

Abstract
Although kidney functions deteriorate with age, little is known about the general morphological alterations and mechanisms of renal senescence. We hypothesized that carbonyl stress causes senescence and investigated the possible role of glyoxalase I (GLO1), which detoxifies precursors of advanced glycation end products in the aging process of the kidney. We observed amelioration of senescence in GLO1-transgenic aged rats (assessed by expression levels of senescence markers such as p53, p21WAF1/CIP1, and p16INK4A) and a positive rate of senescence-associated β-galactosidase (SABG) staining, associated with reduction of renal advanced glycation end product accumulation (estimated by the amount of carboxyethyl lysine). GLO1-transgenic rats showed amelioration of interstitial thickening (observed as an age-related presentation in human renal biopsy specimens) and were protected against age-dependent decline of renal functions. We used GLO1 overexpression or knockdown in primary renal proximal tubular epithelial cells to investigate the effect of GLO1 on cellular senescence. Senescence markers were significantly up-regulated in renal proximal tubular epithelial cells at late passage and in those treated with etoposide, a chemical inducer of senescence. GLO1 cellular overexpression ameliorated and knockdown enhanced the cellular senescence phenotypes. Furthermore, we confirmed the association of decreased GLO1 enzymatic activity and age-dependent deterioration of renal function in aged humans with GLO1 mutation. These findings indicate that GLO1 ameliorates carbonyl stress to retard renal senescence.
Prevention of aging and increase in longevity have long been dreamt of, and many hypotheses to explain the complex process of aging have been advanced. Primary somatic cells grown in vitro do not proliferate indefinitely; rather, after a period of rapid proliferation, the cell division rate slows and ultimately ceases altogether, with the cells becoming unresponsive to mitogenic stimuli, a phenomenon known as replicative senescence or Hayflick's limit. Senescent cells have characteristic features, notably the accumulation of advanced glycation end products (AGEs), wide changes in gene expression (including up-regulation of cell cycle regulators such as p53, p21WAF1/CIP1, and p16INK4A), and increased senescence-associated β-galactosidase (SABG) activity.1, 2 Irrespective of chronological aging, cells exhibiting the combination of these features are considered to be in premature senescence.3
The mechanisms of senescence have been widely investigated. The free radical theory of aging proposes that endogenous reactive oxygen species (ROS) generated in cells result in cumulative damage and subsequent senescence.4 However, attempts to induce longevity by eliminating oxidative stress in genetically engineered mice have been unsuccessful, suggesting that involvement of free radicals is not a sufficient explanation of senescence.5, 6 Another possible mechanism accounting for senescence is carbonyl stress and AGE formation. Carbonyl compound derivatives are generated by carbohydrate oxidation and glycolysis.7 AGE formation is caused by carbonyl compound derivatives as protein modification precursors.8, 9 Among the many carbonyl derivatives, the most reactive are thought to be glyoxal and methylglyoxal, which act as protein modification precursor and synergistically contribute to AGE formation.10 Serum AGE levels were reported to increase with age in a large cohort of normal subjects, and correlated well with levels of established markers of oxidative stress and inflammation.11 AGE accumulation is associated with the development of age-related diseases, such as diabetes mellitus, Alzheimer's disease, and atherosclerosis,12, 13 and carbonyl stress is likely to be, at least in part, a cause rather than a consequence of the aging process.14
Assuming that glyoxal and methylglyoxal accelerate senescence, acceleration of their decomposition might prevent organs from aging. Both compounds are rapidly decomposed by GLO1 and GLO2, the two enzymes of the glyoxalase (GLO) system. GLO1 catalyzes the conversion of glyoxal and methylglyoxal and reduces glutathione (GSH) to S-d-lactoyl-GSH; it is expressed ubiquitously in most tissues, serving as the rate-limiting enzyme of methylglyoxal catabolism.15, 16, 17, 18 In one study, overexpression of GLO1 protected the kidney against ischemia and reperfusion injury by inhibiting the formation of intracellular methylglyoxal adducts and oxidative stress.19
In the present study, we investigated the possible roles of carbonyl stress and of GLO1 in renal senescence.
Materials and Methods
Animal Experimental Protocol
Male Wistar rats [wild type (WT)] and male transgenic Wistar rats overexpressing human GLO1 (Tg-GLO1) were used. The Tg-GLO1 rats were generated as described previously.19, 20 Rats 10 weeks of age or 14 months of age were fed a standard moderate-fat diet and tap water ad libitum (32 total; n = 8 for each age/genotype combination). Food intake was monitored from 2 weeks before sacrifice, and body weight was measured at the time of sacrifice. Systolic blood pressure of awake rats was measured by the tail-cuff method using a Softron BP-98A unit (Softron, Tokyo, Japan). All animal experiments were performed in accordance with the NIH guidelines (7th edition) for use and care of laboratory animals and were approved by the local ethical committees.
Histological Analysis for Age-Related Morphological Change
Tissues from rat and human kidney were fixed in formalin solution and paraffin-embedded. Sections (3 μm thick) were stained with Masson's trichrome staining for rat tissue or with Azan staining for human tissue. Interstitial thickness was measured at a magnification of ×600 as the smallest width of the interstitial area (stained blue) between two intact tubules in 20 sites selected randomly from six fields of renal cortex. This smallest-width measurement was chosen to account for tubules not perpendicular to the cutting plane.
Human renal biopsy specimens obtained from patients admitted to the University of Tokyo Hospital were reviewed with institutional permission (Graduate School of Medicine, University of Tokyo, Permission no. 2671) and with informed consent. Exclusion criteria included confounding factors such as proteinuria [urinary protein > 1 g/g creatinine (Cr)], renal dysfunction (estimated glomerular filtration rate eGFR < 60 mL/min per 1.73 m2), interstitial nephritis of primary or secondary origin, and diabetes mellitus. The human renal biopsy specimens were mostly from patients with IgA nephropathy, and comparison of glomerular change was not performed because this variable was dependent on disease activity. In all, 22 patients were recruited.
Chemical Analysis
Creatinine concentration was measured by the enzyme method using a Kainos Cre kit (Kainos Laboratories, Tokyo, Japan). Protein concentration was measured by the Lowry method, using DC protein assay reagents (Bio-Rad Laboratories, Hercules, CA). Rat blood or urine samples collected for 24 hours in metabolic cages were used after centrifugation for 5 minutes at 3000 × g. Blood glucose level was measured using an automated blood glucose meter (Glutest Ace; Sanwa Chemical, Nagoya, Japan). Blood insulin level was measured using a rat insulin ELISA kit (AKRIN-010T; Shibayagi, Gunma, Japan). HbA1c was measured by high performance liquid chromatography.
Determination of GLO1 Activity and GSH Concentrations
Renal cortex of rats (30 mg) or 104 to 105 renal proximal tubular epithelial cells (RPTECs) were homogenized in, respectively, 0.5 mL or 50 μL of sodium phosphate buffer, pH 7.0, containing 0.02% Triton X-100 surfactant, then sonicated for 1 minute, and finally centrifuged at 20,000 × g for 1 minute at 4°C. The supernatant was used for assessment of GLO1 activity by spectrophotometry, as described previously.21 GSH concentration in renal cortical homogenates from rats was measured with Bioxytech GSH-412 assay reagent (Oxis International, Portland, OR). Values were adjusted by the protein concentration of the same sample.
Determination of Total Superoxide Dismutase Activity
Total superoxide dismutase (SOD) activity in renal tissue was determined using a superoxide dismutase assay kit (Cayman Chemical, Ann Arbor, MI). Sample preparation and measurement were performed according to the manufacturer's instructions. Values were adjusted by the protein concentration of the same sample.
Senescence-Associated β-Galactosidase Staining Assay
Staining for senescence-associated β-galactosidase (SABG), one of the senescence markers, was performed using a Chemicon cellular senescence assay kit (Millipore, Billerica, MA). For tissue staining, frozen 15-μm kidney sections were incubated for 5 hours. The SABG-positive (blue-stained) areas of whole renal cortex were measured at ×100 magnification. The SABG-positive rate of tissue was calculated using ImageJ software (version 1.38) (NIH, Bethesda, MD) with adequate setting of threshold and computed measurement of area fraction, obtained after analyzing particles. Eight areas of each kidney section were used for statistical assessment. For costaining of SABG staining and γ-GTP staining or immunohistochemical staining, frozen 10-μm kidney sections were used. SABG staining, with 12 hours of incubation, was performed prior to immunohistochemical staining.
Immunoblot Analysis
Proteins were separated by 10% to 15% SDS-PAGE under reducing conditions. Monoclonal mouse anti-p53 IgG (Calbiochem Ab-1, 1:100; EMD Chemicals, Cambridge, MA), monoclonal mouse anti-p16INK4A IgG (sc-1661, 1:200; Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal mouse anti-CEL IgG (1:100),22 and polyclonal rabbit anti-β actin IgG (A2066, 1:1000; Sigma-Aldrich, St. Louis, MO) were used as primary antibodies, and horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (170–6516 for anti-mouse and 170–6516 for anti-rabbit, 1:1000; Bio-Rad Laboratories) were used as secondary antibodies. The ECL Plus Western blotting system (GE Healthcare, Piscataway, NJ) was used for detection. Reproducibility was confirmed in three independent experiments; representative data are presented in the figures.
Immunohistochemistry
The following primary antibodies were used: polyclonal rabbit anti-GLO1 IgG (12 μg/mL, obtained from immunization against synthetic peptide of rat GLO1, GIAVPDVYEA, which cross-reacts with the human GLO1 epitope, GIAVPDVYSA); anti-aquaporin2 (AQP2) IgG (Chemicon AB3274) from Millipore; anti-p53 IgG (sc-6243), monoclonal mouse anti-p21WAF1/CIP1 IgG (sc-6246), and anti-p16INK4A IgG (sc-1661) from Santa Cruz Biotechnology; and anti-calbindin-D-28K IgG (c-9848) from Sigma-Aldrich. Polyclonal horse biotinylated anti-mouse IgG antibody or polyclonal goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) was used as secondary antibody. Reproducibility was confirmed in three independent experiments; representative data are reported in the results.
Real-Time Quantitative PCR
The primers used for real-time quantitative PCR were as follows: human GLO1 5′-ATTCGGTCATATTGGAATTGC-3′ (forward), 5′-TTCAATCCAGTAGCCATCAGG-3′ (reverse); human p53 5′-CCTCACCATCATCACACTGG-3′ (forward), 5′-TCTGAGTCAGGCCCTTCTGT-3′ (reverse); human p21WAF1/CIP1 5′-TGGAGACTCTCAGGGTCGAAA-3′ (forward), 5′-GGCGTTTGGAGTGGTAGAAATC-3′ (reverse); human p16INK4A 5′-CAACGCACCGAATAGTTACG-3′ (forward), 5′-AGCACCACCAGCGTGTC-3′ (reverse); human β-actin 5′-TCCCCCAACTTGAGATGTATGAAG-3′ (forward), 5′-AACTGGTCTCAAGTCAGTGTACAGG-3′ (reverse); rat p53 5′-CCTCAATAAGCTGTTCTGCC-3′ (forward), 5′-AAAAGTCTGCCTGTCGTCCA-3′ (reverse); rat p21WAF1/CIP1 5′-ACGTGGCCTTGTCGCTGTCTT-3′ (forward), 5′-TAAGGCAGAAGATGGGGAAGAG-3′ (reverse); rat p16INK4A 5′-ACGAGGTGCGGGCACTG-3′ (forward), 5′-TTGACGTTGCCCATCATCATC-3′ (reverse); rat β-actin 5′-CTTTCTACAATGAGCTGCGTG-3′ (forward), 5′-TCATGAGGTAGTCTGTCAGG-3′ (reverse); rat Mn-SOD 5′-CCGAGGAGAAGTACCACGAG-3′ (forward), 5′-GCTTGATAGCCTCCAGCAAC-3′ (reverse). Amplification specificity was verified by agarose gel electrophoresis of the PCR products. Expression data were normalized to β-actin using the ΔΔCt method.
Cell Culture
Primary human RPTECs derived from a 27-year-old woman of European origin were purchased from Clonetics (Walkersville, MD). The cells were cultured with renal epithelial basal medium (Clonetics). Early- and late-passage cells were compared at passages 3 and 10, respectively, and cells of passage 3 to 4 were used for transfection experiments. Confluent RPTEC monolayers were passaged at 1:4. Each experiment with RPTECs was performed three or more times independently.
Transfection
cDNA of human GLO1 (555 bp) was obtained using the following primers: 5′-CAGGCTAGCCATGGCAGAACCGCAGCC-3′ (forward), 5′-GGAGAATTCTCACAGCACTACATTAAG-3′ (reverse). Plasmid vector pcDNA3.1(−) (Invitrogen, Carlsbad, CA) was used for overexpression of GLO1. GLO1 containing pcDNA3.1(−) was constructed by inserting the complete human form GLO1 cDNA into the EcoRI and NheI sites. For overexpression experiments, 400 ng/cm2 of GLO1-containing vector or empty vector was transfected into RPTECs by 5 hours of incubation with Lipofectamine 2000 transfection reagent (Invitrogen). Cells were exposed to 1 μmol/L etoposide for 24 hours if necessary. Transfection efficiency was confirmed by transfection of LacZ-containing vector followed by in situ β-galactosidase staining solution, using empty vector as control.
Knockdown Study
Knockdown study was performed using Stealth Select RNAi purchased from Invitrogen. Sequences for GLO1 were as follows: 5′-UUAGCGUCAUUCCAAGAACUCUAGU-3′ (siRNA 1), 5′-AAUCCAGUAGCCAUCAGGAUCUUGA-3′ (siRNA 2). Stealth RNAi negative control (universal negative control siRNA) from Invitrogen was used as scrambled siRNA. Knockdown efficiency was confirmed by real-time quantitative PCR with primers of human GLO1.
Cellular Proliferation and Viability Test
The number of viable cells was determined using a colorimetric MTS formazan assay with CellTiter 96 AQueous One Solution cell proliferation assay (Promega, Madison, WI). Cells (103) were seeded with renal epithelial basal medium, with or without etoposide exposure.
For the lactate dehydrogenase (LDH) assay, 104 cells at early or late passage with or without etoposide exposure were seeded into each well of a 24-well plate with renal epithelial basal medium and were incubated for 24 hours. Cells were lysed with 0.1% Triton X-100 surfactant, and 2 μL of cell lysate and 200 μL of culture medium were used for each measurement. LDH activity was measured using a Kainos LDH kit (Kainos Laboratories).
Bromodeoxyuridine (BrdU) uptake was measured using a BrdU detection kit III (Roche Diagnostics, Indianapolis, IN). Cells (104) cells at passage 3 were seeded in each well of a 96-well plate and were cultured with serum-free Dulbecco's modified Eagle's medium for 24 hours, with etoposide added after transfection if necessary.
γ-Glutamyl Transpeptidase Staining
γ-Glutamyl transpeptidase (GTP) staining was performed according to the method of Rutenburg et al.23 Duration of incubation for staining was 10 minutes at room temperature. No fixation was done before staining.
Immunofluorescence Study
Cells were fixed in 50% methanol/50% acetone and then were incubated with anti-CEL antibody (KNH-30; TransGenic, Kobe, Japan) as a primary antibody. Fluorescein isothiocyanate-conjugated polyclonal rabbit anti-mouse IgG (F0261; Dako, Glostrup, Denmark) was used as a secondary antibody. Nuclei were then counterstained with Hoechst 33258 dye (B2883; Sigma-Aldrich).
Renal Function and GLO1 Mutation in Human Schizophrenia Patients
For determination of eGFR, we used data from patients with schizophrenia whose GLO1 genotype had been established previously,24 with permission and informed consent. The local human ethics committee approved all protocols involving patients, and informed written consent was obtained from every patient before enrollment. Three patients had the GLO1 frameshift mutation, with GLO1 activity half that of the nonmutant form,24 and 15 age-matched control patients with WT GLO1 were randomly selected. All medical charts were reviewed for blood pressure, body weight, body height, urinary testing (hematuria, proteinuria), and medication. The eGFR was calculated using the following formula, which estimates GFR in a Japanese population25: eGFR = 194 × Cr−1.094 × Age−0.287 (× 0.739 if female).
Statistical Analysis
Data are expressed as means ± SD. Statistical differences were assessed using the unpaired Student's t-test or single-factor analysis of variance. Significant differences determined on analysis of variance were tested by post hoc comparison using Tukey's method. Values of P < 0.05 were considered statistically significant. In analysis of human renal biopsy, the slope of interstitial thickness against age was calculated by linear regression analysis with Spearman's rank correlation coefficient.
Results
Amelioration of Senescent Status in Aged Kidneys of Tg-GLO1 Rats
To study the biological roles of carbonyl stress and GLO1 in renal senescence, we used young (10-week-old) and aged (14-month-old) Tg-GLO1 and WT rats (32 total; n = 8 for each age/genotype combination).19, 20 No significant differences were seen between the WT and Tg-GLO1 groups, either young or aged, in body weight, food intake per weight, or systolic blood pressure (Table 1). Levels of random (nonfasting) blood glucose did not significantly differ between WT and Tg-GLO1 rats during the experimental period. Normal glucose metabolism in the experimental animals was confirmed by measurements of fasting blood glucose, fasting blood insulin, insulin resistance, and HbA1c. The HOMA-IR (homeostasis model of assessment-insulin resistance) index of insulin resistance was calculated by multiplying fasting blood glucose and fasting blood insulin, then dividing by 405. All of these parameters were equivalent between aged WT and Tg-GLO1 rats (Figure 1, A–E). Similarly, levels of renal cortex GSH, an essential cofactor of GLO1, did not significantly differ between them (Figure 1F). To eliminate the effect of antioxidants, which may ameliorate senescence phenotypes by decreasing ROS levels, total superoxide dismutase (SOD) activity in renal tissue was measured. Total SOD activity was not significantly changed by GLO1 overexpression in either young or aged rats (Figure 1G). In addition, transcript levels of manganese SOD (Mn-SOD, or SOD2) in kidney were measured by quantitative RT-PCR; Mn-SOD regulates only mitochondrial production of ROS because it is present only in mitochondria. No significant differences in Mn-SOD expression were observed between young and aged groups of WT and Tg-GLO1 rats (Figure 1H).
Table 1.
Body Weight, Food Intake, and Systolic Blood Pressure of Young and Elderly Wild-Type and Transgenic Rats
| Young |
Elderly |
|||
|---|---|---|---|---|
| Characteristics | WT | Tg-GLO1 | WT | Tg-GLO1 |
| Body weight (g) | 330 ± 17 | 330 ± 26 | 490 ± 56⁎ | 480 ± 54⁎ |
| Chow intake (mg/day per g weight) | 43 ± 13 | 42 ± 14 | 39 ± 12 | 42 ± 14 |
| Systolic blood pressure (mmHg) | 106 ± 4 | 104 ± 5 | 106 ± 11 | 104 ± 9 |
Significant differences were found in body weight in elderly compared with young rats, but no significant differences were found in those of the same age. No significant differences were observed in food intake or systolic blood pressure; n = 8 in each of the four categories.
P < 0.01 versus young WT.
Figure 1.
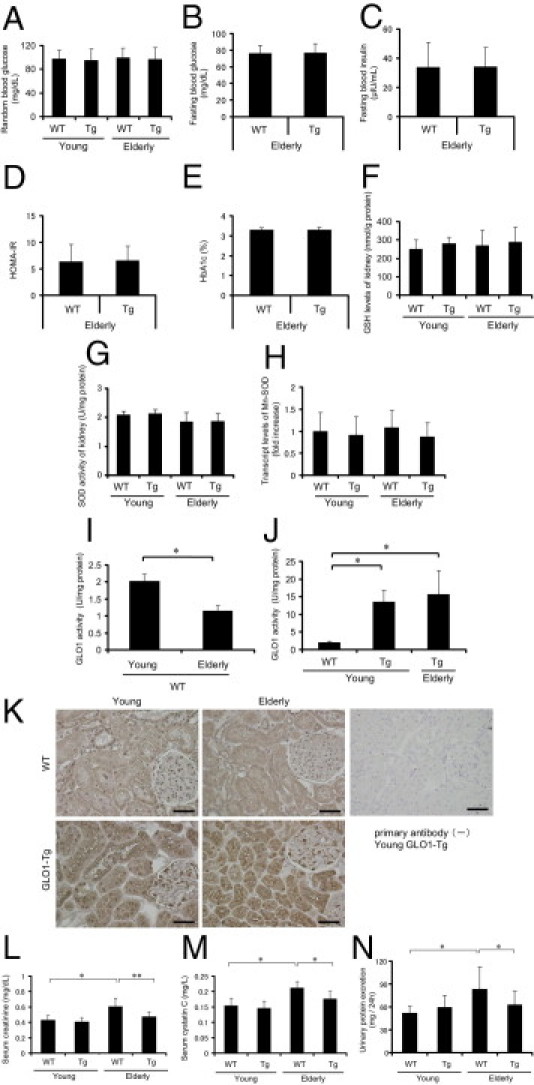
Characteristics of GLO1-overexpressing aged rats and amelioration of senescence-associated renal dysfunction and urinary protein excretion. No significant differences were observed between young and aged WT and Tg-GLO-1 rats for random (nonfasting) blood glucose levels (A), for levels of GSH, an essential cofactor of GLO1, in renal cortex lysate (data adjusted by protein concentration) (F), for total SOD activity of renal cortex lysate (data adjusted by protein concentration) (G), and for relative transcript levels of Mn-SOD (determined by real-time quantitative RT-PCR, with the value for young WT rats set as 1) (H). No significant differences were observed between aged WT and Tg-GLO-1 rats for fasting blood glucose levels (B), for fasting blood insulin levels (C), for HOMA-IR (calculated by multiplying fasting blood glucose and fasting blood insulin, then dividing by 405) (D), and for blood HbA1c levels (E). I: GLO1 activity of renal cortex lysate of WT animals decreased in an age-dependent manner. Data were adjusted by protein concentration. *P < 0.01. J: GLO1 activity of renal cortex lysate in Tg-GLO1 rats was markedly higher than that in WT rats, and did not show an age-dependent decrease. Data were adjusted by protein concentration. *P < 0.01 versus young WT rats. K: Distribution of GLO1 in rat kidney, shown in representative micrographs of immunohistochemical staining of GLO1 in the rat renal cortex. GLO1 was expressed ubiquitously in the kidney of both WT and Tg-GLO1 rats. Methyl Carnoy's fixed specimens (4 μm thick) were counterstained with hematoxylin. Original magnification = ×400. Scale bars: 50 μm. L: Serum creatinine levels determined by enzymatic assay indicate that the age-dependent deterioration of renal function was ameliorated by GLO1 overexpression. *P < 0.01; **P < 0.05. M: Serum cystatin C levels indicate that the age-dependent deterioration of renal function was ameliorated by GLO1 overexpression. *P < 0.01. N: An age-dependent increase in proteinuria was prevented by GLO1 overexpression. Urine was collected for 24 hours using metabolic cages. *P < 0.05.
In WT rats, GLO1 activity was significantly decreased with age, to 0.55-fold the level in young rats (Figure 1I). GLO1 activity of young Tg-GLO1 rats showed a 6.3-fold increase, compared with young WT rats, but did not show an age-dependent decrease (Figure 1J). Immunohistochemical analysis showed that GLO1 was ubiquitously expressed with the same distribution pattern in the kidneys of both young and aged WT rats (Figure 1K). In contrast to the comparison in WT rats which showed age-dependent weakening of signal intensity, GLO1 signal intensity in Tg-GLO1 did not change with aging. Age-dependent deterioration of renal function, as estimated by serum creatinine, cystatin C levels, and urinary protein excretion, were markedly ameliorated by overexpression of GLO1 (Figure 1, L–N).
We assessed renal senescence status in WT and Tg-GLO1 rats by SABG staining. SABG was positive only in the tubular cells of aged rats; glomerular cells remained negative (Figure 2A). The functional improvement in renal senescence described above in Tg-GLO1 rats was associated with a decrease in SABG-positive rates in cortical tubular cells. Amelioration of senescence was confirmed by quantitative analysis of the SABG-positive rate, which was lower in the kidneys of aged Tg-GLO1 rats, compared with aged WT rats (Figure 2B). No significant differences were observed between the kidneys of young Tg-GLO1 and WT rats. To determine which segments of tubules are positive for SABG staining, we performed costaining of SABG and γ-GTP (a marker of proximal tubular cells), calbindin-D-28K (a marker of distal tubules and connecting tubules), or aquaporin2 (AQP2; a marker of connecting tubules and collecting ducts) (Figure 2, C–E). All segments had a propensity toward senescence, with no preponderance in specific tubular segments. The elevated transcript levels of senescence markers such as p53, p21WAF1/CIP1, and p16INK4A in aged rats were significantly suppressed by GLO1 overexpression, compared with aged WT rats, whereas no significant differences were observed between the young groups (Figure 2F). Immunohistochemistry for the senescence markers showed that the nuclei of renal cortical tubules of aged WT rats were stained positive (in staining for p53, cytosol was also positive); these positive staining levels were markedly reduced in aged Tg-GLO1 rats (Figure 2G). Similar results were obtained by immunoblot analysis of renal cortex lysate for p53 and p16INK4A. Protein expression level of p53 and p16INK4A in the young rats was undetectable; however, the increased expression of p53 and p16INK4A with age was reduced by GLO1 overexpression (Figure 2, H and I). In parallel with this suppression of senescence markers, age-dependent accumulation of carboxyethyl lysine (CEL) in the renal cortex was markedly suppressed by overexpression of GLO1 (Figure 2J).
Figure 2.
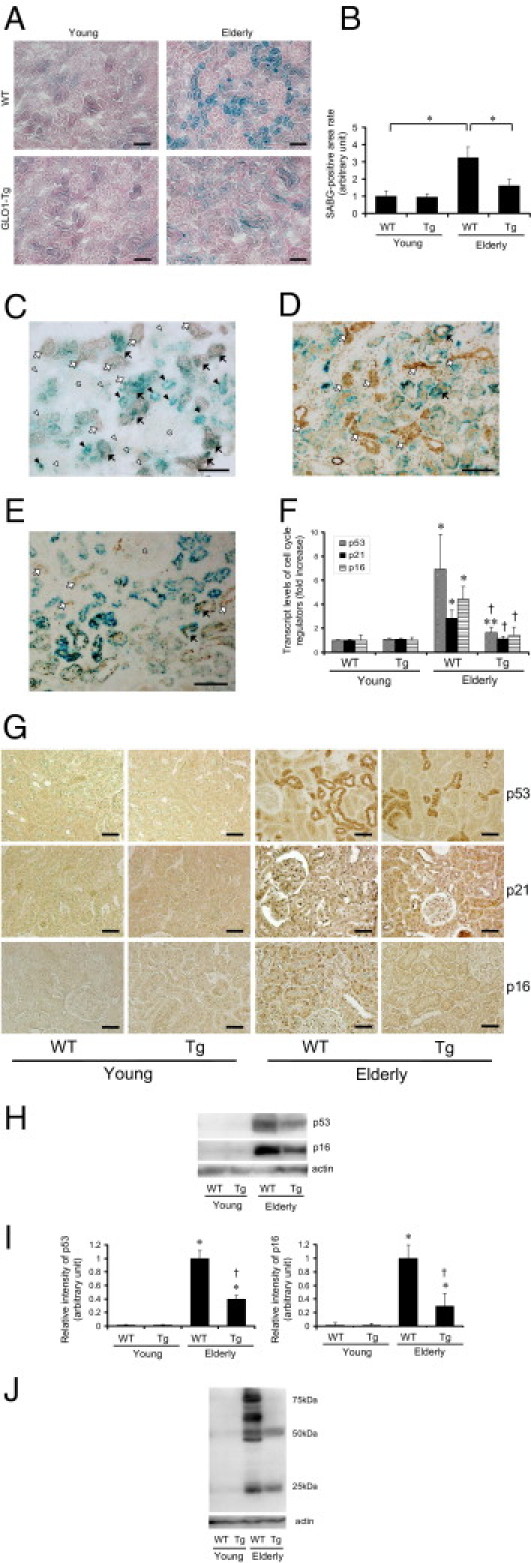
GLO1-overexpressing aged rats showed morphological amelioration in senescence-associated alterations of kidney. A: Light micrographs of rat renal cortex with SABG staining showing senescent tubular cells in aged WT rats. The number of senescent tubules was reduced by overexpression of GLO1. Frozen tissue specimens (15 μm thick) were counterstained with Nuclear Fast Red. Original magnification: ×100. Scale bars: 100 μm. Blue indicates SABG-positive cells. B: Quantitative analysis of SABG-positive area. The positive rate was calculated by dividing the positive area by the total area in each field and is expressed as fold increase, relative to the level of young WT rats. *P < 0.01. C: Representative light micrographs of renal cortex of aged WT rat, costained with SABG (blue) and γ-GTP (pale red). Black arrows mark senescent proximal tubular cells; white arrows mark nonsenescent proximal tubular cells. Black arrowheads mark senescent nonproximal tubular cells; white arrowheads mark nonsenescent nonproximal tubular cells. The letter G marks the glomerulus. Frozen specimens were examined without counterstaining. Original magnification = ×200. Scale bar = 100 μm. D: Representative light micrographs of renal cortex of aged WT rat costained with SABG (blue) and calbindin-D-28K. Black arrows mark senescent distal tubules-connecting tubules; white arrows mark nonsenescent distal tubules-connecting tubules. Frozen specimens were counterstained with hematoxylin. Original magnification = ×200. Scale bar = 100 μm. E: Representative light micrographs of renal cortex of aged WT rat costained with SABG (blue) and AQP2. Black arrows mark senescent connecting tubules-collecting ducts; white arrows mark nonsenescent connecting tubule-collecting ducts. The letter G marks the glomerulus. Frozen specimens were counterstained with hematoxylin. Original magnification = ×200. Scale bar = 100 μm. F: Relative transcript levels of p53, p21WAF1/CIP1, and p16INK4A were determined by real-time quantitative RT-PCR. All were significantly elevated in aged WT. This age-dependent increase was significantly reduced by overexpression of GLO1. The values for young WT rats were set as 1. *P < 0.01; **P < 0.05 versus young WT rats; †P < 0.01 versus aged WT rats. G: Representative micrographs of immunohistochemistry staining for p53, p21WAF1/CIP1, and p16INK4A in the rat renal cortex. In each staining, aged WT tissue exhibited marked increases in positive area in tubules. The extent of positive areas was significantly decreased by overexpression of GLO1. Methyl Carnoy's fixed specimens (4 μm thick) were counterstained with hematoxylin. Original magnification = ×400. Scale bars: 50 μm. H: Protein immunoblot of p53 and p16INK4A of renal cortex lysate. Elevation of p53 and p16INK4A in aged WT rats was attenuated by overexpression of GLO1. Actin served as a control. I: Densitometric quantification of p53 and p16INK4A immunoblot. The values for aged WT rats were set as 1. *P < 0.05 versus young WT rats; †P < 0.05 versus aged WT rats. J: Protein immunoblot of CEL of renal cortex lysate indicates amelioration of carbonyl stress by GLO1 overexpression. Actin served as a control. Many bands were detected as CEL-modified protein, indicating that AGEs were accumulated in renal cortex.
Age-Related Morphological Changes in the Renal Interstitium
To investigate whether amelioration of senescence markers and physiological parameters by overexpression of GLO1 was associated with improvement of pathological changes, we first assessed age-related morphological changes in human renal senescence, with a particular focus on tubulointerstitial changes (Figure 3A), on the basis that SABG staining was observed primarily in the tubules in aged rats. Review of specimens (mostly IgA nephropathy specimens with Azan staining) from 22 patients whose renal functions were preserved (eGFR of 80.4 ± 3.5 mL/min per 1.73m2 and urinary protein excretion of 0.76 ± 0.21 g/day) showed an age-dependent increase in the thickness of the interstitium (Figure 3B). Linear regression analysis showed a strongly positive Pearson's correlation coefficient between age and the thickness of the renal peritubular interstitium (slope, 0.044 μm/year; r2 = 0.91, P < 0.01).
Figure 3.
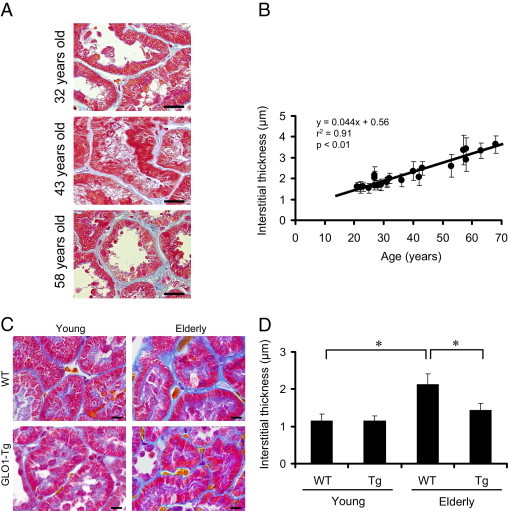
Interstitial thickening was demonstrated to be an age-related morphological change in human and rat kidney. A: Representative figures of human renal biopsy. Specimens were Azan-stained. Proximal tubular epithelial cells stain red and interstitium stains blue. The interstitial areas were thickened in an age-dependent manner. Original magnification = ×600. Scale bars: 20 μm. B: Correlation between age and interstitial thickness. The thickness of renal interstitia (blue) was measured under high-power magnification (×600). Each dot on the univariate linear regression line represents mean thickness (μm); error bars indicate ± SD. The calculation method is described under Materials and Methods. C: Light micrographs of rat renal cortex. Formalin-fixed specimens (3 μm thick) were stained with Masson's trichrome staining. Proximal tubular epithelial cells with brush borders stain red; interstitial areas stain blue (as for the human renal biopsy). The interstitial areas were thickened with age. This age-dependent thickening was attenuated by GLO1 overexpression. Original magnification = ×600. Scale bars: 10 μm. D: Quantitative histological analysis indicates that GLO1 overexpression ameliorated age-dependent interstitial thickening. *P < 0.01; **P < 0.05.
In accord with the findings in humans, we observed tubulointerstitial morphological changes in aged rats. These changes were significantly decreased in aged Tg-GLO1 rats (Figure 3, C and D).
Carbonyl Stress and Cellular Senescence in Primary Cultured Tubular Cells
To confirm the significance of carbonyl compound in age-related alterations in tubular cells, we used primary cultured human RPTECs. First, we investigated the senescence status of RPTECs at early and late passage (passage 3 and 10, respectively). Cell proliferation estimated by the MTS assay showed that RPTECs reached replicative senescence at late passage (Figure 4A). Treatment of early-passaged cells with 1 μmol/L etoposide, a chemical senescence inducer, successfully induced replicative senescence, as was observed in late-passaged cells (Figure 4A). The LDH assay revealed no significant differences in cell viability among the three groups (Figure 4B). The cultured cells retained the phenotypes of proximal tubular cells, as evaluated by γ-GTP staining, a specific marker of proximal tubular cells. In contrast, γ-GTP was negative in rat renal fibroblasts (Figure 4C).
Figure 4.
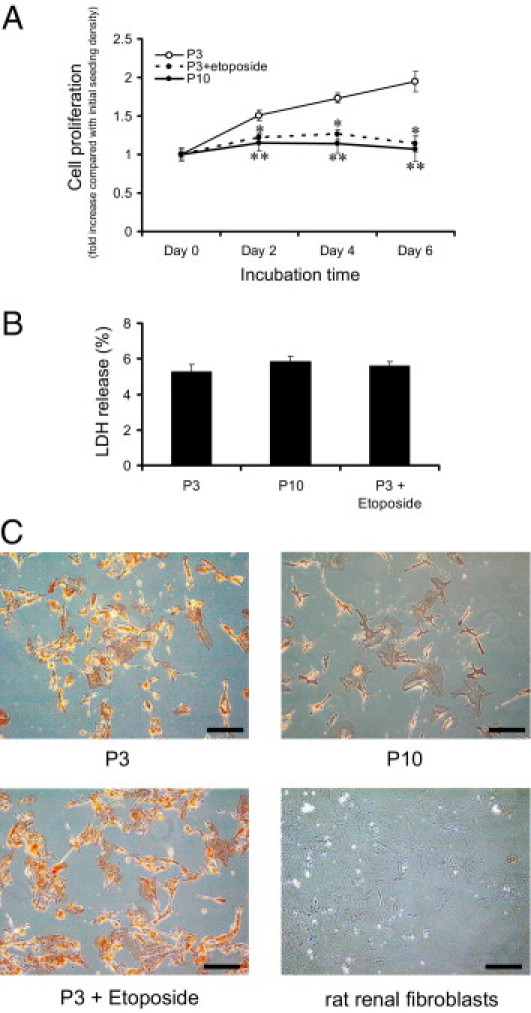
Validation of replicative senescence and senescence induced by etoposide in RPTECs. A: Cell proliferation rate determined by MTS assay showed replicative senescence at late passage (P10) and with exposure to a chemical senescence inducer, etoposide, at early passage (P3). Normal RPTECs doubled their population within 1 week, whereas senescent RPTECs and those exposed to etoposide did not proliferate. Assays were made at 2-day intervals, with the value on day 0 set as 1. *P < 0.01; **P < 0.05 versus day 0 of passage 3. B: LDH release was equivalent among RPTECs at passage 3 with or without etoposide exposure and at passage 10, excluding cells in which proliferation was halted because of cytotoxicity. LDH release = LDH in culture supernatant/(LDH in culture supernatant + LDH in cell lysate) ×100. C: Phase-contrast light micrographs with γ-GTP staining of RPTECs at passage 3 with or without etoposide exposure and at passage 10 and of rat renal fibroblasts confirmed that the cells retained the phenotype of proximal tubular cells. Original magnification = ×100. Scale bars: 200 μm. Red staining indicates cells positive for γ-GTP activity.
We then assessed the change in expression levels of cellular senescence markers in RPTECs at late passage or by etoposide treatment. The number of SABG-positive cells was significantly increased in late-passaged RPTECs and in early-passaged RPTECs treated with etoposide, compared with untreated early-passaged cells, verifying our assumption that the premature senescence induced by this chemical inducer closely mimics cellular senescence (Figure 5, A and B). We further assessed the changes in other senescence markers in the three groups of RPTECs. Transcriptional levels of p53, p21WAF1/CIP1, and p16INK4A were significantly increased in late-passaged RPTECs and etoposide-treated early-passaged RPTECs, compared with untreated early-passaged RPTECs (Figure 5C). Immunoblot analysis followed by densitometry revealed the up-regulation of p53 at the protein level in RPTECs in association with replicative senescence (Figure 5, D and E). Immunofluorescent microscopy showed that late-passaged and etoposide-treated early-passaged cells showed accumulation of CEL, an indicator of carbonyl stress status, but untreated early-passaged cells did not (Figure 5F).
Figure 5.
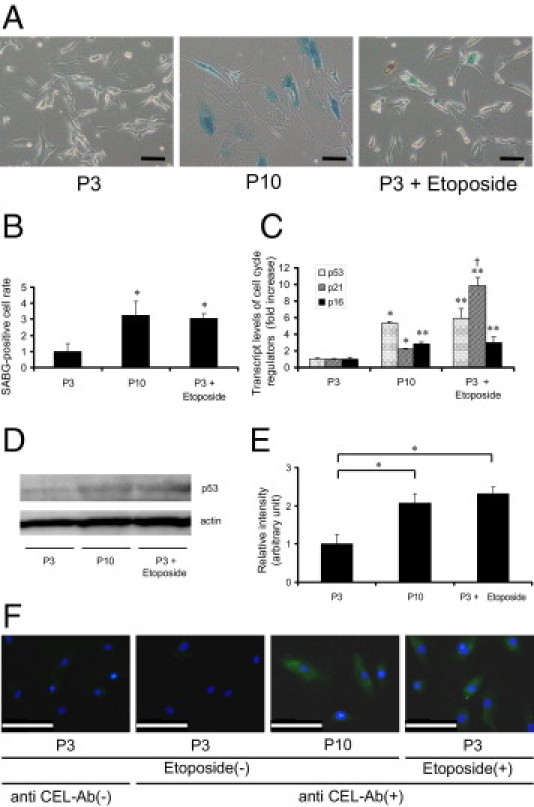
Carbonyl stress and cellular senescence in RPTECs. A: Phase-contrast light micrographs with SABG staining of RPTECs at passage 3 with or without etoposide exposure and at passage 10 show senescence of cells at passage 3 with etoposide exposure and at passage 10. Glutaraldehyde fixation was done before staining. SABG staining was performed by incubation for 5 hours without CO2. Original magnification = ×100. Scale bars: 100 μm. Blue staining indicates positive cells. B: Quantitative analysis of SABG-positive cells per total cells in each field. Most RPTECs were senescent at passage 10 or with etoposide exposure. *P < 0.01 versus day 0 at passage 3. C: Transcript levels of p53, p21WAF1/CIP1, and p16INK4A of RPTECs at passage 3 with or without etoposide exposure and at passage 10 determined by real-time quantitative RT-PCR. All showed significant elevation compared with cells at passage 3 without etoposide exposure. Values of those at passage 3 were set as 1. *P < 0.01; **P < 0.05 versus each at passage 3; †P < 0.05 versus p21WAF1/CIP1 at passage 10. D: Protein immunoblot of p53 of whole lysate of RPTECs at passage 3 with or without etoposide exposure and at passage 10 confirmed the results of real-time quantitative PCR analysis. Cells were lysed in urea buffer under reducing conditions. Actin served as a control. E: Densitometric quantification of p53 immunoblot of RPTECs at early passage (P3) with or without etoposide exposure and at late passage (P10). The level of cells at early passage without etoposide exposure was set as 1. *P < 0.01 versus cells at passage 3 without etoposide exposure. F: Immunofluorescent micrographs for the detection of CEL showed carbonyl stress in senescent RPTECs. Only cells at passage 3 with etoposide exposure and at passage 10 exhibited a cytosolic positive signal. Cells were fixed with methanol 50%:acetone 50%. Green indicates anti-CEL (fluorescein isothiocyanate); blue stain identifies nuclei (Hoechst 33258 dye). Original magnification = ×400. Scale bars: 100 μm.
Beneficial Effect of GLO1 on Senescence of Tubular Cells
We next used RPTECs treated with etoposide for functional studies. First, we performed gain-of-function studies using early-passaged RPTECs overexpressing human GLO1, which is a detoxifying enzyme of reactive carbonyl compounds, including methylglyoxal. Transfection efficiency in RPTECs as estimated using LacZ expression as a reporter gene was 87 ± 6%, and GLO1 activity was up-regulated by 14 ± 2-fold in GLO1-transfected cells, compared with empty vector-transfected cells (Figure 6A). Overexpression of GLO1 did not change the basal expression level of SABG in these transfectants. Of note, empty vector-transfected cells treated with etoposide showed a significant increase in SABG-positive cells, by 2.3 ± 1.0-fold, which was significantly suppressed by GLO1 overexpression (Figure 6, B and C). Transcript expression levels of p53, p21WAF1/CIP1, and p16INK4A, as well as protein expression levels of p53 in whole cellular lysate from etoposide-treated cells, were also significantly decreased in GLO1-transfected cells, compared with empty vector-transfected cells, whereas those without etoposide exposure showed no significant differences (Figure 6, D–F). Accumulation of CEL augmented by etoposide exposure was significantly suppressed by GLO1 overexpression, but remained at the increased levels with transfection of empty vector (Figure 6G).
Figure 6.
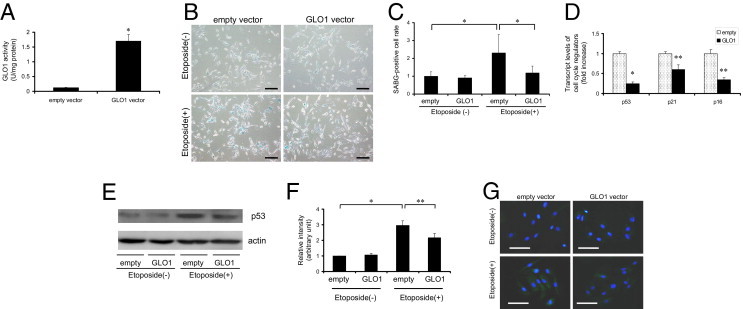
Attenuation of the senescent phenotypes of RPTECs at early passage with etoposide exposure by overexpression of GLO1. A: GLO1 activity assay measured at 24 hours after transfection of each vector confirmed successful overexpression of the transgene. Cells were lysed with sodium phosphate buffer with 1% Triton X-100 surfactant. *P < 0.01 versus empty vector. B: Phase-contrast light micrographs with SABG staining of RPTECs at passages 3 to 4 transfected with empty vector or GLO1-containing vector with or without etoposide exposure showed amelioration of senescence by GLO1. Original magnification = ×100. Scale bars: 200 μm. Blue stain indicates SABG-positive cells. C: Quantitative analysis of SABG-positive cells per total cells in each field. *P < 0.01 versus cells without etoposide exposure and transfected with empty vector. D: Relative transcript levels of p53, p21WAF1/CIP1, and p16INK4A of RPTECs with etoposide exposure and transfected with plasmid vector determined by real-time quantitative RT-PCR. All showed a significant decrease in GLO1 transfectants, which demonstrated that GLO1 protected RPTECs against cellular senescence. Values of those transfected with empty vector were set as 1. *P < 0.01; **P < 0.05 versus cells transfected with empty vector. E: Protein immunoblot of p53 of whole lysate of RPTECs with or without etoposide exposure and transfected with plasmid vectors. p53 level in cells treated with etoposide exposure was ameliorated by GLO1 introduction. Actin serves as a control. F: Densitometric quantification of p53 immunoblot of RPTECs with or without etoposide exposure and transfected with plasmid vectors. The level of those transfected with empty vector without etoposide exposure was set as 1. *P < 0.01; **P < 0.05 versus cells without etoposide exposure and transfected with empty vector. G: Immunofluorescent micrographs of RPTECs with or without etoposide exposure and transfected with plasmid vectors for the detection of CEL accumulation. Cytosolic positive signals in cells exposed to etoposide were decreased by GLO1 introduction, demonstrating that overexpression of GLO1 ameliorated carbonyl stress. Green indicates anti-CEL (fluorescein isothiocyanate); blue stain identifies nuclei (Hoechst 33258 dye). Original magnification = ×400. Scale bars: 100 μm.
We also performed loss-of-function experiments with siRNA targeted to human GLO1 using two independent siRNAs against GLO1, which efficiently knocked down GLO1 expression and activity (Figure 7, A and B). Of note, when GLO1-knocked down cells were treated with etoposide, the number of cells positive for SABG staining was significantly increased, compared with control siRNA-transfected cells (Figure 7, C and D). Other senescence markers, such as transcript expression level of p53, p21WAF1/CIP1, and p16INK4A or protein expression level of p53 in whole cellular lysate, were also markedly augmented by knockdown of GLO1 (Figure 7, E–G). These gain-of-function and loss-of-function studies of GLO1 in cultured cells suggest that GLO1 has a cytoprotective effect against senescence.
Figure 7.
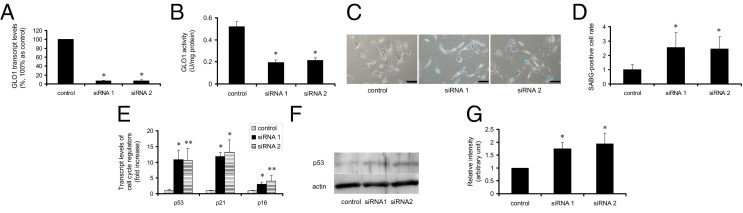
Knockdown of GLO1 exacerbated the senescence phenotype of RPTECs at early passage with etoposide exposure. A: Knockdown of the target gene was confirmed by transcript levels of GLO1 determined by real-time quantitative RT-PCR after 24 hours of transfection. The level of RPTECs transfected with control siRNA was set as 1. *P < 0.01 versus control siRNA. B: GLO1 activity assay measured after 24 hours transfection with siRNA or control RNA was consistent with the decreased transcript levels. *P < 0.01 versus empty vector. C: Phase-contrast light micrographs with SABG staining of RPTECs transfected with siRNA or control RNA with etoposide exposure showed aggravation of senescence by knockdown of GLO1. Original magnification = ×100. Scale bars: 100 μm. Blue stain indicates SABG-positive cells. D: Quantitative analysis of SABG-positive cells per total cells in each field. *P < 0.05 versus cells with etoposide exposure and transfected with control siRNA. E: Relative transcript levels of p53, p21WAF1/CIP1, and p16INK4A of RPTECs with etoposide exposure and transfected with siRNA or control RNA determined by real-time quantitative RT-PCR showed that knockdown of GLO1 aggravated cellular senescence phenotypes. Values of those transfected with control siRNA were set as 1. *P < 0.01; **P < 0.05 versus cells with etoposide exposure and transfected with control siRNA. F: Protein immunoblot of p53 of whole lysate of RPTECs with etoposide exposure and transfected with siRNA or control RNA. The level of p53 from cells exposed to etoposide was aggravated by the knockdown of GLO1. Actin serves as a control. G: Densitometric quantification of p53 immunoblot. The level of cells transfected with control siRNA was set as 1. *P < 0.05 versus cells transfected with control siRNA.
Next, we verified the phenomena observed in the study of premature senescence by examining the status of replicative senescence on treatment with etoposide. Etoposide induced a significantly lower uptake of BrdU in RPTECs with mock transfection, and the degree of inhibition of BrdU uptake by etoposide was equivalent to that of late-passaged RPTECs (Figure 8). In contrast, cells overexpressing GLO1 showed no significant etoposide-induced decrease in the uptake of BrdU. These data demonstrate that the senescence status induced by etoposide, in which growth was inhibited and senescence markers were up-regulated, was ameliorated by GLO1.
Figure 8.
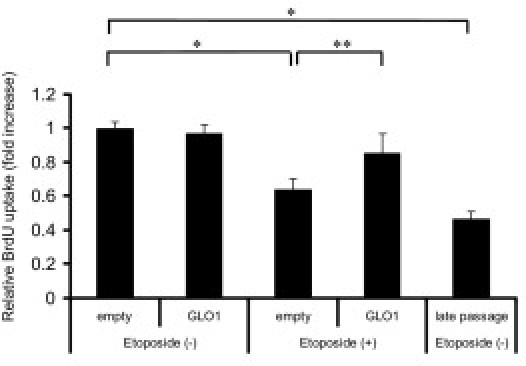
The replicative senescence induced by etoposide exposure was attenuated by transfection with the GLO1-overexpressing vector. BrdU uptake of RPTECs at early passage with or without etoposide exposure and at late passage without etoposide exposure was measured. BrdU uptake was significantly decreased by the addition of etoposide or at late passage, whereas the introduction of GLO1 ameliorated this decrease significantly. The level after transfection with empty vector without etoposide exposure was set as 1. *P < 0.01; **P < 0.05.
Confirmation of Decrease in Renal Function of the Elderly Human Subjects with GLO1 Mutation
We had previously performed a nationwide analysis of GLO1 genotypes in schizophrenia patients and found that a minor subset of patients had frameshift mutations in GLO1.24 Severe and significant decrease in the enzymatic activity was observed in those with frameshift mutations (3.0 ± 0.2 mU/106 red blood cells; n = 3), compared with WT GLO1 (6.8 ± 0.9 mU/106 red blood cells; n = 244). We investigated the effect of GLO1 activity on renal function. In the very rare cases with frameshift mutations, patients were all at the age of 50 to 60 years. We therefore compared the patients at this age, because GFR decreases with age.
Age-matched patients with WT GLO1 (n = 15) and frameshift mutation (n = 3) were analyzed. No clinically significant hematuria or proteinuria was found in any of the patients, with or without mutation. Neither musculature (determined by body mass index) nor systolic blood pressure, which could affect the serum creatinine levels, were significantly different among the three groups (Table 2). None of the patients with or without frameshift mutations were being treated with medication that could deteriorate renal functions. We reconfirmed a decreased activity of GLO1 in the patients with frameshift mutation, compared with the wild type (Figure 9A). Analysis of eGFR of the aged schizophrenia patients who had GLO1 frameshift mutations revealed significantly decreased renal function, compared with the wild type (P < 0.01) (Figure 9B). The data confirmed the protective effect of GLO1 against an age-dependent decrease in renal function.
Table 2.
Physiological Characteristics of Elderly Schizophrenia Patients with or without GLO1 Mutation
|
GLO1 genotype |
||
|---|---|---|
| Characteristics | WT (n = 15) | Frameshift (n = 3) |
| Age (years) | 56 ± 2.7 | 57 ± 4.6 |
| Body mass index | 23 ± 2.5 | 23 ± 4.9 |
| Systolic blood pressure (mmHg) | 110 ± 14 | 110 ± 11 |
No significant differences were observed in mean age, body mass index, or systolic blood pressure.
Figure 9.
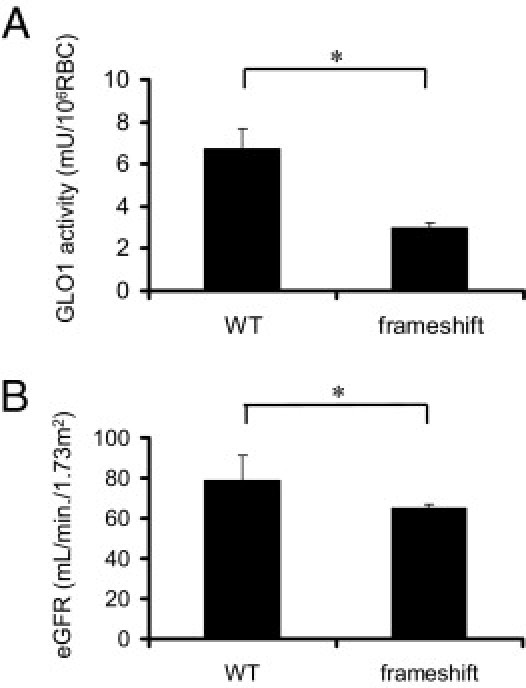
Analysis of human subjects with or without GLO mutations. A: GLO1 activity of red blood cells between the age-matched subjects with WT GLO1 and those with frameshift mutations of GLO1. WT, n = 15; frameshift, n = 3. *P < 0.01. B: eGFR between age-matched subjects with WT GLO1 and those with frameshift mutations of GLO1. WT, n = 15; frameshift, n = 3. *P < 0.01.
Discussion
We found that the aged kidney shows an increase in AGE accumulation in association with a decrease in GLO1 activity, as is seen also in the aged human lens,26 aged human red blood cells,21 and aged human brain.27, 28 Thus, age-dependent acceleration of carbonyl stress may induce renal senescence, at least in part. Accordingly, GLO1 might reduce the senescence phenotype via inhibitory activity against carbonyl stress.9 In the present study, we have demonstrated for the first time both in vitro and in vivo that overexpression of GLO1 ameliorates all phenotypes of renal senescence. The delay in renal senescence in rats overexpressing GLO1 was associated with the preservation of kidney function. Furthermore, knockdown of GLO1 aggravated senescent phenotypes in vitro. Supporting a protective role of GLO1 against senescence, a recent report showed that overexpression of the GLO1 homolog CeGly induced longevity in Caenorhabditis elegans.29
We used 14-month-old rats for the aged group because we observed obvious histological changes associated with senescence in kidney in our preliminary experiments, as reported previously.30 Because the aging process is complex and is likely to involve various mechanisms at different ages, we decided to focus on early senescence.
We focused our studies of kidney senescence on tubular epithelial cells. Functional impairment of the kidney more closely correlates with the degree of tubulointerstitial damage than with that of glomerular injury, and age-related decrease of eGFR is reported to be independent of glomerular change.31 Our approach is consistent with several recent studies that have focused on tubulointerstitial alterations in the aging kidney.30, 32, 33 One reason for the propensity toward senescence in the tubulointerstitium may relate to the metabolic rates of cells, given that podocytes or endothelial cells have far fewer mitochondria than tubular epithelial cells.34
In examination of a human specimen obtained from renal biopsy, we identified interstitial thickening (characterized by augmentation of matrix between tubules) as one of the senescent phenotypes of kidney. This alteration of renal components is distinct from interstitial fibrosis. Interstitial fibrosis is characterized by the replacement of tubules to fibrotic components and is observed in only limited portions of kidney, whereas interstitial thickening is a generalized change in the kidney. More importantly, this age-related phenotype is observed across a wide range of ages; in contrast, thickness of glomerular basement membrane is reported to have different effects at different ages.35 To prevent overestimation of the interstitial thickness for tubules at an angle to the cutting surface, we measured the smallest thickness around tubules.
In the present study, RPTECs reached replicative senescence status at 10 passages, which is earlier than for primary cultured fibroblasts, which do so at approximately 40 passages. This finding is consistent with a previous report,36 and might reflect high metabolic rate, susceptibility to senescence, or the nature of high differentiation. We also used etoposide, a well-established chemical senescence inducer,37, 38, 39, 40 to induce the cultured cells into senescence. The senescent phenotypes induced by etoposide were equivalent to those observed in late-passaged cells (although we note that the mechanism by which etoposide induces senescence has not been clarified).
The expression of senescence markers such as SABG, p16INK4A, p21WAF1/CIP1, and p53 was significantly increased in both late-passaged primary tubular epithelial cells and in the tubules of aged rat kidneys. Staining for p53 was positive not only in the nuclei but also in the cytoplasm, as reported previously.41, 42 These senescent phenotypes were associated in vivo with interstitial thickening in both human and rat kidney. Interstitial thickening might represent an aging-specific morphological change at an early stage of kidney aging. Similar changes were observed in a rodent model that was enhanced to present an age-related phenotype.43, 44
Our analysis of schizophrenia patients demonstrated association of reduction of GLO1 activity and accelerated age-dependent deterioration of renal function in human. Age-dependent decline of renal function in subjects with preserved GLO1 activity was consistent with that described in healthy aged individuals previously (0.64 to 0.75 mL/min per year).45, 46, 47 These results suggest that the difference in deterioration of age-dependent renal function that we observed was not due to improvement of renal function in schizophrenia patients with preserved GLO1 activity but rather was due to acceleration of age-dependent renal dysfunction associated with decreased GLO1 activity. One limitation is that these data are based on a small number of subjects in the GLO1-mutated group (n = 3); however, the incidence rate of this mutation is so low that this difficulty cannot be avoided.
Although excessive carbonyl stress is considered to be a major cause of spontaneous damage to intracellular and extracellular proteins functions,48 detailed mechanisms of the progression of senescence by carbonyl stress remain to be elucidated.
In conclusion, we have established a role of carbonyl stress in senescence and demonstrated that GLO1 can retard renal senescence both in vitro and in vivo.
Footnotes
Supported by Grant-in-Aid for Scientific Research 22590880 (R.I.) and 2139036 (M.N.) from the Japan Society for the Promotion of Science.
References
- 1.Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O., Peacocke M., Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan E.L., Wadhwa R., Kaul S.C. Senescence and immortalization of human cells. Biogerontology. 2000;1:103–121. doi: 10.1023/a:1010000132671. [DOI] [PubMed] [Google Scholar]
- 3.Collado M., Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- 4.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 5.Perez V.I., Bokov A., Remmen H.V., Mele J., Ran Q., Ikeno Y., Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pérez V.I., Van Remmen H., Bokov A., Epstein C.J., Vijg J., Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8:73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtman E.R. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 8.Thorpe S.R., Baynes J.W. Role of the Maillard reaction in diabetes mellitus and diseases of aging. Drugs Aging. 1996;9:69–77. doi: 10.2165/00002512-199609020-00001. [DOI] [PubMed] [Google Scholar]
- 9.Thornalley P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems: role in ageing and disease. Drug Metabol Drug Interact. 2008;23:125–150. doi: 10.1515/dmdi.2008.23.1-2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 11.Uribarri J., Cai W., Peppa M., Goodman S., Ferrucci L., Striker G., Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62:427–433. doi: 10.1093/gerona/62.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Münch G., Shepherd C.E., McCann H., Brooks W.S., Kwok J.B., Arendt T., Hallupp M., Schofield P.R., Martins R.N., Halliday G.M. Intraneuronal advanced glycation endproducts in presenilin-1 Alzheimer's disease. Neuroreport. 2002;13:601–604. doi: 10.1097/00001756-200204160-00013. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed N. Advanced glycation endproducts–role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Chen J., Huang X., Halicka D., Brodsky S., Avram A., Eskander J., Bloomgarden N.A., Darzynkiewicz Z., Goligorsky M.S. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. Am J Physiol Heart Circ Physiol. 2006;290:H1575–H1586. doi: 10.1152/ajpheart.00364.2005. [DOI] [PubMed] [Google Scholar]
- 15.Thangarajah H., Yao D., Chang E.I., Shi Y., Jazayeri L., Vial I.N., Galiano R.D., Du X.L., Grogan R., Galvez M.G., Januszyk M., Brownlee M., Gurtner G.C. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci USA. 2009;106:13505–13510. doi: 10.1073/pnas.0906670106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thornalley P.J. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990;269:1–11. doi: 10.1042/bj2690001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayes J.D., Milner S.W., Walker S.W. Expression of glyoxalase, glutathione peroxidase and glutathione S-transferase isoenzymes in different bovine tissues. Biochim Biophys Acta. 1989;994:21–29. doi: 10.1016/0167-4838(89)90057-5. [DOI] [PubMed] [Google Scholar]
- 18.Thornalley P.J. Glyoxalase I–structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 19.Kumagai T., Nangaku M., Kojima I., Nagai R., Ingelfinger J.R., Miyata T., Fujita T., Inagi R. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol. 2009;296:F912–F921. doi: 10.1152/ajprenal.90575.2008. [DOI] [PubMed] [Google Scholar]
- 20.Inagi R., Miyata T., Ueda Y., Yoshino A., Nangaku M., van Ypersele de Strihou C., Kurokawa K. Efficient in vitro lowering of carbonyl stress by the glyoxalase system in conventional glucose peritoneal dialysis fluid. Kidney Int. 2002;62:679–687. doi: 10.1046/j.1523-1755.2002.00488.x. [DOI] [PubMed] [Google Scholar]
- 21.McLellan A.C., Thornalley P.J. Glyoxalase activity in human red blood cells fractioned by age. Mech Ageing Dev. 1989;48:63–71. doi: 10.1016/0047-6374(89)90026-2. [DOI] [PubMed] [Google Scholar]
- 22.Nagai R., Fujiwara Y., Mera K., Yamagata K., Sakashita N., Takeya M. Immunochemical detection of Nepsilon-(carboxyethyl)lysine using a specific antibody. J Immunol Methods. 2008;332:112–120. doi: 10.1016/j.jim.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 23.Rutenburg A.M., Kim H., Fischbein J.W., Hanker J.S., Wasserkrug H.L., Seligman A.M. Histochemical and ultrastructural demonstration of gamma-glutamyl transpeptidase activity. J Histochem Cytochem. 1969;17:517–526. doi: 10.1177/17.8.517. [DOI] [PubMed] [Google Scholar]
- 24.Arai M., Yuzawa H., Nohara I., Ohnishi T., Obata N., Iwayama Y., Haga S., Toyota T., Ujike H., Arai M., Ichikawa T., Nishida A., Tanaka Y., Furukawa A., Aikawa Y., Kuroda O., Niizato K., Izawa R., Nakamura K., Mori N., Matsuzawa D., Hashimoto K., Iyo M., Sora I., Matsushita M., Okazaki Y., Yoshikawa T., Miyata T., Itokawa M. Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch Gen Psychiatry. 2010;67:589–597. doi: 10.1001/archgenpsychiatry.2010.62. [DOI] [PubMed] [Google Scholar]
- 25.Matsuo S., Imai E., Horio M., Yasuda Y., Tomita K., Nitta K., Yamagata K., Tomino Y., Yokoyama H., Hishida A., Collaborators developing the Japanese equation for estimated G.F.R. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53:982–992. doi: 10.1053/j.ajkd.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 26.Haik G.M., Jr, Lo T.W., Thornalley P.J. Methylglyoxal concentration and glyoxalase activities in the human lens. Exp Eye Res. 1994;59:497–500. doi: 10.1006/exer.1994.1135. [DOI] [PubMed] [Google Scholar]
- 27.Kuhla B., Boeck K., Lüth H.J., Schmidt A., Weigle B., Schmitz M., Ogunlade V., Münch G., Arendt T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol Aging. 2006;27:815–822. doi: 10.1016/j.neurobiolaging.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Kuhla B., Boeck K., Schmidt A., Ogunlade V., Arendt T., Münch G., Lüth H.J. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer's disease brains. Neurobiol Aging. 2007;28:29–41. doi: 10.1016/j.neurobiolaging.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Morcos M., Du X., Pfisterer F., Hutter H., Sayed A.A., Thornalley P., Ahmed N., Baynes J., Thorpe S., Kukudov G., Schlotterer A., Bozorgmehr F., El Baki R.A., Stern D., Moehrlen F., Ibrahim Y., Oikonomou D., Hamann A., Becker C., Zeier M., Schwenger V., Miftari N., Humpert P., Hammes H.P., Buechler M., Bierhaus A., Brownlee M., Nawroth P.P. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7:260–269. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 30.Ding G., Franki N., Kapasi A.A., Reddy K., Gibbons N., Singhal P.C. Tubular cell senescence and expression of TGF-beta1 and p21(WAF1/CIP1) in tubulointerstitial fibrosis of aging rats. Exp Mol Pathol. 2001;70:43–53. doi: 10.1006/exmp.2000.2346. [DOI] [PubMed] [Google Scholar]
- 31.Rule A.D., Am H., Cornell L.D., Taler S.J., Cosio F.G., Kremers W.K., Textor S.C., Stegall M.D. The association between age and nephrosclerosis on renal biopsy among healthy adults. Ann Intern Med. 2010;152:561–567. doi: 10.1059/0003-4819-152-9-201005040-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abrass C.K., Adcox M.J., Raugi G.J. Aging-associated changes in renal extracellular matrix. Am J Pathol. 1995;146:742–752. [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz-Torres M.P., Bosch R.J., O'Valle F., Del Moral R.G., Ramírez C., Masseroli M., Pérez-Caballero C., Iglesias M.C., Rodríguez-Puyol M., Rodríguez-Puyol D. Age-related increase in expression of TGF-beta1 in the rat kidney: relationship to morphologic changes. J Am Soc Nephrol. 1998;9:782–791. doi: 10.1681/ASN.V95782. [DOI] [PubMed] [Google Scholar]
- 34.Pease D.C. Fine structures of the kidney seen by electron microscopy. J Histochem Cytochem. 1955;3:295–308. doi: 10.1177/3.4.295. [DOI] [PubMed] [Google Scholar]
- 35.Shindo S., Yoshimoto M., Kuriya N., Bernstein J. Glomerular basement membrane thickness in recurrent and persistent hematuria and nephrotic syndrome: correlation with sex and age. Pediatr Nephrol. 1988;2:196–199. doi: 10.1007/BF00862590. [DOI] [PubMed] [Google Scholar]
- 36.Glynne P.A. Primary culture of human proximal renal tubular epithelial cells. Methods Mol Med. 2000;36:197–205. doi: 10.1385/1-59259-216-3:197. [DOI] [PubMed] [Google Scholar]
- 37.Krizhanovsky V., Yon M., Dickins R.A., Hearn S., Simon J., Miething C., Yee H., Zender L., Lowe S.W. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.te Poele R.H., Okorokov A.L., Jardine L., Cummings J., Joel S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–1883. [PubMed] [Google Scholar]
- 39.Robles S.J., Buehler P.W., Negrusz A., Adami G.R. Permanent cell cycle arrest in asynchronously proliferating normal human fibroblasts treated with doxorubicin or etoposide but not camptothecin. Biochem Pharmacol. 1999;58:675–685. doi: 10.1016/s0006-2952(99)00127-6. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y., Blandino G., Oren M., Givol D. Induced p53 expression in lung cancer cell line promotes cell senescence and differentially modifies the cytotoxicity of anti-cancer drugs. Oncogene. 1998;17:1923–1930. doi: 10.1038/sj.onc.1202113. [DOI] [PubMed] [Google Scholar]
- 41.Kaserer K., Schmaus J., Bethge U., Migschitz B., Fasching S., Walch A., Herbst F., Teleky B., Wrba F. Staining patterns of p53 immunohistochemistry and their biological significance in colorectal cancer. J Pathol. 2000;190:450–456. doi: 10.1002/(SICI)1096-9896(200003)190:4<450::AID-PATH545>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 42.Ranade K.J., Nerurkar A.V., Phulpagar M.D., Shirsat N.V. Expression of survivin and p53 proteins and their correlation with hormone receptor status in Indian breast cancer patients. Indian J Med Sci. 2009;63:481–490. [PubMed] [Google Scholar]
- 43.Samuel C.S., Zhao C., Bond C.P., Hewitson T.D., Amento E.P., Summers R.J. Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 2004;65:2054–2064. doi: 10.1111/j.1523-1755.2004.00628.x. [DOI] [PubMed] [Google Scholar]
- 44.Floege J., Hackmann B., Kliem V., Kriz W., Alpers C.E., Johnson R.J., Kühn K.W., Koch K.M., Brunkhorst R. Age-related glomerulosclerosis and interstitial fibrosis in Milan normotensive rats: a podocyte disease. Kidney Int. 1997;51:230–243. doi: 10.1038/ki.1997.28. [DOI] [PubMed] [Google Scholar]
- 45.Lindeman R.D., Tobin J., Shock N.W. Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 1985;33:278–285. doi: 10.1111/j.1532-5415.1985.tb07117.x. [DOI] [PubMed] [Google Scholar]
- 46.Rowe J.W., Andres R., Tobin J.D. Letter: age-adjusted standards for creatinine clearance. Ann Intern Med. 1976;84:567–569. doi: 10.7326/0003-4819-84-5-567. [DOI] [PubMed] [Google Scholar]
- 47.Rowe J.W., Andres R., Tobin J.D., Norris A.H., Shock N.W. The effect of age on creatinine clearance in men: a cross-sectional and longitudinal study. J Gerontol. 1976;31:155–163. doi: 10.1093/geronj/31.2.155. [DOI] [PubMed] [Google Scholar]
- 48.Thornalley P.J., Battah S., Ahmed N., Karachalias N., Agalou S., Babaei-Jadidi R., Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]