

Abstract
Cigarette smoke activates the extracellular signal-regulated kinase (ERK) 1/2 mitogen activated-protein kinase pathway, which, in turn, is responsible for early growth response gene-1 (EGR-1) activation. Here we provide evidence that EGR-1 activation can also reactivate ERK 1/2 mitogen activated-protein kinase through a positive feedback loop through its target gene (geranylgeranyl diphosphate synthase) GGPPS. For the first time, the GGPPS gene is identified as a target of EGR-1, as EGR-1 can directly bind to the predicted consensus-binding site in the GGPPS promoter and regulate its transcription. Long-term observations show that there are two ERK 1/2 phosphorylation peaks after cigarette smoke extract stimulation in human lung epithelial Beas-2B cells. The first peak (at 10 minutes) is responsible for EGR-1 accumulation, and the second (at 4 hours) is diminished after the disruption of EGR-1 transcriptional activity. EGR-1 overexpression enhances Ras prenylation and membrane association in a GGPPS-dependent manner, and it augments ERK 1/2 activation. Likewise, a great reduction of the second peak of ERK 1/2 phosphorylation is observed during long-term cigarette smoke extract stimulation in cells where GGPPS is disrupted. Thus, we have uncovered an intricate positive feedback loop in which ERK 1/2-activated EGR-1 promotes ERK 1/2 reactivation through promoting GGPPS transcription, which might affect cigarette smoke-related lung pathological processes.
The transcription factor EGR-1 is the product of early growth response gene-1 (EGR1), which belongs to the immediate-early gene family of proteins characterized by zinc finger domains that recognize the highly conserved consensus GC-rich nucleotide sequences (GCG G/TGG GCG).1, 2, 3, 4 EGR-1 is often rapidly and transiently activated within minutes of a stimulus by a variety of signals, and its activity can decay within hours.4 Many stress signals, such as osmotic pressure variation, heat shock, hypoxia, DNA-damaging agents, radiation, injury, and stretch, can also stimulate EGR1 expression.2, 5, 6, 7, 8, 9, 10, 11 EGR-1 can directly activate the transcription of many genes, including immune effector genes, such as cytokine IL-212 and the pro-inflammation immune mediator tumor necrosis factor,13 and cell surface molecules, such as IL-2 receptors,14 Fas/CD95,15 and intercellular adhesion molecule 1.16 Through the regulation of its target genes, EGR-1 plays important roles in various cellular programs, including cell proliferation, differentiation, and apoptosis.2 We have previously reported that human primary lung fibroblasts can synthesize matrix metalloproteinase (MMP) and chemokines in an EGR-1-dependent manner when exposed to cigarette smoke extract (CSE) stress.17, 18, 19 To further understand the pathological functions of EGR-1 in pulmonary diseases during cigarette smoke-induced stress, we screened new target genes using chromatin immunoprecipitation (ChIP) methods. We have found that geranylgeranyl diphosphate synthase (GGPPS) is one of the novel EGR-1 target genes that mediate the function of EGR-1 during cigarette smoke exposure.
GGPPS is a branch point enzyme in the mevalonate pathway that catalyzes the synthesis of geranylgeranyl diphosphate20, 21 and farnesyl diphosphate,22 which are used for the geranylgeranylation of proteins such as Ras and Ras-related small GTP-binding proteins with a CaaX motif at the C terminus. Prenylation modification of the Ras protein is generally essential for its membrane association and activation.23 Mutation of the prenylation site or blockade of isoprenoid biosynthesis abolishes both the prenylation and membrane association of Ras, and usually leads to a loss of normal cellular function.24, 25 However, EGR-1 function in the regulation of GGPPS production in the Ras signaling pathway has never been evaluated.
As an immediate-early gene, EGR1 is also involved in chronic diseases. Here we present evidence that the transcription factor EGR-1 can regulate the expression of GGPPS in a mitogen-activated protein kinase (MAPK)-mediated manner that involves the extracellular signal-regulated kinase (ERK) 1/2. At the same time, EGR-1 also activates a positive feedback mechanism to regulate the reactivation of MAPK through promoting the transcription of GGPPS, which is responsible for Ras prenylation and membrane association. Thus we have identified a novel feedback pathway that augments EGR-1-induced inflammatory responses in cultured cells and in a mouse model of pulmonary inflammation.26 This study may provide additional insight into the complex processes that lead to cigarette smoke-related damage to respiratory tissues.
Materials and Methods
Preparation and Treatment of CSE
Nonfiltered research reference cigarettes 1R3F were purchased from the University of Kentucky (Lexington, KY). CSE was prepared at a concentration of 1 cigarette/5 mL serum-free Dulbecco's modified Eagle's medium or Dulbecco's minimal essential medium, as previously described27 with modifications. This medium was defined as 100% CSE and was used after adjusting the pH to 7.4 and filtering it through a 0.22-μm filter. Cells were grown to 90% confluence in 10-cm cell culture plates and rendered quiescent in a medium containing 0.5% fetal bovine serum before CSE treatment. The 100% CSE was diluted to 10% before treating the cells. No removal of CSE was performed until sampling.
Plasmids and Cell Culture
Human EGR1 cDNA and dominant negative (Dn)EGR1, which contains only the zinc finger domains, but no transcriptional activity (ie, an effective and selective inhibitor of EGR-1-mediated transcription)28 were gifts from Prof. Jay M. Baraban. The small-interfering RNA (siRNA) purchased from Invitrogen (Carlsbad, CA) was designed to target the following cDNA sequences: 5′-CCTACGCCACCAATTTCGT-3′ (scrambled) and 5′-TCTCCCAGGACAATTGAAATTTGCT-3′ (EGR1). Human GGPPS cDNA was cloned by RT-PCR from the total RNA of HEK293 cells, and its promoter region (−190 to + 98) was cloned from HEK293 genomic DNA. To generate adenoviruses that overexpress EGR-1, DnEGR-1, and GGPPS, the sequences were inserted into the pAdTrack-CMV vector and recombined into the adenovirus vector using the AdEasy System, according to the manufacturer's protocol. To generate siRNA/GGPPS, a unique 19 nucleotide sequence targeting human GGPPS 5′-GTCCCACTGAAGAAGAATA-3′ and the scrambled sequence targeting 5′-TTCTCCGAACGTGTCACGT-3′ were inserted into the pShuttle-H1 vector. The construction of an adenovirus that expresses human GGPPS-siRNA (AdhGGPPS-siRNA) was performed using the AdEasy System according to the manufacturer's protocol. The cells used were cultured in Dulbecco's modified Eagle's medium (Gibco/BRL, Gaithersburg, MD) supplemented with 10% (v/v) fetal bovine serum and penicillin (100 IU/mL)/streptomycin (100 ng/mL) at 37°C in a water-saturated atmosphere with 5% CO2.
Inhibitor Administration
GTI (a GGTase inhibitor) and FTI (an FTase inhibitor) were purchased from Sigma (St. Louis, MO). U0126 (a MEK inhibitor) was purchased from Cell Signaling Technology (Danvers, MA). For GTI or FTI administration, cells infected with an adenovirus for 24 hours were incubated in growth medium containing 540 nmol/L GTI or 560 nmol/L FTI for another 48 hours. The administration of U0126 (10 μmol/L) was performed 2 hours before the cell lysates were collected.
ChIP Clone and Assay
The ChIP assay was performed on HEK293 cells using the SimpleChIP Enzymatic Chromatin IP kit (#9002; Cell Signaling Technology) according to the manufacturer's instructions, and all reagents (unless otherwise specified) were provided in the kit. Antibodies for EGR-1 (sc-110X; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and control normal rabbit IgG (Santa Cruz Biotechnology, Inc.) were used in the immunoprecipitations. After being thoroughly washed with the wash buffer, bound DNA was released by proteinase K digestion. Released DNA was digested with DPN II and cloned into the BglII site of pBScript. The cloned DNA was transformed, and the positive clones were sequenced and identified by Basic Local Alignment Search Tool (BLAST) searches of the nonredundant National Center for Biotechnology Information (NCBI) nucleotide databases. The ChIP DNA and control DNA (2% input) were analyzed using PCR and real-time PCR. Real-time PCR was performed using SYBR-Green master mix (Toyobo Co., Ltd., Osaka, Japan). The PCR ran for 22 cycles to ensure amplification in the linear range,29 and the primers used were as follows: 5′-GAAACAGAAAGTTACGCCTCAA-3′ and 5′-GAAGATCTATCCTTCCCACTATTTCACCAC-3′. The primers used for real-time PCR were 5′-AACAGAAAGTTACGCCTCAA-3′ and 5′-TTGCAGTTCTCGCGGACGAG-3′. Either 0.5 ng ChIP DNA or 0.5 ng control DNA was used as a template. The real-time PCR parameters for cycling were as follows: 95°C for 10 minutes, 40 cycles of PCR at 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds. All reactions were performed in a 20 μL reaction volume in triplicate. Fold enrichments were determined by the 2−▵▵CT method described in the Applied Biosystems user bulletin using RPL30 (60S ribosomal protein L30) as the normalizer.
Nuclear Extract Preparation and Electrophoretic Mobility Shift Assay
Nuclear extract preparation from HEK293 cells and the execution of electrophoretic mobility shift assay (EMSA) were done according to a previously described method.30 Briefly, complementary single-stranded oligonucleotides were annealed, and the resultant double-stranded DNA fragments were labeled with [γ-32P]ATP using T4 Polynucleotide Kinase (NEB, Beverly, MA). 32P-labeled and unlabeled double-stranded oligonucleotides were used as the probe and cold probe, respectively. We directly chose the sequence from the GGPPS promoter containing a consensus EGR-1-binding site as the probe: 5′-CGGTGCGGGGGCGGGGGGGAGGTG-3′. The consensus EGR-1-binding site, underlined, overlaps with a specificity protein 1 (SP1) binding site (GGGGCGGGG), shown in italics.
We also made three mutant probes: Mut1:5′-CGGTAAAGGGGCGGGGGGGAGGTG-3′; Mut2:5′-CGGTGCGGGGAAAGGGGGGAGGTG-3′; Mut3:5′-CGGTGCGGGGGCGAAAGGGAGGTG-3′. Mut1 contained a mutant consensus EGR-1-binding site, Mut3 contained a mutant consensus SP1-binding site and Mut2 contained a mutant consensus-binding site for both EGR-1 and SP1 (mutant nucleotides appear in boldface and the consensus-binding site for EGR-1 is underlined). Binding reaction mixtures contained nuclear extracts, the Gel Shift Binding 5× buffer (Promega, Madison, WI), labeled probe, and a 200-fold molar excess of the competitor cold probe or mutant probe. Anti-EGR1 and anti-SP1 antibodies were also added by a concentration gradient from 0.5 to 4 μg reaction mixtures were subjected to electrophoresis.
Immunoprecipitation
Ras proteins were immunoprecipitated from the cell lysate. A Pan-Ras antibody (sc-32) from Santa Cruz Biotechnology, Inc. was used. The cell lysate was added with the Ras antibody and rotated at 4°C overnight. Protein A/G plus agarose (20 μL) (sc-2003, Santa Cruz Biotechnology Inc.) was then re-suspended in lysis buffer and added with continued rotation at 4°C for 2 hours. The agarose was collected by centrifugation at 18,000 × g for 1 minute, after which immunoprecipitation (IP) buffer (50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1% Nonidet P-40 (NP-40), and 0.5% sodium deoxycholate) was added. The preparation was rotated at 4°C for 5 minutes and then centrifuged at 18,000 × g for 1 minute. This process was repeated 3 times to clear the agarose, which was eventually subjected to Western blot analysis using Pan-Ras (sc-32, Santa Cruz Biotechnology Inc.) and K-Ras (sc-521, Santa Cruz Biotechnology Inc.) antibodies.
Western Blotting
Beas-2B whole cell lysates were prepared according to previously reported standard protocols.19 For Western blotting, equal amounts of protein for each group were resolved on 10% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). The membranes were then incubated with the appropriate primary antibody as indicated. Bound antibody was visualized using alkaline phosphatase-conjugated or horseradish peroxidase-labeled secondary antibodies. The ERK 1/2, phosphorylated-ERK1/2 (p-ERK1/2), and glyceraldehydes-3-phosphate dehydrogenase antibodies were from Cell Signaling Technology. The EGR-1, Pan-Ras, K-Ras, and actin antibodies, and the alkaline phosphatase-conjugated secondary antibody were from Santa Cruz Biotechnology, Inc. The horseradish peroxidase-tagged secondary antibody was from Dako Cytomation (Glostrup, Denmark). Optical densities were analyzed using ImageQuant software (GE Healthcare, Chalfont St. Giles, England), and ERK 1/2 activities are represented as the ratio of p-ERK1/2 to total-ERK1/2 (t-ERK1/2).
Real-Time PCR
Total RNA from lung tissues and treated cells were extracted using the TRIzol reagent (Invitrogen) and reverse transcribed with a ReverTra Ace-α kit (Toyobo). The resulting cDNA was used for real-time PCR performed, which was performed using an ABI-7300. SYBR-Green master mix was purchased from Toyobo. All quantitations were conducted independently in triplicate and normalized to an endogenous 18S RNA control. The sequences of the primer probes for EGR1 (Homo sapiens) are 5′-CAGCAGTCCCATTTACTCAG-3′ and 5′-GACTGGTAGCTGGTATTG-3′; GGPPS (H. sapiens) are 5′-CCAGGTAAACAAGTGAGAACCAA3′ and 5′-CGTCGGAGTTTTGAGTTGTCT-3′; and 18S RNA are 5′-GTCTGTGATGCCCTTAGATG-3′ and 5′-AGCTTATGACCCGCACTTAC-3′.
Luciferase Assay
The wild-type GGPPS promoter or the mutant GGPPS promoter, which contained a mutant consensus EGR-1-binding site (Mut1 in the EMSA assay), was cloned immediately upstream of the luciferase reading frame in the pGL2-basic-vector plasmid. The Beas-2B cells were co-transfected with the luciferase reporters using the calcium phosphate precipitation method. After CSE stimulation for 20 hours, the luciferase expression/activity was measured by the Dual-Luciferase Report assay system (catalog no. E1910, Promega, Madison, WI), and the luminescence was read by a luminometer (TD-20/20) according to the manufacturer's instructions. This system allows the quantification of activities of both firefly luciferase (encoded by wild-type or the mutant GGPPS promoter-pGL2 plasmid construct) and Renilla luciferase (encoded by the pRL-SV40 plasmid). The relative values of firefly luciferase activities were determined by normalizing with Renilla luciferase activities for transfection efficiencies.
The Membrane Association of Ras
To determine the membrane association of Ras, a subcellular membrane fractionation of the treated Beas-2B cells that were infected with the EGR-1 adenovirus and/or small-interfering RNA of GGPPS (siGGPPS) adenovirus was prepared as previously described.31 Briefly, for each sample, 100-mm dishes containing confluent cells were washed with ice-cold PBS, collected in 1.0 mL of PBS and centrifuged at 500 × g for 10 minutes. Cell pellets were swelled with 1225 μL of hypotonic buffer (10 mmol/L Tris-HCl (pH 7.5), 1.0 mmol/L MgCl2, 0.5 mmol/L phenylmethylsulfonyl fluoride, 10 mg/mL leupeptin, 10 mg/mL aprotinin, and 1 mmol/L dithiothreitol) and placed on ice for 10 minutes. Cells were disrupted using an ice-cold Dounce tissue homogenizer, after which 225 μL of 1 M NaCl was added. A total of 450 μL of this solution, as total lysate, was transferred to a microcentrifuge tube and set aside. The remaining 1000 μL was transferred to a polycarbonate ultracentrifuge tube and spun at 100,000 × g for 30 minutes (4°C). The supernatant (S100, representing the cytosolic fraction) was transferred to a new microcentrifuge tube. The pellet (P100, representing the membrane fraction) was re-suspended in 850 μL of hypotonic buffer and 150 μL of 1 M NaCl. Next, 50 μL of 10 × Hi-SDS radioimmunoprecipitation assay buffer was added to the total lysate sample, and 110 μL was added to the S100 and P100 fractions. After incubation on ice for 10 minutes, the lysates were clarified by centrifugation at 25,000 × g for 30 minutes (4°C). Supernatants were transferred to a new microcentrifuge tube, and immunoprecipitations and Western blot detection of the Ras proteins were performed as previously described.
Ras Geranylgeranylation Measurements
Confluent Beas-2B cells were infected with the EGR-1 adenovirus or control virus for 24 hours and were then switched back to Dulbecco's modified Eagle's medium with 10% fetal calf serum (FCS). The cells were then incubated with dimethyl sulfoxide, GTI (a GGTase inhibitor, 540 nmol/L), or FTI (a FTase inhibitor, 560 nmol/L) for another 48 hours. The cells were then lysed in 500 μL of lysis buffer (150 mmol/L NaCl, 5 mmol/L MgCl2, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L dithiothreitol, 1 mmol/L sodium vanadate, 1 mmol/L sodium phosphate, 1% Triton X-100, 0.05% SDS, 10 mg/mL aprotinin, 10 mg/mL leupeptin, 50 mmol/L HEPES, pH 7.4) and centrifuged for 15 minutes at 18,000 × g. The total protein concentration of the resultant supernatant was determined using the bicinchoninic acid assay, and the protein was diluted to 1 mg/mL. Ras geranylgeranylation was measured as previously described.32 Briefly, an equal volume of 4% TritonX-114 was added to the supernatant. TritonX-114 is lipid soluble and can dissolve the cell membrane more easily than the cell cytoplasm because the cell membrane is also lipid soluble. In this manner, the aqueous (upper) phase, which contains enriched intracellular Ras, and the organic (lower) phase, which contains highly enriched membrane-associated Ras, were separated by 4% TritonX-114 at 37°C for 5 minutes. Following the standard immunoprecipitation protocol, the collected samples were subjected to Western blot analysis.
Results
Identification of a New EGR-1 Target Gene, GGPPS
To understand the mechanism of EGR-1-induced inflammation, we screened its target genes in HEK293 cells under CSE stress stimulation. We applied ChIP and subcloning methods to isolate the promoter sequence bound by EGR-1. After sequencing and a BLAST search, we identified one of the cloned DNA fragments located upstream of the GGPPS gene. By analyzing the promoter of GGPPS with the PROSCAN program suite (version 1.7, BioInformatics and Molecular Analysis Section, NIH), an EGR-1 consensus-binding site (−68 to −60) was found in the promoter region of GGPPS (−173 to +77, Figure 1A). To further confirm GGPPS as an EGR-1 target gene, a ChIP assay was performed again in HEK293 cells to demonstrate the binding of EGR-1 to the GGPPS promoter. In this assay, a portion of the cell lysate containing the genomic DNA was directly subjected to the PCR for 22 cycles using the primers for RPL30, and the result was used as the “2% input” or internal control. The rest of the cell lysate was subjected to immunoprecipitation using an EGR-1 antibody to immunoprecipitate the EGR-1-binding genome. Then the immunoprecipitated DNA (ChIP DNA) was released and subjected to PCR for 22 cycles to amplify the GGPPS promoter. Figure 1B shows that the EGR-1 protein could specifically interact with the GGPPS promoter region (confirmed by sequencing the bound DNA). We used real-time PCR to quantify the ChIP results, which are shown as the fold enrichment determined by the 2−▵▵CT method, using RPL30 as the normalizer. Binding was increased by CSE stimulation (Figure 1B, lower panel). The EMSA assay was then applied to determine direct binding of EGR-1 to the GGPPS promoter. A 24 nucleotide GC-rich sequence in the promoter, which contains a predicted EGR-1 consensus-binding site, was synthesized as a probe for EMSA. Figure 1C shows that two DNA protein complexes were formed after CSE stimulation. Competition assays with cold and mutant probes demonstrated that these are the probe-specific-binding bands because the selected GGPPS promoter contained not only the predicted EGR-1-binding site but also a predicted SP1-binding site. To distinguish the two possible probe-protein complexes, we designed three mutant probes. Mutant probe 1 (Mut1) contained only a mutant EGR-1-binding site, mutant probe 3 (Mut3) contained only a mutant SP1-binding site, and mutant probe 2 (Mut2) contained a mutant-binding site for both EGR-1 and SP1. A large decrease of both complex bands emerged in only the SP1 mutant (Mut3), which meant that those bands were not the SP1-probe complex. Although the expected super shift band was not observed, along with the increase in the EGR-1 antibody concentration, both of the probe-protein complexes were decreased while no decrease emerged along with the increase in the SP1 antibody concentration, which confirmed that both of the probe-protein complexes were EGR-1- (not SP1-) forming binding complexes with probe. The results demonstrated that GGPPS is the direct target gene of EGR-1.
Figure 1.
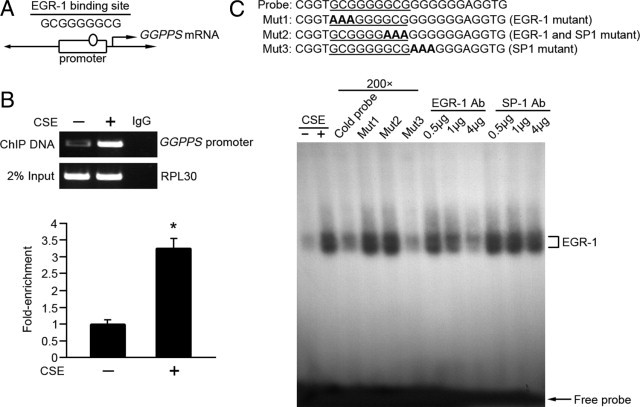
Identification of GGPPS as a new EGR-1 target gene. A: The promoter sequence bound by EGR-1 under 10% cigarette smoke extract (CSE) stress stimulation was acquired by chromatin immunoprecipitation (ChIP) and subcloning methods. The cloned DNA fragments were sequenced and BLAST searched to identify a fragment that was located upstream of the GGPPS promoter. An EGR-1 consensus binding site (−68 to −60) was found at the promoter region of GGPPS (−173 to +77). B: The ChIP assay was repeated to check the binding of EGR-1 to the GGPPS promoter after 10% CSE treatment in HEK293 cells. An EGR-1 antibody was added to the cell lysate to immunoprecipitate the EGR-1-binding genome. The immunoprecipitated DNA (ChIP DNA) was then released and subjected to PCR to amplify the GGPPS promoter. IgG was the immunoglobulin used as the negative control for the EGR-1 antibody groups. The 2% input groups amplified with the RPL30 show that the total genomic DNA subjected to the ChIP was equal. The lower panel is the result of real-time PCR using the ChIP DNA and 2% input control DNA as the template. The fold enrichments were determined by the 2−▵▵CT method using RPL30 as the normalizer. C: EMSA was used to detect the direct binding of EGR-1 with its consensus-binding site of the GGPPS promoter in HEK293 cells treated with 10% CSE for 2 hours. The bands indicate the EGR-1 probe complexes. Competition assays with cold probe indicated a decrease in the probe-protein bands. Mutant probes for the EGR-1-binding site (Mut1, Mut2) showed no decrease in the probe-protein bands, whereas the probe-protein complexes were decreased for the mutant of the consensus SP1-binding site (Mut3). The underlined letters represent the consensus binding site of EGR-1. The bold letters represent the mutant site for EGR-1 only (Mut1), SP1 only (Mut3) and both of EGR-1 and SP1 (Mut2). Along with the increase in the EGR-1 antibody concentration (0.5 to 4 μg), the probe-protein complexes were decreased while no decrease emerged along with the increase in the SP1 antibody concentration (0.5 to 4 μg), which confirmed that both of the bands were EGR-1 (not SP1) forming binding complexes with the probe. (*P < 0.05, compared with the control group without CSE).
EGR-1 Can Promote GGPPS Transcription under CSE Stimulation
It has been reported that CSE can induce EGR-1 accumulation very quickly.17, 18, 19 We found that EGR-1 activation normally precedes GGPPS expression when Beas-2B cells were stimulated with CSE. The accumulation of EGR-1 protein quickly increased within 30 minutes, and the protein level of GGPPS increased 1 to 6 hours after continuous CSE treatment (Figure 2A, upper panel). Besides continuous CSE stimulation, we also pretreated the Beas-2B cells with CSE for only 5 minutes and changed the medium back to the normal to mimic the transient act of smoking, and collected the cell lysates at the indicated times (Figure 2A, lower panel). We found that EGR-1 expression was increased from 10 minutes to 1 hour. This observation implied that the 5 minutes CSE stimulation on the Beas-2B cells could increase EGR-1 expression from 10 minutes and continue to 1 hour. Though 1 hour of EGR-1 expression is a very short time period, it may be long enough to continue EGR-1 expression for a one-pack-per-day smoker as they smoke cigarettes one by one. When we overexpressed exogenous EGR1 with an adenovirus to mimic elevated EGR-1 under CSE stress, we found that GGPPS mRNA and protein were also significantly increased (Figure 2B). To determine whether the transcription of GGPPS was in fact dependent on EGR-1, we exposed cells to CSE for 4 hours after transfection with EGR1 siRNA (the efficiency of the EGR1 siRNA is shown in Figure 2C). The results indicated that GGPPS mRNA production could be markedly inhibited by EGR1 siRNA (Figure 2D). Loss of EGR-1 function (Figure 2E) using dominant negative EGR-1 (DnEGR-1) also confirmed the result of the siRNA (Figure 2E). To further determine whether the induction of GGPPS required the direct interaction of EGR-1 with the GGPPS promoter, a region (−190 to +98) of the GGPPS promoter was cloned upstream of a luciferase reporter construct. Figure 2F shows that GGPPS promoter-driven luciferase activity was significantly increased by CSE treatment, but partially decreased by DnEGR-1 overexpression. The results suggest that the transcription of GGPPS is mostly dependent on the EGR-1 DNA binding activity; however, the increase in GGPPS promoter-driven luciferase activity was not totally blocked by DnEGR-1 overexpression, as with the result of the GGPPS mRNA level (Figure 2E). Thus, we transfected the cells with the mutant GGPPS promoter-driven luciferase reporter, which contained the mutant EGR-1-binding site. After CSE treatment, the mutant GGPPS promoter-driven luciferase activity was also partially decreased compared to the wild type (Figure 2G). Although GGPPS transcription is mostly dependent on EGR-1, this result indicated that there might be other post-transcriptional ways to regulate GGPPS expression.
Figure 2.
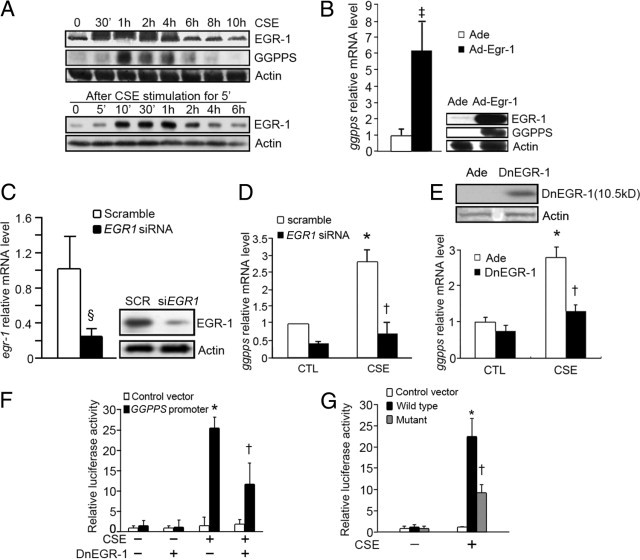
Increased GGPPS transcription under 10% cigarette smoke extract (CSE) treatment was dependent on EGR-1. A: Western blot analysis of EGR-1 and GGPPS during the time course of 10% CSE treatment. Beas-2B cells were treated with continuous 10% CSE until the samples were collected at the indicated times (upper panel). The EGR-1 level increased within 30 minutes after CSE treatment and decayed after 4 hours, and GGPPS increased after EGR-1 at 1 hour. In addition, Beas-2B cells were pretreated with CSE for only 5 minutes and then returned to the normal growth medium. The cells were lysed at the indicated times and subjected to Western blotting (lower panel). The EGR-1 level increased from 10 minutes to 1 hour. B: Overexpression of EGR-1 through Ad-Egr-1 in Beas-2B cells could significantly enhance GGPPS expression as CSE did. Total RNA and protein were collected and subjected to qPCR and Western blot analysis, respectively, after Ad-Egr-1 had been infected for 48 hours. C: The efficiency of EGR1 siRNA in Beas-2B cells. Beas-2B cells were transfected with EGR1 siRNA for 48 hours. Total RNA and protein were extracted and subjected to qPCR and Western blotting, respectively. D and E: Beas-2B cells were transfected with EGR1 siRNA or a scrambled control (D) and infected with DnEGR-1 adenovirus or a control adenovirus (Ade) (E) for 48 hours. Each group was then treated (10% CSE) or not treated (CTL) with CSE for 4 hours. Total RNA was collected and subjected to qPCR. The overexpression efficiency of DnEGR-1 is shown in panel E by Western blot. F: Beas-2B cells were transfected with DnEGR-1 and the luciferase reporter that contained the wild-type GGPPS promoter. The Renilla luciferase plasmid was used as an internal control. After 10% CSE stimulation for 20 hours, luciferase activity was determined. The data were normalized by the Renilla luciferase activity and expressed as a fold-induction with respect to control cells. G: Beas-2B cells were transfected with the luciferase reporter that contained the wild-type or mutant GGPPS promoter. The Renilla luciferase plasmid was used as an internal control. After 10% CSE stimulation for 20 hours, luciferase activity was determined. The data were normalized by the Renilla luciferase activity and expressed as a fold induction with respect to control cells. (*P < 0.05: compared with the control group untreated with CSE; †P < 0.05: compared with CSE treatment only; ‡P < 0.05: compared with the Ade group; and §P < 0.01: compared with the scrambled group).
MAPK ERK 1/2 Is Reactivated in an EGR-1-Dependent Manner
A measurement of the long-term time course activation of ERK 1/2 demonstrated that there were two peaks of ERK 1/2 activation after CSE stimulation: the first at 10 minutes and the second at approximately 4 hours (Figure 3A). It is obvious that the first one was responsible for EGR-1 accumulation, but we did not know what was responsible for the second activation of ERK 1/2. When the transcription activity of EGR-1 was inhibited, it seemed that the entire dynamic response of ERK 1/2 was changed. The first peak of p-ERK 1/2 was delayed from 10 minutes to 1 hour, and the second peak of p-ERK 1/2 at 4 hours was blocked (Figures 3, B and C). These results suggest that EGR-1-promotes GGPPS transcription and might be responsible for the second activation and that inhibition of the transcriptional activity of EGR-1 could not only inhibit the second activation of ERK 1/2 but also the entire dynamic response of ERK 1/2. The U0126 administration assay showed that the accumulation of EGR-1 by CSE at both the early and late times was largely dependent on ERK 1/2 activity (Figure 3D).
Figure 3.
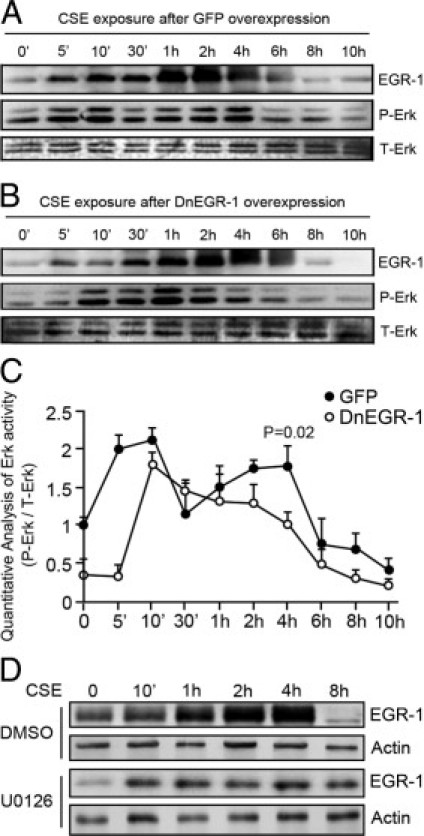
ERK 1/2 was reactivated in an EGR-1-dependent manner. A: Beas-2B cells infected with control adenovirus [green fluorescent protein (GFP)] were exposed to 10% cigarette smoke extract (CSE) for as long as 10 hours, and the protein samples were collected at the indicated times and subjected to Western blotting. We found two peaks of ERK 1/2 activation after CSE stimulation: one at 10 minutes and one at 4 hours. B: When the cells were infected with adenovirus carrying the DnEGR-1 gene before the 10% CSE challenge, the second peak activation of ERK 1/2 at 4 hours disappeared. C: The light density analysis of phosphorylated ERK 1/2 in shown in panels A and B. The data were normalized by total ERK 1/2 expression and expressed as fold induction with respect to the value of the time point at 0 hours. The light density was calculated using ImageQuant software. D: Beas-2B cells were pretreated with dimethyl sulfoxide or U0126 (10 μmol/L) for 2 hours and stimulated by 10% CSE for the indicated times. The cells were lysed and subjected to Western blotting.
Exogenous EGR-1 Might Promote ERK 1/2 Activation through Promoting Ras Membrane Association by GGPPS
GGPPS can catalyze the synthesis of isoprenoid moieties, such as geranylgeranyl diphosphate (GGPP),21, 22 which can be used for the geranylgeranylation of small GTP-binding proteins, including Ras and Ras-related proteins. Prenylation of Ras is generally considered essential for its membrane association and normal activity.24, 25 Herein, we hypothesized that EGR-1 might enhance the Ras signaling pathway by promoting GGPPS expression, which results in increased Ras prenylation and ERK 1/2 reactivation. To prove this hypothesis, we first determined the membrane association of Ras when EGR-1 was overexpressed in Beas-2B cells (Figure 4A) using ultracentrifugation (see Materials and Methods). In this panel, the S100 lanes represent the cytosolic content, which is rich in unbound Ras, whereas the P100 lanes represent the membrane content, which is rich in membrane-bound Ras. As shown in this panel, more K-Ras was binding to the cell membrane in the EGR-1 adenovirus infection (Ad-Egr-1) group compared to the control adenovirus (Ade) group, which means that EGR-1 may enhance the binding of Ras to the membrane. We then measured Ras prenylation using Triton-X114 (Figure 4B) (see Materials and Methods). In this panel, the upper lanes represent the aqueous cytosolic content, which is rich in unprenylated Ras, whereas the lower lanes represent the organic cell membrane content, which is rich in prenylated Ras. In the vehicle group, which overexpressed EGR-1, there is much more prenylated Ras compared to the Ade group, which means that the prenylation levels of Ras can be elevated after EGR-1 overexpression. To further determine which type of prenylation occurred to Ras, GTI (a GGTase inhibitor) and FTI (a FTase inhibitor) were administrated when EGR-1 was overexpressed in Beas-2B cells (see Materials and Methods). The prenylated Ras in the GTI group largely decreased compared to the vehicle group. However, the decrease of the prenylated Ras in the FTI group was much less significant compared to the GTI group. These results showed that it was the geranylation (not the farnesylation modification) that was responsible for Ras prenylation. When we introduced GGPPS siRNA into cells, K-Ras prenylation was largely inhibited (Figure 4C). EGR-1-augmented Ras prenylation is responsible for ERK 1/2 activation because ERK 1/2 activation was inhibited when Ras prenylation was blocked by inhibitor administration (Figure 4D). Also, to determine whether the CSE-stimulated Ras prenylation increasing at 4 hours was dependent on EGR-1 and GGPPS, we infected the cells with the DnEGR-1 or control adenovirus and transfected with scrambled or GGPPS siRNA. After exposure to 10% CSE for 4 hours, membrane-binding K-Ras was increased by this treatment while decreased by DnEGR-1 and siGGPPS administration (Figure 4E).
Figure 4.
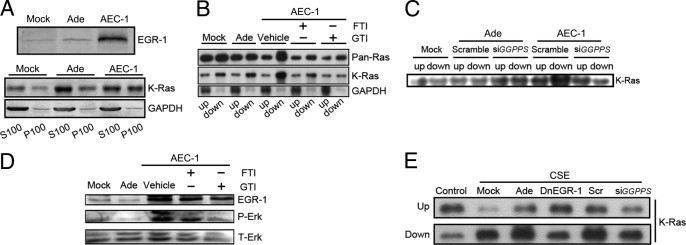
Exogenous expression of EGR-1 could enhance Ras prenylation and membrane association and ERK 1/2 reactivation in Beas-2B cells. A: Beas-2B cells were infected with EGR1 adenovirus infection (Ad-Egr-1) or control adenovirus (Ade). The cells were then lysed and separated into membrane (P100) and cytosolic (S100) portions and subjected to immunoprecipitation and Western blotting against K-Ras and glyceraldehydes-3-phosphate dehydrogenase (GADPH), which was used to show that the separation of membrane and cytosol was successful. The result showed that membrane-associated K-Ras was enhanced by the exogenous expression of EGR-1. B: Beas-2B cells were infected with EGR1 adenovirus infection (Ad-Egr-1) or control adenovirus (Ade). The cell lysates were extracted with Triton-X114, and then the aqueous (upper) and organic (lower) phases were subjected to immunoprecipitation and Western blotting against Pan-Ras, K-Ras, and GAPDH. The GAPDH was used to show that the separation of the aqueous and organic phases was successful. The EGR1 adenovirus-infected cells were also pretreated with FTI, GTI, or vehicle for 48 hours before cell collection. The results showed that prenylated Pan-Ras and K-Ras (lower) were enhanced by exogenous EGR-1 expression. The farnesylation inhibitor FTI has only a slight effect on prenylation, whereas the geranylation inhibitor GTI was able to block the prenylation of both Pan-Ras and K-Ras. This suggested that EGR-1 is able to enhance Ras geranylation. C: Beas-2B cells were co-infected with EGR1 adenovirus (Ad-Egr-1) and a scrambled control or GGPPS siRNA and also co-infected with control adenovirus (Ade). The cell lysate was also subjected to membrane separation through TritonX-114, and then the aqueous (upper) and organic (lower) phases were subjected to immunoprecipitation and Western blotting against K-Ras. EGR-1-enhanced K-Ras prenylation was blocked by GGPPS small-interfering RNA. D: EGR-1 was overexpressed in Beas-2B cells, which was able to activate ERK 1/2. This activation could be inhibited by both GTI and FTI, which suggests that prenylation is crucial to EGR-1-dependent mitogen-activated protein kinase ERK 1/2 activation. E: Beas-2B cells were infected with the DnEGR-1 or control adenovirus and transfected with scrambled or GGPPS small-interfering RNA. After exposure to 10% CSE for 4 hours, the cell lysates were extracted with Triton-X114, and then the aqueous (upper) and organic (lower) phases were subjected to immunoprecipitation and Western blotting against K-Ras. K-Ras membrane binding was increased by 10% CSE treatment for 4 hours while it was decreased by DnEGR-1 and small-interfering GGPPS administration.
MAPK ERK 1/2 Is Reactivated in a GGPPS-Dependent Manner
When GGPPS was knocked-down by siRNA, the second activation of ERK 1/2 at 4 hours was inhibited (Figures 5A–C). However, similar to the results in Figure 3, the entire kinetics of ERK 1/2 appeared to be inhibited after GGPPS was knocked down because the basal prenylation of Ras had already changed. These results suggest that EGR-1-promoted GGPPS transcription was responsible for ERK 1/2 re-activation, and knockdown of GGPPS might inhibit the dynamic response of ERK 1/2 to CSE stimulation. Taken together, our data suggest that MAPK ERK 1/2 might be positively regulated by EGR-1 through increasing Ras prenylation by promoting GGPPS transcription.
Figure 5.
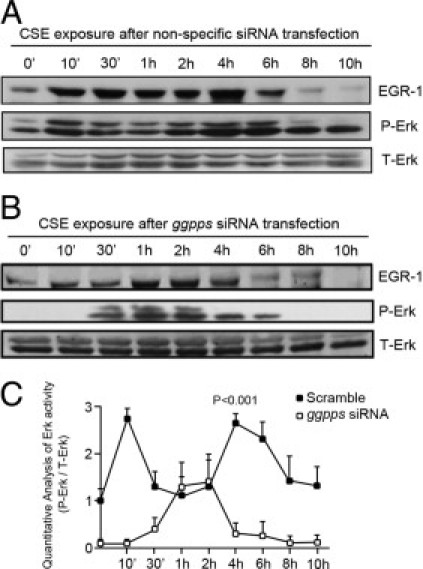
ERK 1/2 was reactivated in a GGPPS-dependent manner. A: Beas-2B cells infected with adenovirus carrying the scrambled control were exposed to 10% cigarette smoke extract (CSE) for as long as 10 hours, and protein samples were collected at the indicated times and subjected to Western blotting. We also found two peaks of ERK 1/2 activation after CSE stimulation: one at 10 minutes and one at 4 hours. B: When the cells were infected with adenovirus carrying the GGPPS small-interfering RNA before the 10% CSE challenge, the second peak activation of ERK 1/2 at 4 hours disappeared. C: The light density analysis of phosphorylated ERK 1/2 in panels A and B. The data are normalized by total ERK 1/2 expression and expressed as a fold induction with respect to the value of the time point at 0 hours. The light density was calculated using ImageQuant software.
Discussion
It has been reported that EGR-1 is regulated by various MAPK pathways, in particular the ERK 1/2 pathway.33, 34, 35, 36, 37, 38 We have suggested that EGR-1 can also stimulate HSP70 elevation in an ERK 1/2-dependent manner, which is thought to regulate cigarette smoke-induced inflammatory processes.18 Here we provide the first evidence that EGR-1 could activate MAPK ERK 1/2 in a positive feedback manner through increasing Ras prenylation by promoting GGPPS transcription in lung epithelial cells during CSE challenge (Figure 6). The sustainably activated ERK 1/2 in this positive feedback loop may lead to the progression of chronic lung inflammation and smoking-related pulmonary diseases.
Figure 6.
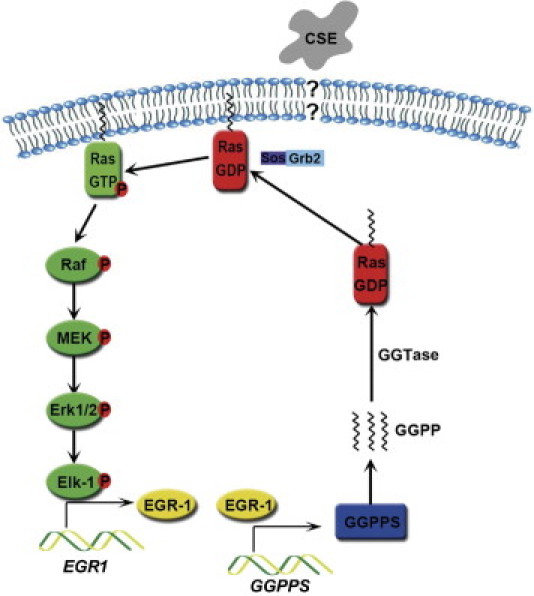
The scheme of the EGR-1/GGPPS/ERK 1/2 pathway. Cigarette smoke extract (CSE) exposure can activate the MAPK pathway, which is responsible for EGR-1 activation. Up-regulated EGR-1 can activate transcription of its target gene GGPPS, which is responsible for geranylation of Ras GTP-binding proteins. Thus, geranylated Ras can bind to the plasma membrane and reactivate the ERK 1/2 MAPK pathway.
Ras-Raf-MAPK cascades are key signaling pathways involved in the regulation of normal cell functions, and they also play roles in a variety of pathological stress-exposure processes.39 Thus, any aberrant regulation of MAPK cascades may contribute to cancer and other human diseases. Among the signaling molecules of Ras-Raf-MAPK cascades, the small GTPase Ras is the most frequently mutated oncogene in human cancers. Mutant Ras then activates Raf, the MEK1/2 dual-specificity protein kinase, which in turn activates ERK 1/2. ERK 1/2 activation also up-regulates expression of EGFR, promoting an autocrine growth loop that is critical for tumor growth.40, 41 Under normal conditions, Ras prenylation by GGPP or farnesyl diphosphate, which is the production of 3-hydroxy-3-methyl-glutaryl-Coenzyme A reductase, is necessary for its membrane association and activation.23 Simvastatin, which is an inhibitor of 3-hydroxy-3-methyl-glutaryl-Coenzyme A reductase, is reported to inhibit CSE-induced ERK 1/2 activation and MMP-9 production in rat alveolar macrophages, and the addition of GGPP or farnesyl diphosphate can reverse simvastatin-induced inhibition.42 Thus, prenylated Ras can mediate CSE-induced ERK 1/2 activation and MMP-9 production, which is a key factor leading to chronic obstructive pulmonary disease (COPD). It has been reported that ERK 1/2 also regulates the activation of EGR-1.33 Here we found that EGR-1 could also provide positive feedback to regulate Ras activation through its target gene GGPPS under CSE stress. The ChIP (Figure 1B) and luciferase assays (Figure 2, F and G) demonstrated that GGPPS is a target gene of EGR-1. However, we did not observe the expected super shift band, which indicated the specific binding of EGR-1 to the GGPPS promoter in the EMSA assay (Figure 1C). We think it is because of the recognition of EGR-1 antibody with its antigen. The binding of EGR-1 antibody may screen EGR-1 from the binding of probe, thus there is few labeled probe in antibody and antigen complex and it is very hard to show the super shift band. But the EGR-1-probe complex was decreased with the concentration of EGR-1 antibody increasing, which suggested that the formed complex did involve EGR-1. Additionally, we hypothesized the reason why two probe-protein bands were observed was that there were some other proteins that could combine with EGR-1 to form a complex, which helped EGR-1 activate GGPPS transcription under CSE stimulation. Thus, we could observe two probe-protein complexes that were both decreased along with the increased concentration of EGR-1 antibody. The mechanism that EGR-1 promoted GGPPS transcription is not specific to cigarette smoke stress and may also be involved in other stresses (eg, hyperinsulinism exposure to adipocytes that enhances insulin resistance development).43
GGPPS is responsible for producing GGPP, and prenylation modification of Ras and increases Ras membrane association. Activated Ras then stimulates the secondary phosphorylation and activation of ERK 1/2, and the Ras-Raf-MAPK signaling cascades are then re-augmented. The most solid evidence we presented in Figure 3, Figure 5 was that ERK 1/2 could be re-activated after 4 hours of CSE stimulation, in which this could be substantially inhibited by the inhibition of EGR-1 transcriptional activity or GGPPS knockdown. Although the elevation of EGR-1 expression by CSE stimulation began from 0.5 to 1 hour (Figure 2A) and could activate the GGPPS promoter during this time frame, the time was still required for GGPPS mRNA to translate into protein, which also needed time to synthesize the GGPP. Thus, the second activation of ERK 1/2 was delayed 2 to 4 hours. However, it appeared that the entire activity of ERK 1/2 was altered. The first p-ERK 1/2 peak was delayed from 10 minutes to 1 hour when DnEGR-1 was overexpressed, and GGPPS knockdown was used. One possibility is that inhibition of EGR-1 transcriptional activity or GGPPS knockdown may inhibit the basal activity and entire dynamic response of ERK 1/2. This response may then lead to delayed ERK 1/2 activation and a reduction in the response to CSE stimulation.
Cigarette smoke is a major risk factor for COPD. Smoking-induced inflammatory reactions in the airways and lung parenchyma composed of neutrophils and alveolar macrophages have long been accepted to be the major cause of the development of airway abnormalities and emphysema, and consequently COPD in susceptible smokers.44, 45, 46, 47 Our previous report showed that cigarette smoke is a key factor in the development of COPD through a transcription factor EGR-1-dependent pathway.19 When pulmonary cells are exposed to CSE, EGR-1 mediates production of MMP-2 and membrane type 1 metalloprotease (MT1-MMP), which are responsible for matrix remodeling in cigarette smoke-related emphysema.17 In addition, EGR-1 can also stimulate HSP70 elevation in an ERK 1/2-dependent manner, which is thought to regulate the cigarette smoke-induced inflammatory processes.18 In this manner, EGR-1 exerts an effect on chronic disease through the direct promotion of COPD-related gene expression under cigarette smoke exposure stress.
In our study, we show that EGR-1 exerts its long-term effects in a different manner by reactivating ERK 1/2 by promoting GGPPS transcription. In this feedback loop, GGPPS, which is a novel target gene of EGR-1, can re-activate ERK 1/2 as long as CSE or cigarette smoke stimulation occurs. However, EGR-1 is usually considered a transiently elevated response factor in short-term stress situations. As shown in Figure 2A, the levels of EGR-1 and GGPPS will diminish over time. Nevertheless, we believe that this short-term stress does not really exist in most situations. A stimulus such as cigarette smoke can frequently and repeatedly affect lung cells. Furthermore, the result of Figure 2A (lower panel) implied that a 5-minute stimulation of CSE on the Beas-2B cells could increase EGR-1 expression from 10 minutes and continued to 1 hour. Though 1 hour of EGR-1 expression is a very short time period, it may be long enough to continue EGR-1 expression for a one-pack-per-day smoker as they smoke cigarettes one by one. Thus, EGR-1 can be continuously activated to induce GGPPS production. Long-term intermittent exposure of CS could cause a constant EGR-1 increase rather than transient elevation, in which the sustained activation of MAPK and/or other proteins may promote the aberrant cell progression that is responsible for cigarette smoke-associated diseases.
Long-term or constantly activated ERK 1/2 can promote chronic inflammation. For example, activated ERK 1/2 is involved in the formation of CSE-induced death-inducing signaling complex in human lung fibroblasts48 and results in sustained lung damage. When we exposed the mice to cigarette smoke for 27 weeks, we found that EGR-1 and GGPPS appeared to be constantly activated and that they promoted inflammatory responses and tissue damage.26 As a novel EGR-1 target gene, GGPPS may mediate the function of EGR-1 during cigarette smoke exposure by driving sustained ERK 1/2 activation. In addition, the transcription of several other EGR-1 target genes (eg, HO-1,49 human IL-2,12 IL-2 receptor,14 Fas/CD95,15 and intercellular adhesion molecule 116) may also be inhibited by the DnEGR-1 adenovirus. Here we showed that as a novel target gene of EGR-1, GGPPS, may mediate the chronic pulmonary inflammation responses which is regulated EGR-1 during cigarette smoke exposure by driving sustained ERK 1/2 activation.
The possibility for the constitutive elevation of EGR-1 is the epigenetic modification of the EGR-1 promoter. Intermittent cigarette smoke exposure might modify this epigenetic status similar to CpG island the cytosine and the guanine covalently bonded by a phosphodiester bond methylation or histone acetylation. In this manner, the acute increase of EGR-1 by CSE could produce much longer-lasting effects, such as inflammation and apoptosis, in lung tissues through reactivating ERK 1/2. These events are the cause of COPD and many other diseases, and cannot be reversed spontaneously and completely by our bodies even when we stop smoking.
Footnotes
Supported by grants from the National Basic Research Program of China (2009CB918703 to C.-J.L.) and the Key Grant of Jiangsu Natural Science Foundation (06KJA31024 to C.-J.L.).
N.S. and Y.S. contributed equally to this work.
Contributor Information
Hua-Qun Chen, Email: hqzy@yahoo.com.
Chao-Jun Li, Email: licj@nju.edu.cn.
References
- 1.Cao X.M., Guy G.R., Sukhatme V.P., Tan Y.H. Regulation of the Egr-1 gene by tumor necrosis factor and interferons in primary human fibroblasts. J Biol Chem. 1992;267:1345–1349. [PubMed] [Google Scholar]
- 2.Chavrier P., Zerial M., Lemaire P., Almendral J., Bravo R., Charnay P. A gene encoding a protein with zinc fingers is activated during G0/G1 transition in cultured cells. EMBO J. 1988;7:29–35. doi: 10.1002/j.1460-2075.1988.tb02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gashler A., Sukhatme V.P. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 4.Yan S.F., Pinsky D.J., Mackman N., Stern D.M. Egr-1: is it always immediate and early? J Clin Invest. 2000;105:553. doi: 10.1172/JCI9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan S.F., Lu J., Zou Y.S., Soh-Won J., Cohen D.M., Buttrick P.M., Cooper D.R., Steinberg S.F., Mackman N., Pinsky D.J., Stern D.M. Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- 6.Rolli M., Kotlyarov A., Sakamoto K.M., Gaestel M., Neininger A. Stress-induced stimulation of early growth response gene-1 by p38/stress-activated protein kinase 2 is mediated by a cAMP-responsive promoter element in a MAPKAP kinase 2-independent manner. J Biol Chem. 1999;274:19559–19564. doi: 10.1074/jbc.274.28.19559. [DOI] [PubMed] [Google Scholar]
- 7.O'Donovan K.J., Tourtellotte W.G., Millbrandt J., Baraban J.M. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–173. doi: 10.1016/s0166-2236(98)01343-5. [DOI] [PubMed] [Google Scholar]
- 8.Liu C., Rangnekar V.M., Adamson E., Mercola D. Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 1998;5:3–28. [PubMed] [Google Scholar]
- 9.Lim C.P., Jain N., Cao X. Stress-induced immediate-early gene, egr-1, involves activation of p 38/JNK 1. Oncogene. 1998;16:2915–2926. doi: 10.1038/sj.onc.1201834. [DOI] [PubMed] [Google Scholar]
- 10.Huang R.P., Adamson E.D. A biological role for Egr-1 in cell survival following ultra-violet irradiation. Oncogene. 1995;10:467–475. [PubMed] [Google Scholar]
- 11.Datta R., Taneja N., Sukhatme V.P., Qureshi S.A., Weichselbaum R., Kufe D.W. Reactive oxygen intermediates target CC(A/T)6GG sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc Natl Acad Sci USA. 1993;90:2419–2422. doi: 10.1073/pnas.90.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skerka C., Decker E.L., Zipfel P.F. A regulatory element in the human interleukin 2 gene promoter is a binding site for the zinc finger proteins Sp1 and EGR-1. J Biol Chem. 1995;270:22500–22506. doi: 10.1074/jbc.270.38.22500. [DOI] [PubMed] [Google Scholar]
- 13.Kramer B., Meichle A., Hensel G., Charnay P., Kronke M. Characterization of an Krox-24/Egr-1-responsive element in the human tumor necrosis factor promoter. Biochim Biophys Acta. 1994;1219:413–421. doi: 10.1016/0167-4781(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 14.Lin J.X., Leonard W.J. The immediate-early gene product Egr-1 regulates the human interleukin-2 receptor beta-chain promoter through noncanonical Egr and Sp1 binding sites. Mol Cell Biol. 1997;17:3714–3722. doi: 10.1128/mcb.17.7.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinkel A., Aicher W.K., Haas C., Zipfel P.F., Peter H.H., Eibel H. Transcription factor Egr-1 activity down-regulates Fas and CD23 expression in B cells. J Immunol. 1997;159:2678–2684. [PubMed] [Google Scholar]
- 16.Maltzman J.S., Carmen J.A., Monroe J.G. Transcriptional regulation of the Icam-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J Exp Med. 1996;183:1747–1759. doi: 10.1084/jem.183.4.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ning W., Dong Y., Sun J., Li C., Matthay M.A., Feghali-Bostwick C.A., Choi A.M. Cigarette smoke stimulates matrix metalloproteinase-2 activity via EGR-1 in human lung fibroblasts. Am J Respir Cell Mol Biol. 2007;36:480–490. doi: 10.1165/rcmb.2006-0106OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C.J., Ning W., Matthay M.A., Feghali-Bostwick C.A., Choi A.M.K. MAPK pathway mediates EGR-1-HSP70-dependent cigarette smoke-induced chemokine production. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1297–L1303. doi: 10.1152/ajplung.00194.2006. [DOI] [PubMed] [Google Scholar]
- 19.Ning W., Li C.J., Kaminski N., Feghali-Bostwick C.A., Alber S.M., Di Y.P., Otterbein S.L., Song R., Hayashi S., Zhou Z. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101:14895–14900. doi: 10.1073/pnas.0401168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rilling H.C., Breunger E., Epstein W.W., Crain P.F. Prenylated proteins: the structure of the isoprenoid group. Science. 1990;247:318–320. doi: 10.1126/science.2296720. [DOI] [PubMed] [Google Scholar]
- 21.Farnsworth C.C., Gelb M.H., Glomset J.A. Identification of geranylgeranyl-modified proteins in HeLa cells. Science. 1990;247:320–322. doi: 10.1126/science.2296721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farnsworth C.C., Wolda S.L., Gelb M.H., Glomset J.A. Human lamin B contains a farnesylated cysteine residue. J Biol Chem. 1989;264:20422–20429. [PMC free article] [PubMed] [Google Scholar]
- 23.Wright L.P., Philips M.R. Thematic review series: lipid posttranslational modifications CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang F.L., Casey P.J. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 25.Glomset J.A., Farnsworth C.C. Role of protein modification reactions in programming interactions between Ras-related GTPases and cell membranes. Annu Rev Cell Biol. 1994;10:181–205. doi: 10.1146/annurev.cb.10.110194.001145. [DOI] [PubMed] [Google Scholar]
- 26.Shen N., Gong T., Wang J.D., Meng F.L., Qiao L., Yang R.L., Xue B., Pan F.Y., Zhou X.J., Chen H.Q. Cigarette smoke-induced pulmonary inflammatory responses are mediated by EGR-1/GGPPS/MAPK signaling. The Am J Pathol. 2011;178:110–118. doi: 10.1016/j.ajpath.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vayssier-Taussat M., Camilli T., Aron Y., Meplan C., Hainaut P., Polla B.S., Weksler B. Effects of tobacco smoke and benzo[a]pyrene on human endothelial cell and monocyte stress responses. Am J Physiol Heart Circ Physiol. 2001;280:H1293–H1300. doi: 10.1152/ajpheart.2001.280.3.H1293. [DOI] [PubMed] [Google Scholar]
- 28.Levkovitz Y., Baraban J.M. A dominant negative inhibitor of the Egr family of transcription regulatory factors suppresses cerebellar granule cell apoptosis by blocking c-Jun activation. J Neurosci. 2001;21:5893–5901. doi: 10.1523/JNEUROSCI.21-16-05893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang A.M., Doyle M.V., Mark D.F. Quantitation of mRNA by the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86 doi: 10.1073/pnas.86.24.9717. 9717–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiribau C.B., Cheng L., Cucoranu I.C., Yu Y.S., Clempus R.E., Sorescu D. FoxO3A regulates peroxiredoxin III expression in human cardiac fibroblasts. J Biol Chem. 2008;283:8211–8217. doi: 10.1074/jbc.M710610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim E., Ambroziak P., Otto J.C., Taylor B., Ashby M., Shannon K., Casey P.J., Young S.G. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J Biol Chem. 1999;274:8383–8390. doi: 10.1074/jbc.274.13.8383. [DOI] [PubMed] [Google Scholar]
- 32.Goalstone M.L., Leitner J.W., Golovchenko I., Stjernholm M.R., Cormont M., Le Marchand-Brustel Y., Draznin B. Insulin promotes phosphorylation and activation of geranylgeranyltransferase II: Studies with geranylgeranylation of rab-3 and rab-4. J Biol Chem. 1999;274:2880–2884. doi: 10.1074/jbc.274.5.2880. [DOI] [PubMed] [Google Scholar]
- 33.Mishra S., Fujita T., Lama V.N., Nam D., Liao H., Okada M., Minamoto K., Yoshikawa Y., Harada H., Pinsky D.J. Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci USA. 2006;103:5191–5196. doi: 10.1073/pnas.0600241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease. Cell Biochem Biophys. 2005;43:167–188. doi: 10.1385/CBB:43:1:167. [DOI] [PubMed] [Google Scholar]
- 35.McNeil L.K., Starr T.K., Hogquist K.A. A requirement for sustained ERK signaling during thymocyte positive selection in vivo. Proc Natl Acad Sci USA. 2005;102:13574–13579. doi: 10.1073/pnas.0505110102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy L.O., MacKeigan J.P., Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mercer B.A., Kolesnikova N., Sonett J., D'Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 2004;279:17690–17696. doi: 10.1074/jbc.M313842200. [DOI] [PubMed] [Google Scholar]
- 38.Baron V., Duss S., Rhim J., Mercola D. Antisense to the early growth response-1 gene (Egr-1) inhibits prostate tumor development in TRAMP mice. Ann NY Acad Sci. 2003;1002:197–216. doi: 10.1196/annals.1281.024. [DOI] [PubMed] [Google Scholar]
- 39.Roberts P.J., Der C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 40.McCubrey J.A., Steelman L.S., Chappell W.H., Abrams S.L., Wong E.W., Chang F., Lehmann B., Terrian D.M., Milella M., Tafuri A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adjei A.A. The role of mitogen-activated ERK-kinase inhibitors in lung cancer therapy. Clin Lung Cancer. 2005;7:221–223. doi: 10.3816/CLC.2005.n.040. [DOI] [PubMed] [Google Scholar]
- 42.Kim S.E., Thuy T.T.T., Lee J.H., Ro J.Y., Bae Y.A., Kong Y., Ahn J.Y., Lee D.S., Oh Y.M., Lee S.D. Simvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extract. Exp Mol Med. 2009;41:277–287. doi: 10.3858/emm.2009.41.4.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen N., Yu X., Pan F.Y., Gao X., Xue B., Li C.J. An early response transcription factor: Egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J Biol Chem. 2011;286:14508–14515. doi: 10.1074/jbc.M110.190165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Black L.F., Kueppers F. Alpha1-antitrypsin deficiency in nonsmokers. Am Rev Respir Dis. 1978;117:421. doi: 10.1164/arrd.1978.117.3.421. [DOI] [PubMed] [Google Scholar]
- 45.Gadek J.E., Fells G.A., Crystal R.G. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science. 1979;206:1315–1316. doi: 10.1126/science.316188. [DOI] [PubMed] [Google Scholar]
- 46.Hunninghake G.W., Crystal R.G. Cigarette smoking and lung destruction: Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis. 1983;128:833–838. doi: 10.1164/arrd.1983.128.5.833. [DOI] [PubMed] [Google Scholar]
- 47.Janus E.D., Phillips N.T., Carrell R.W. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1:152–154. doi: 10.1016/s0140-6736(85)91916-6. [DOI] [PubMed] [Google Scholar]
- 48.Park J.W., Yoon J.Y., Kim Y.J., Kyung S.Y., Lee S.P., Jeong S.H., Moon C. Extracellular signal-regulated kinase (ERK) inhibition attenuates cigarette smoke extract (CSE) induced-death inducing signaling complex (DISC) formation in human lung fibroblasts (MRC-5) cells. J Toxicol Sci. 2010;35:33–39. doi: 10.2131/jts.35.33. [DOI] [PubMed] [Google Scholar]
- 49.Chen H., Wang L., Gong T., Yu Y., Zhu C., Li F., Wang L., Li C. EGR-1 regulates Ho-1 expression induced by cigarette smoke. Biochem Biophys Res Commun. 2010;396:388–393. doi: 10.1016/j.bbrc.2010.04.102. [DOI] [PubMed] [Google Scholar]