

Abstract
Cholangitis arising from biliary infection dominates the prognosis in Caroli's disease. To clarify the influences of bacterial infection on the biliary cystogenesis, in vivo and in vitro studies were performed using the polycystic kidney (PCK) rat as an animal model of Caroli's disease. Cholangitis became a frequent histological finding in aged PCK rats, and neovascularization around the bile ducts also increased in aged PCK rats. Immunohistochemistry revealed that expression of vascular endothelial growth factor (VEGF) was increased in PCK rat biliary epithelium. In vitro, PCK cholangiocytes overexpressed VEGF, and the supernatant of cultured PCK cholangiocytes significantly increased the proliferative activity, migration, and tube formation of cultured rat vascular endothelial cells. Stimulation with lipopolysaccharide (LPS) further induced VEGF expression in PCK cholangiocytes, which might be mediated by signaling pathways involving phosphatidylinositol 3-kinase (PI3K)-Akt and c-Jun N-terminal kinase (JNK). Both LPS and VEGF increased cell proliferative activity in PCK cholangiocytes, and siRNA against VEGF significantly reduced LPS-induced cell proliferation. Thus, LPS-induced overexpression of VEGF in the biliary epithelium may lead to hypervascularity around the bile ducts; concurrently, LPS and VEGF act as cell proliferation factors for cholangiocytes. Biliary infection may thus exacerbate biliary cystogenesis in PCK rats.
Caroli's disease is characterized by progressive, multiple cystic dilation of intrahepatic bile ducts; it is frequently associated with portal fibrosis, corresponding to congenital hepatic fibrosis.1, 2 Caroli's disease belongs to a group of congenital hepatorenal fibrocystic syndromes, and is a well-known hepatic manifestation of autosomal recessive polycystic kidney disease (PKD).3
Caroli's disease with congenital hepatic fibrosis presents clinical features consisting of bouts of cholangitis and cholelithiasis and portal hypertension. Clinical progression and presentation are highly variable, and symptoms may appear early or late in life. Bile stagnation due to bile duct dilation and hepatolithiasis explains the recurrent cholangitis that dominates the clinical course and is the principal cause of morbidity and mortality. Cholangitis can lead to hepatic abscess and sepsis, and a large number of patients die within 5 to 10 years after cholangitis occurs in Caroli's disease.4
Human and experimental data suggest several potential mechanisms that could lead to cystic dilation of intrahepatic bile ducts of Caroli's disease. These include increased cell proliferation and apoptosis, enhanced fluid secretion, abnormal cell-matrix interactions, and abnormal ciliary structure or function.5, 6, 7, 8, 9 Although the clinical significance of cholangitis due to biliary infection is well recognized in Caroli's disease, the effect of biliary infection on disease pathogenesis and progression has not been studied previously.
Lipopolysaccharide (LPS) is a bacterial component that is a proximal mediator in the initiation of local inflammation and sepsis. LPS induces production of proinflammatory cytokines and permeability factors. In certain types of cells, LPS induces angiogenic factors, such as vascular endothelial growth factor (VEGF).10, 11 Because cholangiocytes express the receptor of LPS, Toll-like receptor 4 (TLR4),12, 13 LPS may also be able to induce angiogenic growth factors in cholangiocytes.
VEGF is overexpressed in human and rodent cholangiocytes from polycystic liver diseases, including Caroli's disease, and has been implicated in disease pathogenesis.14 Overexpression of VEGF in cholangiocytes indicates that VEGF may induce portal neovascularization, which leads to cholangiocyte overgrowth due to abundant vascular supply, in turn resulting in the exacerbation of cystic bile duct dilation. In addition, a recent study showed that VEGF stimulates cholangiocyte proliferation via an autocrine mechanism.15
Using the polycystic kidney (PCK) rat as an animal model of Caroli's disease,5 we conducted the present study to clarify the influences of bacterial infection on the biliary cystogenesis with regard to VEGF expression in cholangiocytes and portal neovascularization.
Materials and Methods
Animals
The PCK rats were maintained at the laboratory animal institute of Kanazawa University Graduate School of Medicine. Normal (Crj:CD) rats were purchased from Charles River Japan (Sagamihara, Japan). Animal studies were performed in accordance with guidelines for the care and use of laboratory animals at the Takara-machi Campus of Kanazawa University.
Liver Specimens
Livers were removed from 3-week-old, 2-month-old, and 10-month-old rats. Human liver tissues of Caroli's disease with congenital hepatic fibrosis (5 cases) were obtained at time of surgery. As controls, normal or subnormal human livers were used (n = 5). Liver tissues were immersed in 10% formalin neutral buffer solution (pH 7.4) and then embedded in paraffin. More than 10 serial sections (4 μm thick) cut from each paraffin block were subjected to histological analysis. Experiments using human subjects were performed with the approval of the local ethics committee of Kanazawa University under patient informed consent.
Cell Culture of Cholangiocytes
Cholangiocytes were isolated, purified, and cultured from the intrahepatic large bile ducts of normal and PCK rats as described previously.6 Cells were set on cell culture dishes covered with a standard growth medium, composed of Dulbecco's modified Eagle's medium/F-12 (Gibco, Grand Island, NY) containing 10% bovine growth serum (HyClone, Logan, UT), 5 μmol/L forskolin (Wako Pure Chemical Industries, Osaka, Japan), 20 ng/mL of epidermal growth factor (Upstate Biotechnology; Millipore, Billerica, MA) and 1% antibiotic-antimycotic (Gibco) at 37°C in an atmosphere of 5% CO2.
At subconfluent state, cholangiocytes were incubated with standard medium containing LPS (Ultra-Pure E. coli LPS; InvivoGen, San Diego, CA). Cell signaling inhibition studies were performed using NF-κB inhibitor (isohelenin; 30 μmol/L; Calbiochem, La Jolla, CA), VEGF receptor tyrosine kinase inhibitor (SU5614; 10 μmol/L; Sigma-Aldrich, St. Louis, MO), phosphatidylinositol 3-kinase (PI3K) inhibitor (LY294002; 20 μmol/L; Merck, Darmstadt, Germany), c-Jun N-terminal kinase (JNK) inhibitor I (2 μmol/L; Merck), and JNK inhibitor II (100 nmol/L; Merck).
Cell Culture of Vascular Endothelial Cells
Rat aorta endothelial cells (RAOECs) purchased from Cell Applications (San Diego, CA) were maintained with endothelial growth medium (rat endothelial cell basal medium; Cell Applications).
To determine their angiogenic effects, cholangiocytes were incubated with a standard medium for 3 days, and the culture supernatant was added to the basal medium of RAOECs at a concentration of 20%.
Cell Proliferation Assay
Cell proliferative activity was determined using a WST-1 assay according to the manufacturer's instructions (Roche, Mannheim, Germany). Cholangiocytes were treated with appropriate concentrations of LPS (InvivoGen) and recombinant VEGF (R&D Systems, Minneapolis, MN), and cell proliferative activity was determined at specified time intervals. Cell proliferative activity of RAOECs was assessed after 72 hours after stimulation with the cholangiocyte culture supernatant. WST-1 reagent was added and incubated for 2 hours before the plate was read. Each assay was performed in eight sets.
Cell Migration Assay
Cell migration activity of RAOECs was examined using a BioCoat cell migration chamber (BD Biosciences, Bedford, MA). In the upper chamber, a total of 5 × 104 cells in serum-free culture medium were seeded. In the lower chamber, rat endothelial cell basal medium (Cell Applications) or the basal medium containing 20% rat cholangiocyte culture supernatant with and without 24 hour-LPS treatment at the concentration of 1 μg/mL was placed. After 48 hours, cells were fixed in 100% methanol and were stained with hematoxylin. Cells that had migrated to the bottom side of the membrane were visualized under a light microscope, and the number of cells was counted in five randomly selected fields.
Tube Formation Assay
Tube formation assay was performed with use of growth-factor-reduced Matrigel (BD Biosciences). Matrigel was dispensed into a 24-well plate, and a total of 5 × 104 cells of RAOECs were placed on the gel. The cells were incubated with serum-free basal medium for 24 hours. The medium was changed then changed to the basal medium or to basal medium containing 20% rat cholangiocyte culture supernatant with and without 24 hour-LPS treatment at the concentration of 1 μg/mL; cells were then further incubated for 18 hours. The number of branching points of the cells was counted in five randomly selected fields.
RT-PCR and Real-Time Quantitative PCR
RT-PCR was performed using total RNA (1 μg) extracted from the cholangiocytes. Total RNA was used to synthesize cDNA with reverse transcriptase (ReverTra Ace; Toyobo, Osaka, Japan). Primer sequences and PCR conditions are given in Table 1. The PCR products were subjected to 2% agarose gel electrophoresis and were stained with ethidium bromide.
Table 1.
Primer Sequences and PCR Conditions
| Gene | Sequences, forward and reverse | Annealing temperature (°C) | PCR cycles | Product size (bp) |
|---|---|---|---|---|
| VEGF | 5′-AGTGGTCCCAGGCTGCAC-3′ | 60 | 40 | 70 |
| 5′-TCCATGAACTTCCCACTTCGT-3′ | ||||
| Flk-1 | 5′-AAGGACCTCAGACGCAAGAA-3′ | 55 | 40 | 237 |
| 5′-CATCCCAACACACAAAGCAC-3′ | ||||
| Flt-1 | 5′-AATCATTCCGAAGCAAGGTG-3′ | 55 | 40 | 360 |
| 5′-TTTCTTCCCACAGTCCCAAC-3′ | ||||
| TLR4 | 5′-GCCGGAAAGTTATTGTGGTG-3′ | 60 | 30 | 205 |
| 5′-TCCCACTCGAGGTAGGTGTT-3′ | ||||
| GAPDH | 5′-GAGTCAACGGATTTGGTCGT-3′ | 60 | 40 | 240 |
| 5′-TTGATTTTGGAGGGATCTC-3′ |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TLR, Toll-like receptor; VEGF, vascular endothelial growth factor.
Quantitative real-time PCR was performed according to a standard protocol using SYBR Green PCR master mix (Toyobo Co.) and an ABI Prism 7700 sequence detection system (PerkinElmer-Applied Biosystems, Warrington, UK). Cycling conditions were incubation at 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Fold difference relative to GAPDH was calculated. Each assay was performed in five sets.
Western Blotting
Total proteins were extracted from cholangiocytes using Pierce T-PER protein extraction reagent (Thermo Fisher Scientific, Rockford, IL). The protein (40 μg) was subjected to 10% SDS-PAGE and then was electrophoretically transferred onto a nitrocellulose membrane. The membrane was incubated with primary antibodies against Akt, phosphorylated Akt (p-Akt), JNK-stress-activated protein kinase (SAPK), p-JNK-SAPK, extracellular signal-regulated kinase 1/2 (ERK1/2), p-ERK1/2, p38 mitogen-activated protein kinase (MAPK), and p-p38 MAPK. All primary antibodies (rabbit monoclonal) were purchased from Cell Signaling Technology (Danvers, MA) and were used at 1:1000 dilution. Protein expression was detected using a peroxidase-labeled polymer (EnVision+; DakoCytomation, Glostrup, Denmark), and 3,3′-diaminobenzidine tetrahydrochloride (DAB) was used as the chromogen.
ELISA
The VEGF level in the culture supernatant of the cholangiocytes was determined using an enzyme-linked immunosorbent assay (ELISA) kit (Quantikine rat VEGF immunoassay; R&D Systems). Samples were added to a 96-well plate coated with an antibody for VEGF and were incubated for 2 hours. After a washing, the plate was incubated with an anti-VEGF antibody conjugated to horseradish peroxidase for 2 hours. Color development was performed using a substrate solution for 30 minutes, and absorbance at 450 nm was measured. Each assay was performed in five sets.
Immunofluorescence Confocal Microscopy
Cells were fixed with 4% paraformaldehyde for 15 minutes and permeabilized for 3 minutes with 0.1% Triton X-100 surfactant. After blocking, the cells were incubated for 1 hour at room temperature with a primary antibody against VEGF (1:100, mouse monoclonal; Abcam, Cambridge, MA), NF-κB p50 (5 μg/mL, rabbit polyclonal; Immuno-Biological Laboratories, Fujioka, Japan), and NF-κB p65 (5 μg/mL, rabbit polyclonal; Immuno-Biological Laboratories). Alexa Fluor 488 (10 μg/mL, Invitrogen-Molecular Probes, Eugene, OR) was used as a secondary antibody. Nuclei were stained with DAPI.
NF-κB Activation
The activation of NF-κB was measured by the DNA binding capacity of NF-κB using a TransAM NF-κB kit according to the manufacturer's instructions (Active Motif, Carlsbad, CA). Briefly, cholangiocytes were treated with LPS (10 μg/mL) for 30 minutes, and the cell extract was added to an oligonucleotide-coated 96-well plate and incubated for 1 hour. After a washing, the NF-κB antibodies were added and incubated for 1 hour. The plate was then incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour. Color development was performed using a developing solution for 30 minutes, and absorbance at 450 nm was measured. Each assay was performed in three sets.
VEGF Inhibition Studies
Synthetic VEGF siRNA and nonsilencing (negative control) siRNA were purchased from Qiagen (Tokyo, Japan). Transfections of siRNA were performed using HiPerFect transfection reagent (Qiagen) according to the manufacturer's instructions. Briefly, a total of 2 × 104 cholangiocyte cells were seeded on a 96-well plate and incubated for 24 hours with a standard growth medium. After removal of the standard medium, the cells were incubated with 200 μL of Dulbecco's modified Eagle's medium/F-12 (Gibco) containing premixed siRNA (10 nmol/L) and 0.75 μL of HiPerFect transfection reagent for 48 hours. Gene silencing was monitored using RT-PCR at the mRNA level and using immunofluorescence confocal microscopy at the protein level. Transfected cells were treated with LPS (0.5 μg/mL) for 48 hours. Cell proliferative activity was determined using the WST-1 assay.
Immunohistochemistry
The antibodies used were anti-VEGF antibody for rat (1:100, mouse monoclonal) from Abcam and anti-VEGF antibody for human (1:200, rabbit polyclonal), anti-CD31 antibody (1:100, rabbit polyclonal), anti-Flk-1 antibody (1:100, mouse monoclonal), anti-Flt-1 antibody (1:100, rabbit polyclonal), and anti-TLR4 antibody (1:100, rabbit polyclonal) from Santa Cruz Biotechnology (Santa Cruz, CA). After deparaffinization of the sections, antigen retrieval was performed by microwaving in 10 mmol/L citrate buffer pH 6.0. After blocking endogenous peroxidase, sections were incubated overnight at 4°C with individual primary antibodies. The sections were then incubated with secondary antibody conjugated to the EnVision+ system (DakoCytomation). Color development was performed using DAB, and the sections were counterstained with hematoxylin. For immunostaining of rat liver sections with anti-CD31 antibody, protein expression was detected using an alkaline phosphatase-labeled polymer (HISTOFINE system; Nichirei, Tokyo, Japan); color development was performed using a Vector Red alkaline phosphatase substrate kit (Vector Laboratories, Burlingame, CA). Control sections were evaluated by substitution of the primary antibodies with nonimmunized serum, resulting in no signal detection.
Microvessel Density
Liver sections stained with the anti-CD31 antibody were analyzed. The number of microvessels around the bile ducts was counted for five randomly selected fields in each section at ×400 magnification, and the value was defined as microvessel density.
Statistical Analysis
The mean ± SD was calculated for all parameters. Statistical differences were determined using t-test and analysis of variance. A P value of <0.05 was accepted as the level of statistical significance.
Results
Portal Neovascularization and Cholangitis in the PCK Liver
Portal neovascularization was examined using immunostained liver sections of 3-week-old, 2-month-old, and 10-month-old rats with anti-CD31 antibody. In normal rats of any age, vascular structures in the portal tract are composed mainly of portal vein, hepatic artery, and peribiliary vascular plexus; other vascular structures were unremarkable (Figure 1A). By contrast, portal neovascularization was evident in the PCK rats, and it tended to be increased in the aged rat (10-month-old; Figure 1A). Microvessel density was significantly higher in the PCK rats, compared with that of normal rats at any age examined; it reached the highest level in 10-month-old PCK rats (Figure 1B).
Figure 1.
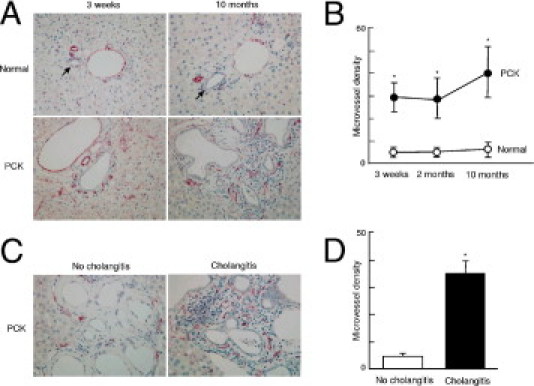
Portal neovascularization and cholangitis in the PCK liver. A: Vascular structures were visualized by immunohistochemical staining with anti-CD31. B: Microvessel density was significantly higher in the PCK liver, compared with that of normal rats; the value was highest in 10-month-old PCK liver. C and D: In 10-month-old PCK liver, portal neovascularization tended to be more densely seen at the sites of cholangitis, compared with sites of no cholangitis (C); analysis of microvessel density confirmed this tendency (D). Arrows indicate interlobular bile ducts of normal rats. *P < 0.01. Original magnification, ×400.
Chronic and/or suppurative cholangitis due to biliary infection became frequent histological findings in the PCK rats during aging. Cholangitis was rare in 3-week-old PCK rats, but it was frequently observed in the 10-month-old PCK rats. In 10-month-old PCK rats, portal neovascularization was more dense at the sites of cholangitis, compared with sites without cholangitis (Figure 1, C and D), suggesting a close correlation between portal neovascularization and biliary infection.
Expression of VEGF and Its Receptors in the PCK Liver
Immunohistochemical expression of VEGF was examined using liver sections of 3-week-old, 2-month-old, and 10-month-old rats. In normal rats, weak expression of VEGF was observed in the bile duct epithelium, and hepatocytes also showed diffuse and weak positive signals of VEGF (Figure 2A). More intense expression of VEGF was observed in the bile duct epithelium of PCK rats, and such staining tended to increase with aging (Figure 2A). In PCK rats, VEGF expression of the bile duct epithelium was not uniform in a single section; in some parts, the immunohistochemical labeling of VEGF of the bile duct epithelium was accentuated at sites of intense cholangitis, relative to sites of less inflammatory cell infiltrates (Figure 2B).
Figure 2.
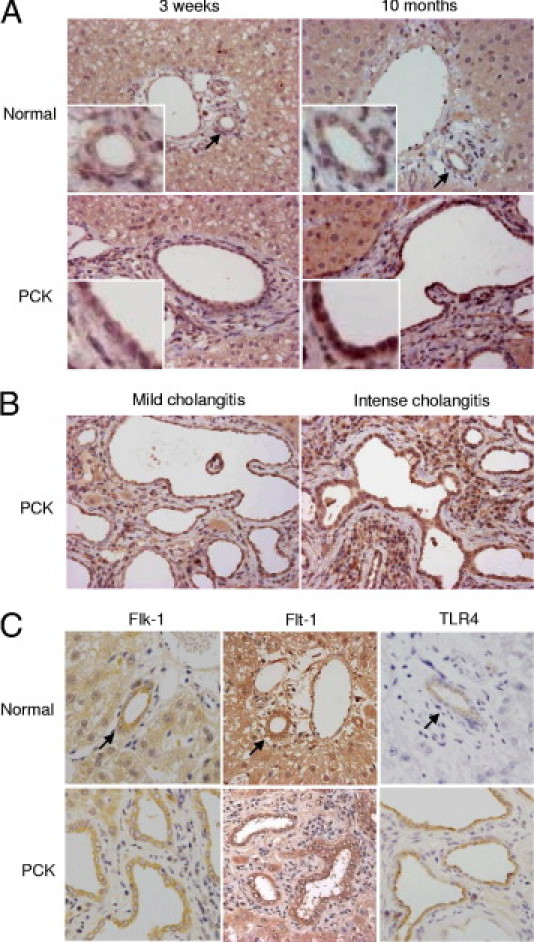
Expression of VEGF and its receptors in the PCK liver. A: The immunohistochemical expression of VEGF was more intensely observed in the bile duct epithelium of the PCK rats than in that of normal rats, and such staining tended to be increased along with aging of the PCK rats. B: In the PCK liver of 10-month-old age, there were foci in which the immunohistochemical labeling of VEGF of the bile duct epithelium was accentuated at the sites of intense cholangitis. C: Immunohistochemical analysis showed that the bile duct epithelium expressed the VEGF receptors Flk-1 and Flt-1 in both normal and PCK rats. The LPS receptor TLR4 was also expressed in the bile duct epithelium of both rats, and expression tended to be more intense in the PCK rats. Arrows indicate interlobular bile ducts of normal rats. Original magnification: ×400, main images; ×1000, insets.
Immunohistochemical analysis showed that the bile duct epithelium constitutively expressed the VEGF receptors Flk-1 and Flt-1, in both normal and PCK rats (Figure 2C). The extent of immunohistochemical expression of Flk-1 and Flt-1 in the bile duct epithelium appeared to be similar between normal and PCK rats. The LPS receptor, TLR4, was also expressed in the bile duct epithelium of both normal and PCK rats, with a tendency toward more intense expression in the PCK rats (Figure 2C).
Portal Neovascularization and VEGF Expression in the Liver of Caroli's Disease
Around the intrahepatic bile ducts of Caroli's disease, the microvessels examined using liver sections immunostained with CD31 antibody were well developed (Figure 3A). The analysis of microvessel density confirmed this tendency (Figure 3B). The immunohistochemical expression of VEGF of the bile duct epithelium appeared to be increased in Caroli's disease, compared with that of normal liver (Figure 3C).
Figure 3.
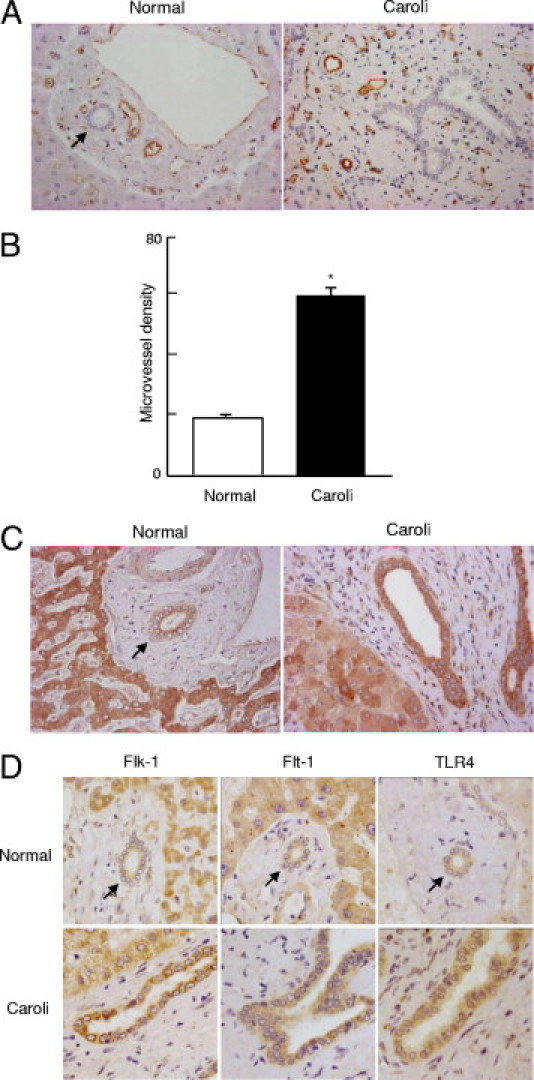
Portal neovascularization and VEGF expression in liver in Caroli's disease. A: Similar to findings from PCK rats, the microvessels were well developed around the intrahepatic bile ducts in human Caroli's disease, as examined using liver sections immunostained with CD31 antibody. B: Microvessel density was significantly higher in liver of Caroli's disease, compared with normal liver. C: Immunohistochemical expression of VEGF of the bile ducts appeared to be increased in liver of Caroli's disease. D: The bile duct epithelium was positive for immunohistochemical expression of Flk-1, Flt-1, and TLR4 in normal liver and in Caroli's disease; their expression tended to be more intense in liver of Caroli's disease. Arrows indicate interlobular bile ducts of normal liver. *P < 0.01. Original magnification, ×400.
The bile duct epithelium was positive for immunohistochemical expression of Flk-1 and Flt-1, and expression was more intense in several cases of Caroli's disease, relative to that of normal liver, which is consistent with previous report.14 In both normal liver and in Caroli's disease, TLR4 was expressed in bile duct epithelium; more intense immunohistochemical labeling was observed in liver of Caroli's disease (Figure 3D). These results were almost identical to those for PCK rats, except for immunohistochemical expression of Flk-1 and Flt-1, suggesting that pathogenesis might be similar in the rodent model and in human disease.
VEGF Expression and Effects of LPS on Its Expression in PCK Cholangiocytes
To examine the angiogenic effects of PCK cholangiocytes in association with biliary infection, cultured cholangiocytes were treated with LPS, and the expression of VEGF was examined. RT-PCR analysis confirmed that the cholangiocytes expressed TLR4, as well as Flk-1 and Flt-1 (Figure 4A). Even in the absence of LPS stimulation, PCK cholangiocytes expressed a significantly high level of VEGF mRNA, and LPS further induced the expression of VEGF mRNA in cholangiocytes (Figure 4B).
Figure 4.
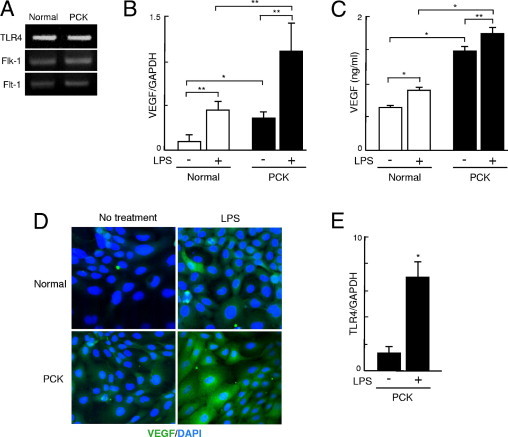
VEGF expression and effects of LPS on its expression of PCK cholangiocytes. A: Cholangiocytes were treated with LPS (10 μg/mL), and expression of VEGF was examined. RT-PCR analysis confirmed that cholangiocytes expressed TLR4 as well as Flk-1 and Flt-1. B: Real-time quantitative PCR analysis showed that PCK cholangiocytes initially overexpressed VEGF mRNA, and LPS significantly induced VEGF mRNA expression in both normal and PCK cholangiocytes at 3 hours after stimulation. C: ELISA analysis showed that the culture supernatant of PCK cholangiocytes contained a significantly high level of VEGF protein, and treatment with LPS for 24 hours further increased the VEGF level in the culture supernatant of both normal and PCK cholangiocytes. D: Immunofluorescence confocal microscopy also showed that LPS induced VEGF protein expression in both cell lines at 24 hours after LPS stimulation. E: LPS was also able to induce the expression of TLR4 mRNA of PCK cholangiocytes. *P < 0.01; **P < 0.05. Original magnification, ×1000.
The ELISA analysis showed that the culture supernatant of PCK cholangiocytes contained significantly high levels of VEGF protein, and that treatment with LPS further increased the VEGF level in the culture supernatant of both normal and PCK cholangiocytes (Figure 4C). Consistent with these findings, analysis using immunofluorescence confocal microscopy showed that LPS induced VEGF expression in both cell lines (Figure 4D).
In addition to the effects on VEGF induction in cholangiocytes, LPS was able to induce the expression of TLR4 mRNA in PCK cholangiocytes (Figure 4E).
Effect of LPS and VEGF on Cell Proliferation in PCK Cholangiocytes
Cell proliferative activity was determined at 24, 72, and 120 hours after stimulation with LPS and VEGF, using the WST-1 assay. At 72 hours after LPS stimulation, cell proliferative activity was significantly higher in PCK cholangiocytes, compared with that of the untreated groups (Figure 5A). In both normal and PCK cholangiocytes, LPS significantly induced cell proliferative activity at 120 hours after stimulation (Figure 5A).
Figure 5.
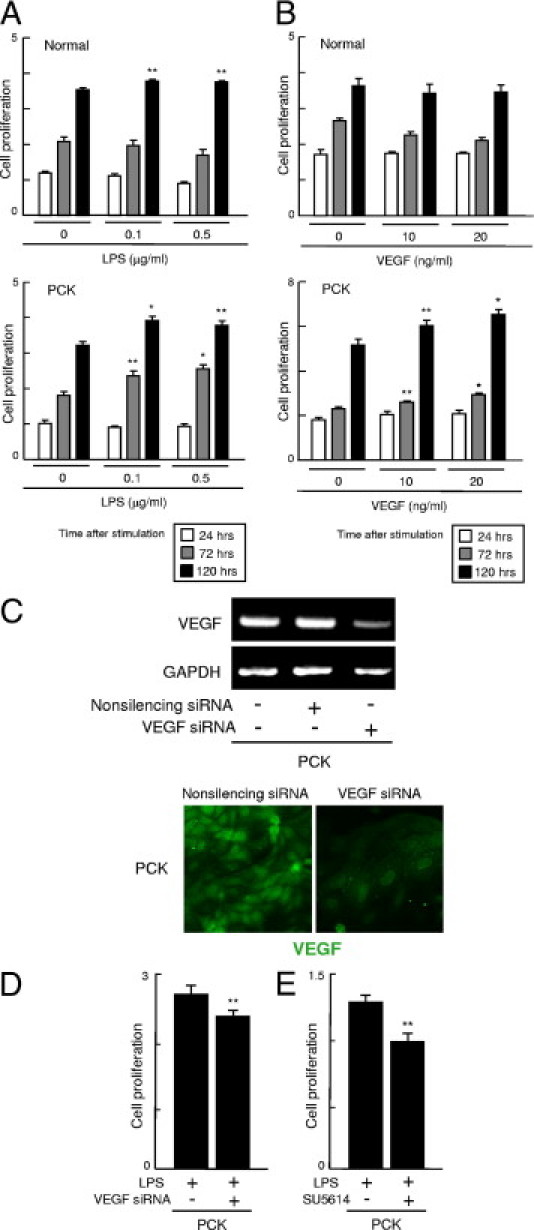
Effect of LPS and VEGF on cell proliferation of PCK cholangiocytes. A: Normal and PCK cholangiocytes were treated with LPS and VEGF at the concentrations indicated, and cell proliferative activity was determined using the WST-1 assay. In both normal and PCK cholangiocytes, LPS significantly induced cell proliferative activity of PCK cholangiocytes at 72 hours after stimulation, and it was significantly increased in both normal and PCK cholangiocytes at 120 hours. B: VEGF significantly increased cell proliferative activity of PCK cholangiocytes at 72 and 120 hours after stimulation, but did not affect cell proliferation in normal cholangiocytes at the concentrations of VEGF tested. C: VEGF siRNA reduced the expression of VEGF mRNA and VEGF protein in PCK cholangiocytes, which was evaluated using RT-PCR and immunofluorescence confocal microscopy, respectively. D: Cell proliferative activity was significantly inhibited by treatment with VEGF siRNA at 48 hours after LPS treatment. E: Treatment of PCK cholangiocytes with the VEGF receptor tyrosine kinase inhibitor SU5614 also significantly reduced cell proliferative activity at 48 hours after LPS treatment. *P < 0.01; **P < 0.05 versus untreated experimental groups at the same time period after stimulation (A and B); **P < 0.05 versus untreated experimental groups with VEGF siRNA (D) and SU5614 (E). Original magnification, ×1000 (C).
VEGF also significantly increased cell proliferative activity of PCK cholangiocytes at 72 and 120 hours after stimulation, but cell proliferative activity was not increased in normal cholangiocytes at the concentrations of VEGF tested (Figure 5B).
To determine the effects of VEGF on cell proliferative activity, siRNA against VEGF was used. siRNA reduced VEGF mRNA and protein expression of the cholangiocytes, although their expression was not completely diminished (Figure 5C). As expected, cell proliferative activity of PCK cholangiocytes was significantly inhibited by VEGF siRNA (Figure 5D). The effects of VEGF on the induction of cell proliferative activity was further confirmed by studies using an inhibitor of VEGF receptor tyrosine kinase, SU5614 (Figure 5E).
Angiogenic Effects of PCK Cholangiocytes
The culture supernatant of PCK cholangiocytes contained significantly high levels of VEGF (Figure 4C). To further address the angiogenic effects of PCK cholangiocytes, the culture supernatant of cholangiocytes of normal and PCK rats was added to the dishes in which RAOECs were cultured, and the cell proliferative activity of the RAOECs was determined. Both normal and PCK culture supernatant significantly induced cell proliferative activity of RAOECs, compared with that of the basal medium only; the culture supernatant of PCK cholangiocytes had more prominent effects on the induction of cell proliferative activity, compared with normal cholangiocyte supernatant (Figure 6A).
Figure 6.
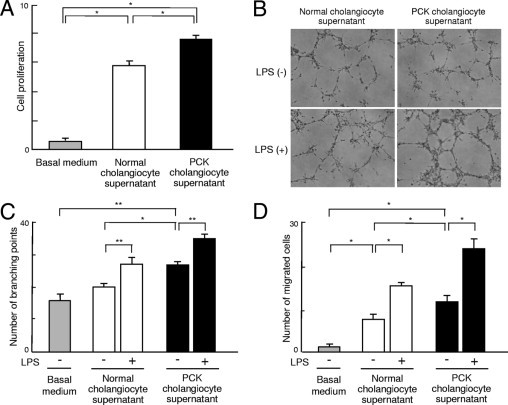
Angiogenic effects of PCK cholangiocytes. A: Rat aorta endothelial cells (RAOECs) were cultured in the presence of the culture supernatant of normal and PCK cholangiocytes, and cell proliferative activity was measured using the WST-1 assay. The culture supernatant of PCK cholangiocytes had more prominent effects on the induction of cell proliferative activity of RAOECs compared with that of normal rats. B and C: Tube formation assay using RAOECs demonstrated that the addition of PCK cholangiocyte culture supernatant increased the branching pattern of growth of RAOECs (B) (phase-contrast microscopy), and quantitative analysis of the number of branching points confirmed this tendency (C). D: Analysis using the cell migration chamber showed that the cholangiocyte culture supernatant significantly increased cell migration activity; the PCK culture supernatant had more prominent effects. The addition of LPS-treated cholangiocyte culture supernatant further increased both branching (B and C) and cell migration activity (D) of RAOECs. *P < 0.01; **P < 0.05. Original magnification, ×100.
The tube formation assay using RAOECs demonstrated that the addition of PCK cholangiocyte culture supernatant induced the branching pattern of growth of RAOECs (Figure 6B), and the quantitative analysis of the number of branching points showed that the PCK cholangiocyte culture supernatant significantly increased this pattern, compared with the normal supernatant (Figure 6C). Notably, the treatment of RAOECs with the cholangiocyte culture supernatant obtained after 24-hour LPS treatment further accelerated the branching of RAOECs (Figure 6, B and C).
In the analysis using a cell migration chamber, the addition of cholangiocyte culture supernatant significantly increased cell migration activity of RAOECs; the PCK culture supernatant had more prominent effects than the normal supernatant (Figure 6D). Again, the addition of LPS-treated cholangiocyte culture supernatant further increased the migration activity of RAOECs (Figure 6D).
Cell Signaling Pathways Involved in VEGF Expression in PCK Cholangiocytes
In both normal and PCK cholangiocytes, LPS induced NF-κB activation (Figure 7A). Under immunofluorescence confocal microscopy, nuclear expression of NF-κB p50 and p65 was observed in both cell lines after LPS stimulation, and the NF-κB inhibitor isohelenin inhibited nuclear translocation of NF-κB p50 and p65 (Figure 7B). Despite inactivation of NF-κB by isohelenin, the expression of VEGF induced by LPS was unaffected (Figure 7C).
Figure 7.
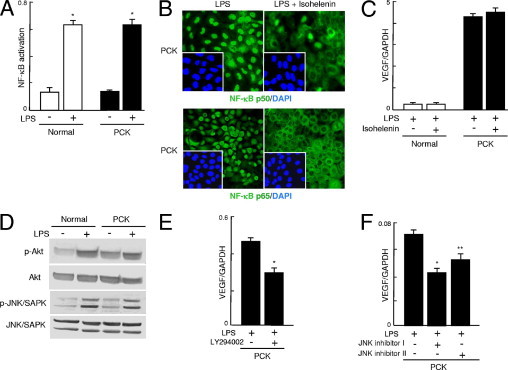
Cell signaling pathways involved in the VEGF expression of PCK cholangiocytes. A: After stimulation with LPS (10 μg/mL), activation of NF-κB was examined as described under Materials and Methods. LPS induced NF-κB activation in both normal and PCK cholangiocytes at 30 minutes after stimulation. B: Under immunofluorescence confocal microscopy, nuclear expression of NF-κB p50 and p65 was observed in both cell lines after LPS stimulation, and the NF-κB inhibitor isohelenin inhibited nuclear translocation of NF-κB p50 and p65; results shown are for PCK cholangiocytes. C: Real-time quantitative PCR showed that the expression of VEGF induced by LPS was unaffected by isohelenin, despite inactivation of NF-κB. D: Western blot analysis showed that LPS induced the phosphorylation of Akt and JNK/SAPK in both cell lines. E and F: The PI3K inhibitor LY294002 reduced the expression of VEGF mRNA after LPS stimulation in PCK cholangiocytes (E), and JNK inhibitor I and JNK inhibitor II also significantly reduced the LPS-induced VEGF mRNA expression (F), as determined using the real-time quantitative PCR at 3 hours after stimulation. *P < 0.01; **P < 0.05. Original magnification, ×1000.
Western blot analysis showed that LPS induced phosphorylation of Akt and JNK/SAPK in both cholangiocyte cell lines (Figure 7D), but phosphorylation of ERK1/2 and p38 MAPK was unaffected (data not shown). The increased phosphorylation of Akt was observed in PCK cholangiocytes even in the absence of LPS stimulation (Figure 7D). The PI3K inhibitor LY294002 reduced the expression of VEGF after LPS stimulation in PCK cholangiocytes (Figure 7E). JNK inhibitor I and JNK inhibitor II also significantly reduced the LPS-induced VEGF expression in PCK cholangiocytes (Figure 7F), demonstrating the involvement of PI3K-Akt and JNK pathways in the induction of VEGF by LPS.
Discussion
In the present study, we elucidated the pathogenic significance of biliary infection in the progression of cystic dilation of the intrahepatic bile ducts in PCK rats. Microvessels around intrahepatic bile ducts were well developed in PCK rats, and this vascular development tended to be associated with pathological progression of cholangitis. LPS-induced overexpression of VEGF in PCK cholangiocytes seemed to have a close correlation with portal neovascularization, as well as with cholangiocyte overgrowth. Increased portal neovascularization and VEGF overexpression in the biliary epithelium were also observed in Caroli's disease.
The overexpression of VEGF in cholangiocytes has been demonstrated in human PKD and in a rodent model of autosomal dominant PKD.14, 16, 17 Liver cyst fluid of autosomal dominant PKD contains elevated levels of VEGF, and the cyst fluid induces vascular endothelial cell proliferation.18, 19 The contribution of signaling pathways involving ERK1/2 and mammalian target of rapamycin has been implicated in liver cyst progression of autosomal dominant PKD.16, 17 Similarly, in the kidney, overexpression of VEGF in the renal cyst epithelium has been implicated in cyst pathogenesis and pericystic hypervascularity in human PKD and also in a rodent model of PKD (Han:SPRD rat).20 Although the significance of VEGF in PKD pathogenesis is being established, the association between VEGF expression and biliary infection has not been previously studied in autosomal recessive PKD or in autosomal dominant PKD.
LPS induced VEGF overexpression in PCK cholangiocytes in the present study. Because LPS induces the expression of VEGF through the TLR4-NF-κB signaling pathway in certain types of cells,21, 22 we examined the involvement of the signaling pathway in the induction of VEGF in cholangiocytes. As expected, LPS induced the activation of NF-κB in cholangiocytes. However, the NF-κB inhibitor isohelenin failed to inhibit the LPS-induced overexpression of VEGF in PCK cholangiocytes, even though it inhibited the nuclear translocation of NF-κB p50 and p65. These results suggest the involvement of the signaling pathways other than the TLR4-NF-κB pathway in the induction of VEGF expression in cholangiocytes.
Our data indicate that LPS-induced VEGF expression is mediated by PI3K-Akt and JNK. Although it is unclear whether the LPS-induced VEGF expression is a direct or an indirect effect of LPS, one possibility is that LPS induces bioactive molecules that in turn act as an inducer of VEGF in cholangiocytes. Tumor necrosis factor α (TNF-α) and cyclooxygenase-2 are two such candidate molecules that can induce VEGF via the phosphorylation of Akt.23, 24, 25 Indeed, we have confirmed the up-regulation of TNF-α mRNA in PCK cholangiocytes after LPS stimulation (unpublished data). Hypoxia-inducible transcription factors may also be associated with the induction of VEGF in cholangiocytes.20 Another possibility is that interferon regulatory factor 3, another downstream regulator of TLR4, may be involved in the process of VEGF induction in cholangiocytes.26
Several previous reports have shown that LPS stimulation can up-regulate TLR4 expression in various epithelial cells.27 Consistent with these results, LPS induced TLR4 expression in PCK cholangiocytes in vitro, and immunohistochemical analysis showed that TLR4 was up-regulated in the biliary epithelium of the PCK rats. Thus, modulation of the signaling pathways through up-regulation of TLR4 may contribute to biliary pathogenesis of PCK rats.
Cholangitis was a negligible histological finding in liver of 3-week-old PCK rats. However, the microvessel density around bile ducts was significantly higher in the 3-week-old PCK rats than in normal rats. The PCK cholangiocytes initially overexpressed VEGF in the absence of LPS stimulation, and had angiogenic effects on the vascular endothelial cell growth (Figure 4). Overexpression of VEGF was accompanied by increased phosphorylation of Akt in the cells (Figure 7D). Although the mechanism of this spontaneous overexpression of VEGF in PCK cholangiocytes remains to be examined, these findings may explain the increased microvessel density around bile ducts without cholangitis in the PCK liver.
A recent study showed that VEGF stimulates proliferation of normal rat cholangiocytes via an autocrine mechanism by phosphorylating ERK1/2.15 Although our data showed that the increase in cell proliferative activity of PCK cholangiocytes after VEGF stimulation was not mediated by phosphorylation of ERK1/2, VEGF expressed in the biliary epithelium may not only induce portal neovascularization of vascular endothelial cells via a paracrine mechanism, but may also increase cell proliferative activity of cholangiocytes via an autocrine/paracrine mechanism. Indeed, LPS induced PCK cholangiocyte proliferation in the present study, and biliary mitogens such as interleukin-6 as well as VEGF induced in cholangiocytes by LPS might be involved in the process.28
In summary, the present study demonstrated that biliary epithelium of PCK rats overexpresses VEGF, and LPS was identified as one of the factors leading to VEGF up-regulation in cholangiocytes. VEGF secreted from the biliary epithelium by LPS may lead to overgrowth of cholangiocytes due to hypervascularity around the bile ducts; concurrently, LPS and VEGF act as a cell proliferative factor for cholangiocytes. Thus, biliary infection is a possible exacerbating factor for biliary cystogenesis through the induction of VEGF in cholangiocytes of the PCK rats. Similar mechanisms may also exist in the pathogenesis of Caroli's disease.
References
- 1.Nakanuma Y., Terada T., Ohta G., Kurachi M., Matsubara F. Caroli's disease in congenital hepatic fibrosis and infantile polycystic disease. Liver. 1982;2:346–354. doi: 10.1111/j.1600-0676.1982.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 2.Nakanuma Y., Harada K., Sato Y., Ikeda H. Recent progress in the etiopathogenesis of pediatric biliary disease, particularly Caroli's disease with congenital hepatic fibrosis and biliary atresia. Histol Histopathol. 2010;25:223–235. doi: 10.14670/HH-25.223. [DOI] [PubMed] [Google Scholar]
- 3.Harris P.C., Torres V.E. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yonem O., Bayraktar Clinical characteristics of Caroli's syndrome. World J Gastroenterol. 2007;13:1934–1937. doi: 10.3748/wjg.v13.i13.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanzen T., Harada K., Yasoshima M., Kawamura Y., Ishibashi M., Nakanuma Y. Polycystic kidney rat is a novel animal model of Caroli's disease associated with congenital hepatic fibrosis. Am J Pathol. 2001;158:1605–1612. doi: 10.1016/S0002-9440(10)64116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato Y., Harada K., Kizawa K., Sanzen T., Furubo S., Yasoshima M., Ozaki S., Ishibashi M., Nakanuma Y. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli's disease. Am J Pathol. 2005;166:49–60. doi: 10.1016/S0002-9440(10)62231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato Y., Harada K., Furubo S., Kizawa K., Sanzen T., Yasoshima M., Ozaki S., Isse K., Sasaki M., Nakanuma Y. Inhibition of intrahepatic bile duct dilation of the polycystic kidney rat with a novel tyrosine kinase inhibitor gefitinib. Am J Pathol. 2006;169:1238–1250. doi: 10.2353/ajpath.2006.051136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yasoshima M., Sato Y., Furubo S., Kizawa K., Sanzen T., Ozaki S., Harada K., Nakanuma Y. Matrix proteins of basement membrane of intrahepatic bile ducts are degraded in congenital hepatic fibrosis and Caroli's disease. J Pathol. 2009;217:442–451. doi: 10.1002/path.2472. [DOI] [PubMed] [Google Scholar]
- 9.Onori P., Franchitto A., Mancinelli R., Carpino G., Alvaro D., Francis H., Alpini G., Gaudio E. Polycystic liver disease. Dig Liver Dis. 2010;42:261–271. doi: 10.1016/j.dld.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim C.O., Huh A.J., Kim M.S., Chin B.S., Han S.H., Choi S.H., Jeong S.J., Choi H.K., Choi J.Y., Song Y.G., Kim J.M. LPS-induced vascular endothelial growth factor expression in rat lung pericytes. Shock. 2008;30:92–97. doi: 10.1097/SHK.0b013e31815d19ad. [DOI] [PubMed] [Google Scholar]
- 11.Meng X., Brown J.M., Ao L., Shames B.D., Banerjee A., Harken A.H. Reduction of infarct size in the rat heart by LPS preconditioning is associated with expression of angiogenic growth factors and increased capillary density. Shock. 1999;12:25–31. doi: 10.1097/00024382-199907000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Harada K., Ohira S., Isse K., Ozaki S., Zen Y., Sato Y., Nakanuma Y. Lipopolysaccharide activates nuclear factor-kappaB through toll-like receptors and related molecules in cultured biliary epithelial cells. Lab Invest. 2003;83:1657–1667. doi: 10.1097/01.lab.0000097190.56734.fe. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda H., Sasaki M., Ishikawa A., Sato Y., Harada K., Zen Y., Kazumori H., Nakanuma Y. Interaction of Toll-like receptors with bacterial components induces expression of CDX2 and MUC2 in rat biliary epithelium in vivo and culture. Lab Invest. 2007;87:559–571. doi: 10.1038/labinvest.3700556. [DOI] [PubMed] [Google Scholar]
- 14.Fabris L., Cadamuro M., Fiorotto R., Roskams T., Spirli C., Melero S., Sonzogni A., Joplin R.E., Okolicsanyi L., Strazzabosco M. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 15.Gaudio E., Barbaro B., Alvaro D., Glaser S., Francis H., Ueno Y., Meininger C.J., Franchitto A., Marzioni M., Taffetani S., Fava G., Stoica G., Venter J., Rechenbach R., De Morrow S., Summers R., Alpini G. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–1282. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 16.Spirli C., Okolicsanyi S., Fiorotto R., Fabris L., Cadamuro M., Lecchi S., Tian X., Somlo S., Strazzabosco M. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2010;138 doi: 10.1053/j.gastro.2009.09.005. 360–371.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spirli C., Okolicsanyi S., Fiorotto R., Fabris L., Cadamuro M., Lecchi S., Tian X., Somlo S., Strazzabosco M. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology. 2010;51:1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols M.T., Gidey E., Matzakos T., Dahl R., Stiegmann G., Shah R.J., Grantham J.J., Fitz J.G., Doctor R.B. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 19.Brodsky K.S., McWilliams R.R., Amura C.R., Barry N.P., Doctor R.B. Liver cyst cytokines promote endothelial cell proliferation and development. Exp Biol Med (Maywood) 2009;234:1155–1165. doi: 10.3181/0903-RM-112. [DOI] [PubMed] [Google Scholar]
- 20.Bernhardt W.M., Wiesener M.S., Weidemann A., Schmitt R., Lechler P., Campean V., Ong A.C., Willam C., Gretz N., Eckardt K.U. Involvement of hypoxia-inducible transcription factors in polycystic kidney disease. Am J Pathol. 2007;170:830–842. doi: 10.2353/ajpath.2007.060455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pei Z., Li H., Guo Y., Jin Y., Lin D. Sodium selenite inhibits the expression of VEGF, TGFbeta(1) and IL-6 induced by LPS in human PC3 cells via TLR4-NF-(k) B signaling blockage. Int Immunopharmacol. 2010;10:50–56. doi: 10.1016/j.intimp.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Novotny N.M., Markel T.A., Crisostomo P.R., Meldrum D.R. Differential IL-6 and VEGF secretion in adult and neonatal mesenchymal stem cells: role of NFkB. Cytokine. 2008;43:215–219. doi: 10.1016/j.cyto.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 23.Park D.W., Baek K., Lee J.G., Park Y.K., Kim J.H., Kim J.R., Baek S.H. Activation of toll-like receptor 4 modulates vascular endothelial growth factor synthesis through prostacyclin-IP signaling. Biochem Biophys Res Commun. 2007;362:1090–1095. doi: 10.1016/j.bbrc.2007.08.119. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida S., Ono M., Shono T., Izumi H., Ishibashi T., Suzuki H., Kuwano M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol. 1997;17:4015–4023. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koyama S., Sato E., Tsukadaira A., Haniuda M., Numanami H., Kurai M., Nagai S., Izumi T. Vascular endothelial growth factor mRNA and protein expression in airway epithelial cell lines in vitro. Eur Respir J. 2002;20:1449–1456. doi: 10.1183/09031936.02.00089802. [DOI] [PubMed] [Google Scholar]
- 26.Korherr C., Gille H., Schafer R., Koenig-Hoffmann K., Dixelius J., Egland K.A., Pastan I., Brinkmann U. Identification of proangiogenic genes and pathways by high-throughput functional genomics: tBK1 and the IRF3 pathway. Proc Natl Acad Sci USA. 2006;103:4240–4245. doi: 10.1073/pnas.0511319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei Z., Lin D., Song X., Li H., Yao H. TLR4 signaling promotes the expression of VEGF and TGFβ1 in human prostate epithelial PC3 cells induced by lipopolysaccharide. Cell Immunol. 2008;254:20–27. doi: 10.1016/j.cellimm.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Park J., Gores G.J., Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–1043. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]