

Abstract
To explore the role of antigen-specific CD4+ T cells in glomerulonephritis, we administered ovalbumin 323–339 peptide conjugated to glomerular-binding polyclonal antibody and induced disease in RAG1−/− mice with CD4+ T cells from OT2 × RAG1−/− mice. These OT2 × RAG1−/− mice have a transgenic T-cell receptor specific for this peptide. When CD4+ T cells were primed in vivo, crescentic glomerulonephritis developed after 21 days in mice given peptide-conjugated glomerular-binding antibody but not unconjugated antibody control. We then investigated the relative roles of TH1 and TH17 cells, using Fab2 fragments of glomerular-binding antibody to exclude a role for antibody in this model. T cells from OT2 × RAG1−/− mice were polarized in vitro, and TH1 or TH17 cell lines were injected into mice that were also given peptide-conjugated Fab2 or unconjugated Fab2 control, giving four experimental groups. After 21 days crescentic glomerulonephritis was seen in mice receiving TH17 cells and peptide-conjugated Fab2 but in none of the other three groups. These results suggest that TH17 but not TH1 cells can induce crescentic glomerulonephritis.
The ability of passively administered antibody to cause glomerulonephritis has been long established,1 and studies in Fc receptor–deficient mice have confirmed a role for immune complexes in both autologous nephrotoxic nephritis and lupus models.2, 3 Evidence for T-cell–mediated, antibody-independent disease was suggested in early studies of lymphocyte transfer4 and more recently in murine autologous nephrotoxic nephritis using B-cell–deficient mice.5 T-cell effector mechanisms have also been demonstrated in a rat model of anti–glomerular basement membrane (GBM) disease6 and in a mouse model of antineutrophil cytoplasmic antibody–associated vasculitis.7, 8 The subset of TH17 CD4+ T cells has recently emerged as a key mediator of tissue damage in a number of systems where TH1 cells were previously thought to play a prominent role.8 Their pathogenicity has been proven in colitis, multiple sclerosis, and arthritis models.9, 10, 11, 12 However, the suggestion that TH17 cells are pathogenic in all cases and TH1 cells are not is likely to be simplistic,13 and data have shown that both TH1 and TH17 cells can induce disease in uveitis and multiple sclerosis.14, 15
Work based largely on studies in the autologous nephrotoxic nephritis model has suggested that TH1 CD4+ effector mechanisms are central in crescentic glomerulonephritis,16 although importantly studies in IL-12–deficient mice were performed in mice deficient for the p40 subunit shared by IL-12 and IL-23.17 Therefore, the relative ability of TH17 and TH1 effector CD4+ T cells to induce glomerulonephritis was not clear. Further data have suggested that IL-17 is a key mediator in this model,18 although several cell types produce IL-17 and direct evidence of a role for TH17 cells was not presented in this study. A recent report addressed the role of TH17 cells using mice with a transgenic T-cell receptor specific for ovalbumin and a model in which ovalbumin was planted on the GBM.19 In this article, both TH1 and TH17 cells were shown to induce glomerulonephritis, but only TH1 cells caused crescent formation. We have used a similar experimental system and have found that only TH17 cells induce glomerulonephritis with crescent formation and that TH1 cells do not induce significant disease.
Materials and Methods
Mice
OT2 mice20 on a C57BL/6 background were obtained from Charles River (Paris, France) and then bred in house. RAG1−/− mice21 also on a C57BL/6 background were obtained from the Jackson Laboratory (Bar Harbor, ME) and bred in house. OT2 and Rag1−/− mice were interbred to obtain OT2 × RAG1−/− mice with mice genotyped by PCR. Mice were bred under specified pathogen-free conditions, and all experiments were performed according to local and UK home office regulations.
Preparation and Conjugation of Glomerular-Binding Antibody
Polyclonal glomerular-binding serum was raised in sheep as previously described.22 For initial experiments, this serum was purified with protein G chromatography. A conjugation strategy was developed in which sulfhydryl groups from cysteine on the peptide would react with maleimide groups on the antibody. Modified ovalbumin 323–339 was commercially synthesized (Cambridge Research Biochemicals, Billingham, UK) as a 19–amino acid peptide with an additional cysteine residue at the N terminus and an additional biotinylated lysine added to the C terminus. The addition of biotin was to allow characterization of the final product. Antibody was maleimide activated by reaction with sulfosuccinimidyl-4-(N maleimidomethyl) cyclohexane-1-carboxylate. After buffer exchange by gel filtration, the peptide was reacted with the maleimide-activated antibody. The product was dialyzed into PBS. The final product was then characterized. The molar biotin:IgG ratio of the product was assessed because each peptide contained one biotin. 4′-Hydroxyazobenzene-2-carboxylic acid undergoes a color change with biotin, and using a standard curve of known biotin concentrations, we calculated the biotin:IgG ratio of our conjugated antibody. In later experiments, we prepared Fab2 fragments of the glomerular-binding antibody. This was performed based on a previously published method.23 Digested fragments were removed using a centrifugal device with a 30-kDa membrane (Vivaspin 20). Repeated concentration and dilution in 150 mmol/L NaCl and 20 mmol/L piperazine (pH 6) were performed. The Fab2 preparation was then passed through Hitrap Q and protein G columns (GE Healthcare, Little Chalfont, UK) to remove any residual pepsin or whole antibody, respectively. The run through was then concentrated (Vivaspin 20) and buffer exchanged into PBS with 1 mmol/L EDTA for conjugation to modified ovalbumin 323–339 as described for whole antibody. At the end of the conjugation, the product was buffer exchanged to 150 mmol/L NaCl and 20 mmol/L piperazine (pH 6) using gel filtration. SDS-PAGE confirmed that there was no detectable whole antibody in the preparation (Figure 1D).
Figure 1.
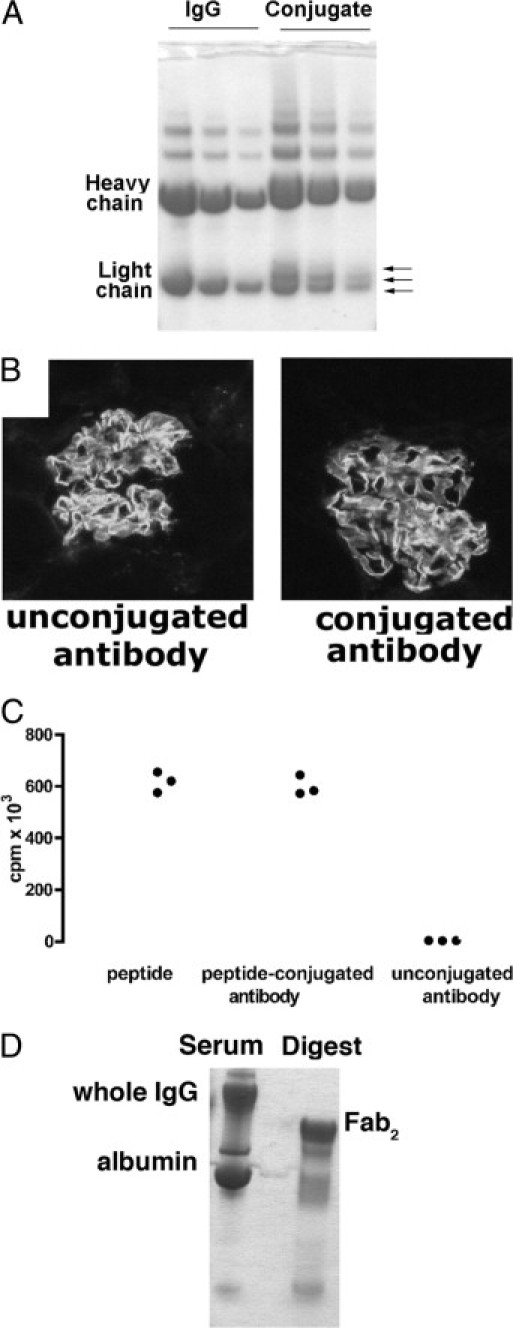
A: Coomassie-stained SDS-PAGE (reduced) of glomerular-binding IgG before and after conjugation with peptide (three dilutions of each). The heavy chain of the conjugate has a higher molecular weight, and three bands are visible for the light chain, each corresponding to a light chain with differing numbers of peptide molecule added (arrows). B: Immunofluorescence staining for sheep IgG was performed on kidney from mice injected with conjugated or unmodified IgG, and the intensities were similar. C: CD4+ T cells from OT2 × RAG1-/- mice proliferated in vitro when cultured with OT2 peptide and peptide-conjugated IgG, but not with IgG alone. Data are from a 3H thymidine proliferation assay using 2 × 105 spleen cells/mL with 2 mg/mL peptide and conjugated IgG containing equimolar amounts of peptide. D: Coomassie-stained SDS-PAGE (nonreduced) of glomerular-binding IgG before and after digestion to yield Fab2 fragments. There was no detectable intact IgG.
Induction and Assessment of Disease
RAG1−/− mice were injected with 106 splenocytes from OT2 × RAG1−/− mice. Seven days later, they were injected with 0.132 mg/g of peptide-conjugated glomerular-binding antibody or unconjugated antibody. They were sacrificed 21 days later, with the last 24 hours spent in metabolic cages for urine collection. At the end of the experiment, spleens were cultured in complete medium [Dulbecco's modified Eagle's medium; HEPES, 15 mmol/L; 2-beta-mercaptohethanol, 50 μmol/L; and fetal calf serum, 10% (Invitrogen, Paisley, UK)] for 48 hours, and cytokines were assessed by enzyme-linked immunosorbent assay (ELISA) in supernatants. ELISA kits were from R&D Systems (Abingdon, UK). Disease was assessed by measuring albuminuria with radial immunodiffusion and serum creatinine with electrospray mass spectrometry as previously described.22, 24 Kidneys were fixed in Bouin's fixative and stained with PAS for assessment of histologic features. Neutrophils were identified by their characteristic nuclear morphologic findings. Samples were also fixed in phosphate-lysine-periodate and then frozen for immunofluorescence staining. CD4 (clone L3T4; BD Biosciences, Oxford, UK) and CD68 (clone FA11; AbD Serotec, Oxford, UK) staining was detected using fluorescein isothiocyanate (FITC) mouse anti-rat IgG (Jackson Immunoresearch Laboratories Inc, West Grove, PA) followed by Alexa fluor 488 goat anti-FITC (Invitrogen). For quantitation of glomerular cells, at least 40 glomerular cross-sections (GCSs) were assessed, and for interstitial cells, at least five randomly selected fields at ×400 magnification were assessed.
T-Cell Lines
Single-cell suspensions were prepared from spleens of OT2 × RAG1−/− mice in complete medium (as above). Cells were cultured with ovalbumin 323–339 peptide at 2 μg/mL at 5 × 106/mL in polarizing conditions as follows. For TH1 development, 10 ng/mL of recombinant murine IL-12 (BD Biosciences) and 5 μg/mL of neutralizing anti–IL-4 antibody (11B11; BD Biosciences) were used. For TH17 development, 25 ng/mL of recombinant murine IL-6 (Peprotech EC, London, UK), 3 ng/mL of human transforming growth factor-β (R&D), 10 ng/mL of recombinant murine IL-23 (R&D), and 10 μg/mL of anti–interferon (IFN)-γ (XMG1.2; BD Biosciences) were used. At day 3, 10 ng/mL of IL-2 and IL-23 were added for TH1 and TH17 culture, respectively. For the day 21 experiment, at day 6 cells were restimulated with irradiated splenocytes at a ratio of 5:1 in fresh medium and polarizing conditions. Cells were harvested after 3 days (total of 9 days in culture). For the day 14 experiment, cells were used after 7 days without restimulation. In this experiment, cells from OT2 rather than OT2 × RAG1−/− mice were used because of the availability of the former. All cells used in vivo were analyzed as described under intracellular cytokine staining. To induce glomerulonephritis with the T-cell lines, RAG1−/− mice were injected i.v. with 5 × 106 cells per mouse. Immediately after this, they were given 0.25 mg/g of ovalbumin peptide–conjugated Fab2 glomerular-binding antibody or unconjugated Fab2 kidney binding antibody. They were sacrificed up to 21 days later, with the last 24 hours spent in metabolic cages for urine collection. At the end of the experiment, single cell suspensions were prepared from spleens. For each mouse in the day 21 experiment, one-third of each kidney was taken and digested using a previously published method.25 After digestion, cells were further purified using a Ficoll separation.
Intracellular Cytokine Staining
Restimulation and intracellular staining was performed as previously described except that Brefeldin rather than Monesin was used.26 The same method was used for analysis of the polarized T-cell lines or spleen and kidney cells at the end of the experiments using these T-cell lines. Antibodies used for flow cytometry were from BD Biosciences as follows: phycoerythrin (PE) and IL-17 (TC11-18H10), FITC and IFN-γ (XMG1.2), and PECy5 and CD4 (H129.19). Analysis was performed on a Cyan (Dako Cytomation, Ely, UK) or a BD FACScalibur flow cytometer (BD Biosciences).
Statistical Analyses
Statistical analyses were performed using GraphPad Prism Software (GraphPad Software Inc, San Diego, CA). An unpaired Student's t-test was used to compare two groups. If more than two groups were compared, a one-way analysis of variance with Tukey's posttest was used. If the variances were significantly different, a logarithmic transformation was performed before analysis.
Results
Characterization of Conjugated Antibody and Fab2 Fragments
We conjugated the ovalbumin 323–339 peptide recognized by OT2 T cells to glomerular-binding sheep IgG to target this to the glomerulus (Figure 1). The heavy chain of the conjugate has a higher molecular weight, and three bands are visible for the light chain, corresponding to different numbers of peptide molecules added (Figure 1A). When conjugated and unconjugated antibody was injected intravenously, binding was similar on immunofluorescence staining kidney section (Figure 1B). We confirmed that peptide from the conjugate could be presented and stimulated OT2 cells in vitro (Figure 1C). Fab2 fragments were generated by digestion, and we confirmed that they did not contain whole IgG (Figure 1D). The conjugates were assayed for biotin, and the biotin:IgG ratio (and hence peptide:IgG ratio) of our conjugated whole IgG antibody was found to be 4.7. For the Fab2 conjugate, there were 3.25 biotins and thus 3.25 peptides per Fab2 molecule. We also confirmed that peptide-conjugated Fab2 bound to the glomerular capillary wall when injected in vivo (not shown).
Antigen-Specific CD4+ T Cells Induce Glomerulonephritis
In initial experiments, we aimed to show that antigen-specific OT2 T cells could induce glomerulonephritis using this approach. We transferred spleen cells from OT2 × RAG1−/− mice into RAG1−/− mice and 7 days later injected them with glomerular-binding IgG that had been conjugated with OT2 peptide or with unconjugated glomerular-binding IgG as a control. These mice had no CD8+ cells or B cells, and all CD4+ T cells had a receptor specific for the ovalbumin 323–339 peptide. After 21 days there was significant albuminuria in mice given peptide-conjugated antibody but not unconjugated antibody as shown in Figure 2A. No significant difference was found in serum creatinine concentrations between groups with levels of less than 15 μmol/L in all mice. Histologic parameters also showed disease in mice given peptide-conjugated antibody but not control (Figure 2, B-F), with increased crescent formation, glomerular CD4+ T cells, glomerular CD68+ macrophages, interstitial CD4+ T cells, and interstitial CD68+ macrophages. There were few glomerular neutrophils seen, and these were less than 0.08 per GCS in all mice. Representative light microscopy and immunofluorescence staining for CD4 and CD68 is shown in Figure 2G. Splenocytes from experimental animals were restimulated with ovalbumin 323–339 peptide, and production of IL-17, IFN-γ, and IL-4 was greater in mice given peptide-conjugated antibody than in those given unconjugated antibody control, suggesting that an immune response, including TH1, TH2, and TH17 cells, had been generated and that any of these cells could have caused glomerular injury (Figure 2H). In these experiments, unconjugated antibody did not induce significant disease, and the disease seen in mice given ovalbumin peptide-conjugated antibody was therefore caused by the OT2 × RAG1−/− CD4+ T cells that recognized this peptide.
Figure 2.
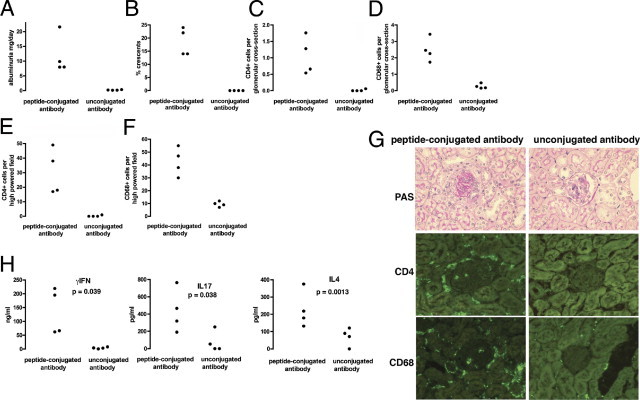
Disease was induced in RAG1−/− mice reconstituted with OT2 × RAG1−/− spleen cells as described in Materials and Methods. A–F: In mice given peptide-conjugated glomerular-binding IgG there was albuminuria and crescent formation, with glomerular and interstitial CD68+ and CD4+ cell infiltrates, which were significantly greater than in mice given an equal amount of unconjugated glomerular-binding IgG (P < 0.001 for all parameters). G: Representative histologic analysis on PAS-stained sections from the experiment shown in A–F, with immunofluorescence staining for CD68+ macrophages and CD4+ cells. The glomeruli are darker than the surrounding autofluorescent tubules. H: Splenocytes were restimulated with peptide, and there was production of IFN-γ, IL-4, and IL-17 in cells from mice given peptide-IgG, which was not seen in mice given IgG alone. For each mouse, spleen cells were also cultured without peptide, and cytokine levels were below the detection of the ELISA in all cases. Each symbol in A–H represents data from an individual mouse.
TH17 but not TH1 Cells Induce Crescentic Glomerulonephritis
To explore the relative roles of TH1 and TH17 cells in this model, we prepared polarized CD4+ T-cell lines in vitro, using T cells from OT2 × RAG1−/− mice. In the initial experiments, we could not exclude the possibility that disease was partially dependent on the glomerular-binding antibody used to target the peptide to the glomerulus. As a further refinement to our experimental system, we therefore prepared Fab2 fragments of glomerular-binding antibody and used these to target ovalbumin peptide to the glomerulus (Figure 1D). This system therefore totally excluded an antibody-mediated component, and glomerulonephritis was entirely mediated by OT2 CD4+ T cells. TH1 or TH17 cells were injected intravenously, immediately followed by an injection of peptide-conjugated Fab2 fragments or unconjugated Fab2 fragments as a control, giving four experimental groups. Three independent cell lines of TH1 or TH17 polarity were generated. Any given donor OT2 × RAG1−/− mouse contributed cells to one TH1 and one TH17 culture only, so that no two TH1 or TH17 cultures derived from the same donors, and we had three independent TH1 and TH17 cultures, derived from three groups of donors. Each of these six cell lines were given to one mouse that received peptide-conjugated Fab2 and to one mouse that received unconjugated Fab2 control. The data from this experiment are shown in Figure 3. Figure 3A shows that TH1 and TH17 cells of the desired polarity were obtained for all six cell lines used. A significant increase in glomerular CD4+ T cells and CD68+ macrophages, crescent formation, and increased interstitial CD4+ T cells were seen in mice given TH17 cells and peptide conjugated Fab2 but in none of the other groups (Figure 3, B-E). Crescent formation was also only seen in this group, and interstitial CD4+ T cells were increased. There was a trend toward more macrophages in both groups given TH17 cells, but this did not reach statistical significance. We assessed glomerular neutrophil numbers, and these numbers were small with no differences between groups. For mice given TH1 cells with conjugated Fab2, TH1 cells with unconjugated Fab2, TH17 cells with conjugated Fab2, and TH17 cells with unconjugated Fab2, the numbers (mean ± SEM as cells per GCS) were 0.17 ± 0.15, 0.06 ± 0.06, 0.24 ± 0.19, and 0.11 ± 0.08, respectively. Variable proteinuria was found, but this was not associated with the histologic changes and was not greater in mice given peptide-conjugated Fab2 compared with unconjugated Fab2. Albuminuria levels (mean ± SEM) for TH1, TH1 control, TH17, and TH17 control groups were 87 ± 23, 77 ± 19, 100 ± 75, and 356 ± 176 μg/d, respectively. In addition, no significant difference was found in serum creatinine concentrations between groups with levels of less than 11 μmol/L in all mice. We performed intracellular cytokine staining on CD4 T cells from spleens and kidneys at day 21 and confirmed that transferred polarized T cells had retained their polarity in vivo (Figure 3, H-I).
Figure 3.
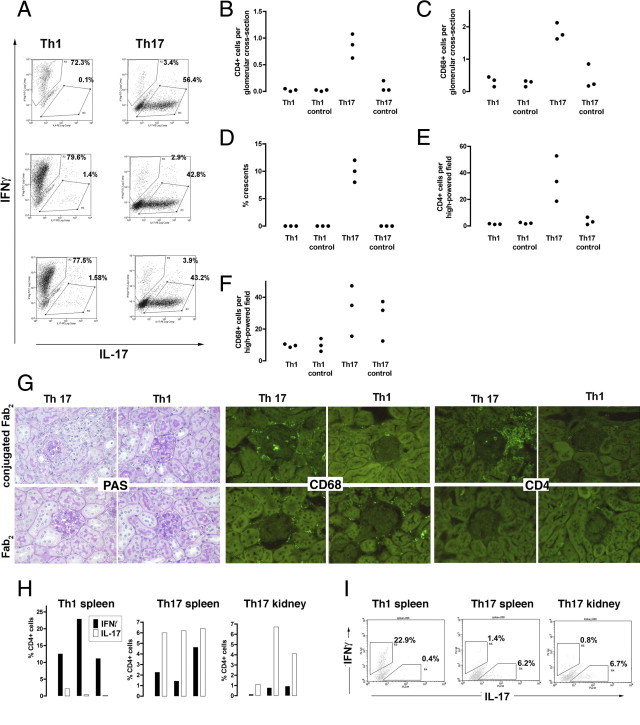
A: OT2 × RAG1−/− splenocytes were cultured in polarizing conditions and FACS staining showed that CD4+ cells of the desired polarity were obtained using these methods. The plots shown are from three independently cultured cell lines (derived from different mice) of each type that were used in this experiment. B–F: Polarized TH1 or TH17 CD4+ T cells were given to RAG1−/− mice followed by an injection of peptide-conjugated Fab2 nephrotoxic antibody or unconjugated Fab2 antibody. Each mouse in each group was given a different cell line and so is an independent data point, and each symbol is an individual mouse. The data show that at day 21 glomerular CD68+ macrophage and CD4+ T-cell infiltration with crescent formation developed only in mice given peptide-conjugated antibody and TH17 cells but not in mice given TH17 or TH1 cells with unconjugated Fab2 or in mice given TH1 cell with peptide-conjugated Fab2 (P < 0.001 for all comparisons of glomerular histologic analysis). The interstitial CD4+ cell infiltrate was also increased in this group (P < 0.01). The interstitial macrophage infiltrate was greater after TH17 cells in both the presence and absence of peptide (but not significant when all four groups were compared). G: Representative histologic analysis on PAS-stained sections from the experiment shown in B–F, with immunofluorescence staining for CD68+ macrophages and CD4+ cells. The glomeruli are darker than the surrounding autofluorescent tubules. H: Intracellular cytokine staining to examine the polarity of CD4+ T cells in mice that had been given TH1 or TH17 cells and peptide-conjugated Fab2. Each bar is a separate mouse from the experiment depicted. We also digested kidneys from mice that had been given TH17 cells and peptide-conjugated Fab2 and performed intracellular cytokine staining on isolated CD4+ T cells, which showed that they had retained their polarity. No data are shown for the other groups because kidneys did not contain significant T-cell numbers (B and E). I: Representative flow cytometry plots gated on CD4+ cells. Although the percentages are lower than those seen in the original cell lines (A), this probably represents technical issues around isolating splenic and renal T cells and performing intracellular cytokine staining, and the data clearly show that polarity had been retained.
We confirmed the increased pathogenicity of TH17 cells in a second experiment in which mice were sacrificed at day 14 after injection of TH1 or TH17 cells. In this experiment OT2 rather than OT2 × RAG1−/− cells were used, and two cell lines of each type (TH1 or TH17) were generated, with each mouse receiving one of these cell lines followed by an injection of peptide-conjugated Fab2 fragments or unconjugated Fab2 fragments as a control. The polarity of the injected cells is shown in Figure 4A. Glomerular CD4+ T cells and CD68+ macrophages, crescent formation, and interstitial CD4+ T cells were again greater in mice given TH17 cells and peptide-conjugated Fab2 as shown in Figure 4, B-F. In addition, interstitial macrophage numbers were higher in this group (Figure 4E). We again assessed glomerular neutrophil numbers, and these numbers were small with no differences between groups (mean, <0.1 cells per GCS for all groups). In this experiment, albuminuria was measured at day 14 but was less than 50 μg/mL (the detection limit of the assay used) in all mice. In this second experiment, we assessed absolute CD4+ T-cell numbers in spleens at day 14 (which we had not done in the first experiment in shown in Figure 4) and found no significant differences among any of the four groups (Figure 4G). This finding meant that the increased pathogenicity of TH17 cells was not due to increased survival or proliferation. We also performed intracellular cytokine staining on CD4+ T cells from spleens taken at the end of this experiment and again confirmed that transferred CD4+ T cells retained their polarity (Figure 4H).
Figure 4.
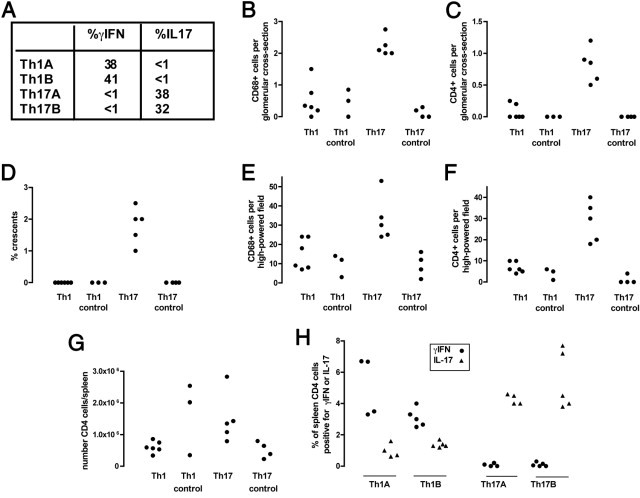
A further experiment was performed with two TH1 and two TH17 OT2 cell lines (TH1A, TH1B, TH17A, TH17B) that were generated and injected into RAG1−/− mice followed by an injection of peptide-conjugated Fab2 nephrotoxic antibody or unconjugated Fab2 antibody. A: The polarity obtained in vitro for each cell line before injection is shown. Cell lines denoted A or B were derived from different donor OT2 mice, and each recipient mouse received one of these four cell lines. B–F: Histologic data at day 14 after induction of disease. Significantly more glomerular macrophages (B) and CD4 T cells (C) were found in mice given TH17 cells and peptide-conjugated Fab2 compared with other groups (P < 0.001 for all comparisons). D: There were occasional crescents seen but only in the TH17 group (P < 0.001 compared with other groups). E: There were more interstitial macrophages in mice given TH17 cells (P < 0.05 compared with both TH1 groups and P < 0.01 compared with the TH17 control group). F: There were also more interstitial CD4 T cells in mice given TH17 cells (P < 0.001 compared with other groups). G: There were no differences in absolute numbers of CD4+ cells in spleens taken from mice at day 14. H: Intracellular cytokine staining from spleen cells for IL-17 and IFN-γ showed that TH1 and TH17 cells retained their polarity with data grouped according to the cell line received. B–H: Each symbol represents data from an individual mouse.
Discussion
These data established that crescentic glomerulonephritis could be induced by TH17 but not TH1 CD4+ T cells. Significant albuminuria was induced by CD4+ T cells that were sensitized in vivo in the initial experiment shown in Figure 2, but albuminuria was not significantly different from controls when in vitro sensitized TH1 or TH17 cells were injected. There are a number of possible reasons for this finding. In the initial experiment, we used intact whole IgG to plant ovalbumin peptide in the glomerulus, whereas Fab2 fragments were used with in vitro polarized T cells. It is therefore possible that the additive effect of the antibody, which was not pathogenic on its own, contributed to the proteinuria. Alternatively, the in vitro polarized T cells may have been of a lower pathogenicity than the in vivo sensitized cells. An additional possibility is that full expression of disease with proteinuria requires both TH1 and TH17 subsets. In keeping with our data, proteinuria may be mild in other models in which crescentic glomerulonephritis is not induced by immune complexes, at least in C57BL/6 mice. For example, significant crescent formation may be seen in antineutrophil cytoplasmic antibody vasculitis models in C57BL/6 mice in association with minimal proteinuria. Despite this limitation, histologic crescentic glomerulonephritis was clearly induced by TH17 and not TH1 cells. Recent data have shown that in certain circumstances CD4 T-cell subsets have plasticity and that TH17 cells may convert to TH1 cells.27 Therefore, it was important to show that in this system transferred CD4 T cells had retained polarity in both the spleen (Figure 3, H–I, and Figure 4H) and kidney (Figure 3, H–I).
The precise mechanism by which TH17 cells localize to the glomerulus to induce glomerulonephritis is not yet defined. Previous work has shown that major histocompatibility complex class II expression on both leukocytes and parenchymal renal cells is required for glomerulonephritis.28 We could therefore speculate that our peptide-Fab2 conjugate is taken up by both renal antigen-presenting cells and non–bone marrow–derived renal cells. This leads to localization of TH17 cells in the kidney with the release of proinflammatory cytokines and the development of glomerulonephritis. A range of proinflammatory cytokines are produced by TH17 cells and act on both immune and nonimmune cells. These cytokines include IL-21, IL-22, IL-17A, IL-F, tumor necrosis factor, IL-6, and granulocyte-macrophage colony-stimulating factor.29 The current study has not defined which of these cytokines lead to crescentic glomerulonephritis, although it is likely to be a combination of several. It does not appear to require a prominent neutrophil influx because this was not seen. It is not clear why TH17 but not TH1 cells localized to the glomerulus in our model. Because all of the transferred CD4+ cells expressed the OT2 T-cell receptor, they had the same antigen specificity and the numbers of TH1 and TH17 cells in the spleens of recipient mice were similar at day 14 (Figure 4G). In a recent study, IFN-γ–secreting CD4 T cells were isolated from renal tissue in the nephrotoxic nephritis model.30 It is possible that inflammation (initiated by immune complexes in the nephrotoxic nephritis model) is necessary to provide the chemokine secretion required to retain TH1 cells in the kidney. This requirement may not be present or as stringent for TH17 cells.
Our results contrast with a recent report in which both TH1 and TH17 cells induced mild proteinuria and only TH1 cells caused crescent formation.19 A similar experimental system was used, but there were some differences. A monoclonal antibody with specificity for the Goodpasture antigen was used to plant whole ovalbumin protein on the GBM. In this previous report, the effect of unconjugated antibody, together with TH1 or TH17 cells, on histologic changes or proteinuria was not assessed as a control. Activated OT2 T cells will proliferate when given to RAG1−/− mice even in the absence of antigen. This effect could cause a systemic cytokine release that, combined with a nonpathogenic dose of unconjugated GBM-binding monoclonal antibody, could induce proteinuria, which may have been of a similar magnitude to the proteinuria seen in mice given ovalbumin-conjugated monoclonal antibody and polarized T cells. The differences in antigen preparation and targeting strategy in this previous report may also be relevant. The binding sites of the antibodies within the kidney would have been different, with a broader localization for our polyclonal antibody compared with the monoclonal antibody. In addition, the use of whole monoclonal antibody may have allowed uptake and presentation through Fc and complement receptor-mediated mechanisms, and this would not have been the case with our Fab2 fragment. Therefore, the OT2 peptide may have been targeted to different cells. Although the IgG1 monoclonal used was not itself pathogenic, it may have had a synergistic effect with the CD4+ T cells because it would have bound to macrophage FcγRIII and hence could have contributed to disease.
Whatever the reasons for the differences among these studies, together they show that both TH1 and TH17 T cells are able to induce crescentic glomerulonephritis under appropriate experimental conditions. Further work will be required to explore the interactions of these CD4+ T-cell subsets and to precisely define the mechanisms by which they induce disease.
Acknowledgments
We are grateful to Richard Smith for help with the conjugation and to Neil Dalton and Charles Turner for creatinine measurements.
Footnotes
Supported by Kidney Research UK (RP15/2009) and Guy’s and St. Thomas’ Charity (R041003).
References
- 1.Lerner R.A., Glassock R.J., Dixon F.J. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967;126:989–1004. doi: 10.1084/jem.126.6.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clynes R., Dumitru C., Ravetch J.V. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 3.Park S.Y., Ueda S., Ohno H., Hamano Y., Tanaka M., Shiratori T., Yamazaki T., Arase H., Arase N., Karasawa A., Sato S., Ledermann B., Kondo Y., Okumura K., Ra C., Saito T. Resistance of Fc receptor- deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhan A.K., Schneeberger E.E., Collins A.B., McCluskey R.T. Evidence for a pathogenic role of a cell-mediated immune mechanism in experimental glomerulonephritis. J Exp Med. 1978;148:246–260. doi: 10.1084/jem.148.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S., Holdsworth S.R., Tipping P.G. Antibody independent crescentic glomerulonephritis in mu chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- 6.Wu J., Hicks J., Borillo J., Glass W.F., II, Lou Y.H. CD4(+) T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J Clin Invest. 2002;109:517–524. doi: 10.1172/JCI13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruth A.J., Kitching A.R., Kwan R.Y., Odobasic D., Ooi J.D., Timoshanko J.R., Hickey M.J., Holdsworth S.R. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1940–1949. doi: 10.1681/ASN.2006020108. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E., Korn T., Kuchroo V.K. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y., Langrish C.L., McKenzie B., Joyce-Shaikh B., Stumhofer J.S., McClanahan T., Blumenschein W., Churakovsa T., Low J., Presta L., Hunter C.A., Kastelein R.A., Cua D.J. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langrish C.L., Chen Y., Blumenschein W.M., Mattson J., Basham B., Sedgwick J.D., McClanahan T., Kastelein R.A., Cua D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy C.A., Langrish C.L., Chen Y., Blumenschein W., McClanahan T., Kastelein R.A., Sedgwick J.D., Cua D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yen D., Cheung J., Scheerens H., Poulet F., McClanahan T., McKenzie B., Kleinschek M.A., Owyang A., Mattson J., Blumenschein W., Murphy E., Sathe M., Cua D.J., Kastelein R.A., Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinman L. A rush to judgment on Th17. J Exp Med. 2008;205:1517–1522. doi: 10.1084/jem.20072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luger D., Silver P.B., Tang J., Cua D., Chen Z., Iwakura Y., Bowman E.P., Sgambellone N.M., Chan C.C., Caspi R.R. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroenke M.A., Carlson T.J., Andjelkovic A.V., Segal B.M. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology: CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tipping P.G., Kitching A.R. Glomerulonephritis: Th1 and Th2: what's new? Clin Exp Immunol. 2005;142:207–215. doi: 10.1111/j.1365-2249.2005.02842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitching A.R., Holdsworth S.R., Tipping P.G. IFN-gamma mediates crescent formation and cell-mediated immune injury in murine glomerulonephritis. J Am Soc Nephrol. 1999;10:752–759. doi: 10.1681/ASN.V104752. [DOI] [PubMed] [Google Scholar]
- 18.Paust H.J., Turner J.E., Steinmetz O.M., Peters A., Heymann F., Holscher C., Wolf G., Kurts C., Mittrucker H.W., Stahl R.A., Panzer U. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20:969–979. doi: 10.1681/ASN.2008050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Summers S.A., Steinmetz O.M., Li M., Kausman J.Y., Semple T., Edgtton K.L., Borza D.B., Braley H., Holdsworth S.R., Kitching A.R. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. 2009;20:2518–2524. doi: 10.1681/ASN.2009030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnden M.J., Allison J., Heath W.R., Carbone F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 21.Mombaerts P., Iacomini J., Johnson R.S., Herrup K., Tonegawa S., Papaioannou V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 22.Brown H.J., Lock H.R., Sacks S.H., Robson M.G. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol. 2006;177:1925–1931. doi: 10.4049/jimmunol.177.3.1925. [DOI] [PubMed] [Google Scholar]
- 23.Jones R.G., Landon J. A protocol for ‘enhanced pepsin digestion’: a step by step method for obtaining pure antibody fragments in high yield from serum. J Immunol Methods. 2003;275:239–250. doi: 10.1016/s0022-1759(03)00005-x. [DOI] [PubMed] [Google Scholar]
- 24.Brown H.J., Sacks S.H., Robson M.G. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J Am Soc Nephrol. 2006;17:1931–1939. doi: 10.1681/ASN.2005111167. [DOI] [PubMed] [Google Scholar]
- 25.Anders H.J., Banas B., Linde Y., Weller L., Cohen C.D., Kretzler M., Martin S., Vielhauer V., Schlondorff D., Grone H.J. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: role of TLR9-mediated expression of chemokines and chemokine receptors. J Am Soc Nephrol. 2003;14:317–326. doi: 10.1097/01.asn.0000042169.23931.73. [DOI] [PubMed] [Google Scholar]
- 26.Giorgini A., Noble A. Blockade of chronic graft-versus-host disease by alloantigen-induced CD4+CD25+Foxp3+ regulatory T cells in nonlymphopenic hosts. J Leukoc Biol. 2007;82:1053–1061. doi: 10.1189/jlb.0407227. [DOI] [PubMed] [Google Scholar]
- 27.Annunziato F, Romagnani S: The transient nature of the Th17 phenotype. Eur J Immunol 40:3312–3316 [DOI] [PubMed]
- 28.Li S., Kurts C., Kontgen F., Holdsworth S.R., Tipping P.G. Major histocompatibility complex class II expression by intrinsic renal cells is required for crescentic glomerulonephritis. J Exp Med. 1998;188:597–602. doi: 10.1084/jem.188.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korn T., Bettelli E., Oukka M., Kuchroo V.K. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 30.Turner J.E., Paust H.J., Steinmetz O.M., Peters A., Riedel J.H., Erhardt A., Wegscheid C., Velden J., Fehr S., Mittrucker H.W., Tiegs G., Stahl R.A., Panzer U. CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J Am Soc Nephrol. 2010;21:974–985. doi: 10.1681/ASN.2009070741. [DOI] [PMC free article] [PubMed] [Google Scholar]