from 24 to 48 hours. Such phosphorylation levels decreased from day 3 to day 5 and reverted to control levels at day 5 after injury. In contrast, in the mutant liver the pErk1/2 induction level was lower (∼threefold induction compared to untreated controls) but such lower phosphorylation levels were prolonged from 24 hours to day 5 after injury (Figure 5C). There were no obvious differences in the induction patterns and levels in the phosphorylation of Akt between control and mutant liver (Figure 5C). Immunohistochemical observation revealed that the number of pErk1/2-positive cells in the mutant liver was significantly lower compared with controls at 72 hours after injury, whereas the number of pAkt-positive cells did not show significant differences between two groups (Figure 5D). These findings indicate that FXIIIA deficiency causes delayed but not defective hepatocyte regeneration phenotype largely mediated by Erk pathway, and strongly suggest that FXIII plays a significant role in protecting hepatocyte cell death.
Inappropriate cell-ECM interactions and environment induce apoptosis.29 Plasma fibronectin can support cell survival through ECM receptor integrin-mediated signals.16 p53-p21–Mediated hepatocyte apoptosis plays a crucial role in the response to some liver injuries such as cholestasis.30 Subsequent analysis of p53 revealed that FXIIIA-null liver showed a markedly high induction of p53 expression and its activation, as evidenced by increased p21 expression levels at 24 and 48 hours after injury by Western analysis (see Supplemental Figure S2A at http://ajp.amjpathol.org). Furthermore, the number of nuclear p53 positive hepatocytes significantly increased in FXIII-null liver at 24 hours after injury (see Supplemental Figure S2B at http://ajp.amjpathol.org). Thus, these findings suggest that the lack of FXIIIA causes the defect in plasma fibronectin and fibrin(ogen) covalent cross-linking complexes after liver injury (Figures 1 and 2), which could lack FXIII-mediated matrix-survival signals and induces p53-mediated hepatocyte apoptosis in the initial stages of liver damage, which in turn could result in the delay in hepatocyte proliferation.
Discussion
FXIII belongs to the transglutaminase family of proteins and has been postulated to participate in the stabilization of the provisional matrix and cell-matrix interactions in the adult tissue repair process.12 However, the role FXIII plays in adult tissue remodeling remains to be defined. In the present study, the functional requirement of FXIII-mediated provisional matrix formation during adult tissue remodeling was investigated. We provide evidence using a mouse model of acute liver injury that: i) FXlllA deficiency results in the decreased covalent cross-linking activities between plasma fibronectin and/or fibrin in the initial stage after injury; ii) The absence of FXIIIA does not affect hepatic stellate cell activation or collagen fibrillogenesis during the initial stages of tissue remodeling; and iii) FXIIIA deficiency causes significantly increased hepatocyte apoptosis and a delay in hepatocyte proliferation after injury, which are accompanied by a significantly high induction of p53 expression. Thus, these findings indicate the possible functional links between FXIII and cell survival signaling during adult tissue remodeling.
ECM remodeling in response to wound healing is a complex process.12 Fibronectin, a component of the provisional matrix, exists in two isoforms: as a soluble isoform in plasma (plasma fibronectin, produced solely by hepatocytes) and as an insoluble isoform in tissue ECM (cellular fibronectin, produced by a variety of cells).5, 6 Although cellular fibronectin is less efficiently incorporated into fibrin clots in vitro31, its contribution in the early stages of tissue repair is not fully understood. Here, acute liver injury induced by the cytotoxic reagent CCl4 was used as a current tissue repair model since it has several benefits when compared to general wound healing models such as skin wound healing. First, the liver tissue subjected to CCl4-induced injury does not start cellular fibronectin synthesis until 24 hours after the treatment.32 Second, in CCl4-induced liver injury without bleeding, it leads to the extravasation of plasma but not platelets or monocytes/macrophages, which could be the source of cellular FXIIIA and cellular fibronectin, until 18 hours after injury.33 Therefore, the formation and any potential role of the provisional matrix in the initial stage of CCl4-induced liver injury is dependent exclusively on plasma derived components: plasma fibronectin, fibrin(ogen), and plasma FXIII. Indeed, it has been demonstrated in this study that both the exudated fibrin and plasma fibronectin at 6 hours after tissue damage forms high-molecular-weight complexes and their nonreducible covalent cross-linking is largely mediated by FXlll.
The livers lacking FXIIIA markedly upregulated TG2 mRNA compared to controls at 3 days after injury, suggesting a compensatory mechanism as observed by others in FXIIIA-deficient platelets.34 The TG2-null liver treated with CCl4 fails to clear the necrotic tissue and exhibits a progressive accumulation of ECM with an elevated number of both activated hepatic stellate cells and inflammatory cells, suggesting a protective role for TG2 in liver injury.35 Thus, both FXIII and TG2 are apparently involved in adult tissue injury and remodeling,25 and the precise contribution of each molecule to these processes needs to be defined.
The surprising finding in the present study was that the lack of FXIII-mediated provisional matrix did not interfere with normal collagen organization in response to injury, even though there are several lines of evidence that a preformed fibronectin matrix is crucial for type III and type I collagen–containing fibril formation in culture and that the cross-linking of fibronectin to type III and type I collagens is mediated by FXIII in vitro36, 37, 38, 39. Thus, covalently and/or non-covalently formed fibronectin aggregates19, 26, 27 observed in the current in vivo system could sufficiently nucleate collagen fibril formation in adult tissue remodeling.
We have previously shown that plasma fibronectin supports neuronal survival after transient cerebral ischemia through integrin-mediated signals.16 However, the functional requirement of FXIII for this process has not yet been defined. Although non-FXIII mediated high-molecular-weight fibronectin complexes were detected in the mutant liver after injury, they did not compensate and protect FXIIIA-null hepatocyte apoptosis. ECM-mediated cell survival pathway is transduced by integrins and subsequent activation of focal adhesion kinase (FAK), which suppresses p53-p21–mediated apoptosis in serum free conditions in vitro.40, 41 FAK inactivation results in p53-p21–dependent cell growth arrest during development.42 Taken together, these findings suggest that the FXIII-mediated covalently cross-linked plasma fibronectin and fibrin plays a crucial role as a survival factor in response to liver damage and that ECM survival signals for hepatocytes could be transduced through the FAK-p53 signaling axis. The delayed wound healing in patients with FXIIIA deficiency7 could be in part because of the defect in such a cell survival signaling. Which cross-linked complex mediated by FXIII is the most likely to be responsible for a survival factor after acute liver injury? FXIII-mediated fibrin-fibrin complexes are unlikely, as certain cell types such as fibroblasts neither attach nor spread on cross-linked fibrin clots and they are not able to invade in fibronectin-depleted plasma clots.43, 44 In contrast, FXIII-mediated fibronectin-fibrin matrices have the ability to support adhesion and spreading of fibroblasts in vitro45, 46. Thus, FXIII-mediated fibrin-fibronectin matrices are the most likely to play a pivotal role as a survival factor during adult tissue remodeling.
Acknowledgments
We thank Dr. Takako Sasaki (University of Erlangen-Nuernberg, Erlangen, Germany) for type III and type I collagen antibodies and Emma Stephenson for editorial assistance.
Footnotes
Supported by NIH research grant DK074538 (T.S.).
Disclosures: G.D. is employed by CSL Behring. None of the other authors disclosed any conflict of interest.
Supplemental material for this article can be found at http://ajp.amjapthol.org or at doi:10.1016/j.ajpath.2011.08.037.
Supplementary data
Characterization of FXIIIA-null mice. (A) Histological analysis of the intact livers from 8-week old adult control and Factor XIIIA-null mice with hematoxylin/eosin (HE, upper panels), periodic acid-schiff (PAS, middle panels) and mallory-azan (Azan, lower panels) staining. Scar = 50 μm. (B) Western blot analysis of plasma fibronectin (FN) and fibrinogen (FG) levels in intact control and FXlllA-null mice. The same samples were stained with Coomassie brilliant blue (CBB) to confirm that comparable amounts of protein were loaded.
The FXIII-null liver shows increased expression levels of p53 and p21 following acute liver injury. (A) Western blot analysis of p53, p21 and Hsc70 (loading control) in response to acute liver injury. Pooled samples from 3 mouse livers in control and mutant mice were used for the analysis. (B) Left panels: Pronounced induction of p53 in FXIIIA-null hepatocyte nuclei at 24 hrs after injury by immunofluorescent staining (p53 in red; DAPI [cell nuclei] in blue). Scale bar = 25 μm. Right panel: Analysis of p53 positive hepatocytes (Hepatocyte nuclei are ∼2- to 3-fold larger than those of non-parenchymal cells as shown in Fig. 5B). Data are the means ± S.D (cells/field; field = 0.036 mm2 [n = 5 for each group]). *, P < 0.05.
References
- 1.Lorand L., Graham R.M. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 2.Lorand L. Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann NY Acad Sci. 2001;936:291–311. doi: 10.1111/j.1749-6632.2001.tb03516.x. [DOI] [PubMed] [Google Scholar]
- 3.Mosesson M.W. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 4.Muszbek L., Yee V.C., Hevessy Z. Blood coagulation factor XIII: structure and function. Thromb Res. 1999;94:271–305. doi: 10.1016/s0049-3848(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 5.Mosher D.F. Academic Press; San Diego: 1989. Fibronectin. [Google Scholar]
- 6.Hynes R.O. Springer-Verlag; New York: 1990. Fibronectins. [Google Scholar]
- 7.Iismaa S.E., Mearns B.M., Lorand L., Graham R.M. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 2009;89:991–1023. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- 8.Adany R., Bardos H. Factor XIII subunit A as an intracellular transglutaminase. Cell Mol Life Sci. 2003;60:1049–1060. doi: 10.1007/s00018-003-2178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernal W., Auzinger G., Dhawan A., Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 10.Pathikonda M., Munoz S.J. Acute liver failure. Ann Hepatol. 2010;9:7–14. [PubMed] [Google Scholar]
- 11.Bataller R., Brenner D.A. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark R.A.F. Plenum Press; New York: 1996. The molecular and cellular biology of wound repair. [Google Scholar]
- 13.Inbal A., Lubetsky A., Krapp T., Castel D., Shaish A., Dickneitte G., Modis L., Muszbek L. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005;94:432–437. doi: 10.1160/TH05-04-0291. [DOI] [PubMed] [Google Scholar]
- 14.Lauer P., Metzner H.J., Zettlmeissl G., Li M., Smith A.G., Lathe R., Dickneite G. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88:967–974. [PubMed] [Google Scholar]
- 15.Moriya K., Bae E., Honda K., Sakai K., Sakaguchi T., Tsujimoto I., Kamisoyama H., Keene D.R., Sasaki T., Sakai T. A fibronectin-independent mechanism of collagen fibrillogenesis in adult liver remodeling. Gastroenterology. 2011;140:1653–1663. doi: 10.1053/j.gastro.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai T., Johnson K.J., Murozono M., Sakai K., Magnuson M.A., Wieloch T., Cronberg T., Isshiki A., Erickson H.P., Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 17.Sechler J.L., Takada Y., Schwarzbauer J.E. Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol. 1996;134:573–583. doi: 10.1083/jcb.134.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosher D.F., Johnson R.B. In vitro formation of disulfide-bonded fibronectin multimers. J Biol Chem. 1983;258:6595–6601. [PubMed] [Google Scholar]
- 19.Chen H., Mosher D.F. Formation of sodium dodecyl sulfate-stable fibronectin multimers: Failure to detect products of thiol-disulfide exchange in cyanogen bromide or limited acid digests of stabilized matrix fibronectin. J Biol Chem. 1996;271:9084–9089. doi: 10.1074/jbc.271.15.9084. [DOI] [PubMed] [Google Scholar]
- 20.Sakai T., Li S., Docheva D., Grashoff C., Sakai K., Kostka G., Braun A., Pfeifer A., Yurchenco P.D., Fassler R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda K., Sakaguchi T., Sakai K., Schmedt C., Ramirez A., Jorcano J.L., Tarakhovsky A., Kamisoyama H., Sakai T. Epidermal hyperplasia and papillomatosis in mice with a keratinocyte-restricted deletion of csk. Carcinogenesis. 2007;28:2074–2081. doi: 10.1093/carcin/bgm112. [DOI] [PubMed] [Google Scholar]
- 22.Constandinou C., Henderson N., Iredale J.P. Modeling liver fibrosis in rodents. Methods Mol Med. 2005;117:237–250. doi: 10.1385/1-59259-940-0:237. [DOI] [PubMed] [Google Scholar]
- 23.Mirza A., Liu S.L., Frizell E., Zhu J., Maddukuri S., Martinez J., Davies P., Schwarting R., Norton P., Zern M.A. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am J Physiol. 1997;272:G281–G288. doi: 10.1152/ajpgi.1997.272.2.G281. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann B.R., Annis D.S., Mosher D.F. Reactivity of the amino-terminal region of fibronectin to transglutaminase 2 and factor XIIIa. J Biol Chem. 2011;286:32220–32230. doi: 10.1074/jbc.M111.255562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aeschlimann D., Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi T., Erickson H.P. Revisiting the mystery of fibronectin multimers: the fibronectin matrix is composed of fibronectin dimers cross-linked by non-covalent bonds. Matrix Biol. 2009;28:170–175. doi: 10.1016/j.matbio.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langenbach K.J., Sottile J. Identification of protein-disulfide isomerase activity in fibronectin. J Biol Chem. 1999;274:7032–7038. doi: 10.1074/jbc.274.11.7032. [DOI] [PubMed] [Google Scholar]
- 28.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 29.Gilmore A.P. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 30.Amaral J.D., Castro R.E., Steer C.J., Rodrigues C.M. p53 and the regulation of hepatocyte apoptosis: implications for disease pathogenesis. Trends Mol Med. 2009;15:531–541. doi: 10.1016/j.molmed.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Wilson C.L., Schwarzbauer J.E. The alternatively spliced V region contributes to the differential incorporation of plasma and cellular fibronectins into fibrin clots. J Cell Biol. 1992;119:923–933. doi: 10.1083/jcb.119.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odenthal M., Neubauer K., Meyer zum Buschenfelde K.H., Ramadori G. Localization and mRNA steady-state level of cellular fibronectin in rat liver undergoing a CCl4-induced acute damage or fibrosis. Biochim Biophys Acta. 1993;1181:266–272. doi: 10.1016/0925-4439(93)90031-u. [DOI] [PubMed] [Google Scholar]
- 33.Hirata K., Ogata I., Ohta Y., Fujiwara K. Hepatic sinusoidal cell destruction in the development of intravascular coagulation in acute liver failure of rats. J Pathol. 1989;158:157–165. doi: 10.1002/path.1711580211. [DOI] [PubMed] [Google Scholar]
- 34.Jobe S.M., Leo L., Eastvold J.S., Dickneite G., Ratliff T.L., Lentz S.R., Di Paola J. Role of FcRgamma and factor XIIIA in coated platelet formation. Blood. 2005;106:4146–4151. doi: 10.1182/blood-2005-03-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nardacci R., Lo Iacono O., Ciccosanti F., Falasca L., Addesso M., Amendola A., Antonucci G., Craxi A., Fimia G.M., Iadevaia V., Melino G., Ruco L., Tocci G., Ippolito G., Piacentini M. Transglutaminase type II plays a protective role in hepatic injury. Am J Pathol. 2003;162:1293–1303. doi: 10.1016/S0002-9440(10)63925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velling T., Risteli J., Wennerberg K., Mosher D.F., Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 37.Sottile J., Shi F., Rublyevska I., Chiang H.Y., Lust J., Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293:C1934–C1946. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- 38.Sottile J., Hocking D.C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mosher D.F., Schad P.E. Cross-linking of fibronectin to collagen by blood coagulation Factor XIIIa. J Clin Invest. 1979;64:781–787. doi: 10.1172/JCI109524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ilic D., Almeida E.A., Schlaepfer D.D., Dazin P., Aizawa S., Damsky C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Almeida E.A., Ilic D., Han Q., Hauck C.R., Jin F., Kawakatsu H., Schlaepfer D.D., Damsky C.H. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J Cell Biol. 2000;149:741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim S.T., Chen X.L., Lim Y., Hanson D.A., Vo T.T., Howerton K., Larocque N., Fisher S.J., Schlaepfer D.D., Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corbett S.A., Wilson C.L., Schwarzbauer J.E. Changes in cell spreading and cytoskeletal organization are induced by adhesion to a fibronectin-fibrin matrix. Blood. 1996;88:158–166. [PubMed] [Google Scholar]
- 44.Knox P., Crooks S., Rimmer C.S. Role of fibronectin in the migration of fibroblasts into plasma clots. J Cell Biol. 1986;102:2318–2323. doi: 10.1083/jcb.102.6.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grinnell F., Feld M., Minter D. Fibroblast adhesion to fibrinogen and fibrin substrata: requirement for cold-insoluble globulin (plasma fibronectin) Cell. 1980;19:517–525. doi: 10.1016/0092-8674(80)90526-7. [DOI] [PubMed] [Google Scholar]
- 46.Corbett S.A., Schwarzbauer J.E. Fibronectin-fibrin cross-linking: a regulator of cell behavior. Trends Cardiovasc Med. 1998;8:357–362. doi: 10.1016/s1050-1738(98)00028-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of FXIIIA-null mice. (A) Histological analysis of the intact livers from 8-week old adult control and Factor XIIIA-null mice with hematoxylin/eosin (HE, upper panels), periodic acid-schiff (PAS, middle panels) and mallory-azan (Azan, lower panels) staining. Scar = 50 μm. (B) Western blot analysis of plasma fibronectin (FN) and fibrinogen (FG) levels in intact control and FXlllA-null mice. The same samples were stained with Coomassie brilliant blue (CBB) to confirm that comparable amounts of protein were loaded.
The FXIII-null liver shows increased expression levels of p53 and p21 following acute liver injury. (A) Western blot analysis of p53, p21 and Hsc70 (loading control) in response to acute liver injury. Pooled samples from 3 mouse livers in control and mutant mice were used for the analysis. (B) Left panels: Pronounced induction of p53 in FXIIIA-null hepatocyte nuclei at 24 hrs after injury by immunofluorescent staining (p53 in red; DAPI [cell nuclei] in blue). Scale bar = 25 μm. Right panel: Analysis of p53 positive hepatocytes (Hepatocyte nuclei are ∼2- to 3-fold larger than those of non-parenchymal cells as shown in Fig. 5B). Data are the means ± S.D (cells/field; field = 0.036 mm2 [n = 5 for each group]). *, P < 0.05.