

Abstract
Hepatic stellate cells (HSCs) interact with fibrillar collagen through the discoidin domain receptor 2 (DDR2) in acute hepatic injury, generating increased fibrosis. However, the contribution of DDR2 signaling to chronic liver fibrosis in vivo is unclear, despite its relevance to chronic human liver disease. We administered carbon tetrachloride (CCl4) to DDR2+/+ and DDR2−/− mice twice weekly, and liver tissues and isolated HSCs were analyzed. In contrast to changes seen in acute injury, after chronic CCl4 administration, DDR2−/− livers had increased collagen deposition, gelatinolytic activity, and HSC density. Increased basal gene expression of osteopontin, transforming growth factor-β1, monocyte chemoattractant protein-1, and IL-10 and reduced basal gene expression of matrix metalloproteinase-2, matrix metalloproteinase-13, and collagen type I in quiescent DDR2−/− HSCs were amplified further after chronic CCl4. In concordance, DDR2−/− HSCs isolated from chronically injured livers had enhanced in vitro migration and proliferation, but less extracellular matrix degradative activity. Macrophages from chronic CCl4-treated DDR2−/− livers showed stronger chemoattractive activity toward DDR2−/− HSCs than DDR2+/+ macrophages, increased extracellular matrix degradation, and higher cytokine mRNA expression. In conclusion, loss of DDR2 promotes chronic liver fibrosis after CCl4 injury. The fibrogenic sinusoidal milieu generated in chronic DDR2−/− livers recruits more HSCs to injured regions, which enhances fibrosis. Together, these findings suggest that DDR2 normally orchestrates gene programs and paracrine interactions between HSCs and macrophages that together attenuate chronic hepatic fibrosis.
Hepatic fibrosis is a wound-healing response to chronic injury characterized by failure of regeneration and inflammatory responses that provoke deposition of a fibrous collagen-rich extracellular matrix (ECM).1, 2, 3 Hepatic stellate cell (HSC) activation orchestrates the fibrogenic and inflammatory response to chronic liver injury. Activation of HSCs is initiated by paracrine stimulation from injured neighboring cells.4, 5, 6 In particular, soluble factors released by liver-associated macrophages within the fibrotic milieu render HSCs susceptible to a wide number of cytokines, chemokines, and growth factors that promote their migration and accumulation at the sites of tissue repair. Activated HSCs synthesize the collagenous scar tissue, and secrete both gelatinases that degrade the normal ECM matrix [matrix metalloproteinase (MMP)-2 and MMP-9], and collagenases that degrade fibrillar collagen (MMP-1, MMP-8, and MMP-13 in humans and MMP-13 in mice).7, 8 They also contribute to the immunoregulatory response to injury through the release of chemokines and cytokines.9
Chronic fibrosis develops as autocrine stimuli, perpetuating HSC synthesis of excessive collagenous ECM. As a result, signaling of ECM receptors in the HSCs is altered and contributes to a vicious cycle of inflammatory and fibrogenic stimulation.3, 10 HSCs express two types of collagen receptors: integrins 11 and discoidin domain receptors (DDRs).12 DDR2 is an unusual receptor because, although it belongs to the tyrosine kinase family, it signals in response to fibrillar collagen instead of soluble growth factors.13, 14 Increased DDR2 expression occurs in humans during development and in pathologic processes, such as osteoarthritis, cancer, and cirrhosis,15, 16 and DDR2 expression also is induced in experimental alcoholic liver fibrosis.17
Based on DDR2's unique responsiveness to collagen ligands and potential role in transducing signals from the accumulating collagen in liver injury, we compared the responses of wild-type (DDR2+/+) and DDR2-deficient (DDR2−/−) mice with chronic liver injury by CCl4. Although our earlier studies explored the contribution of DDR2 receptors to HSC responses after acute injury, their characterization in a chronic model of injury was essential to model human liver disease. Our results indicate that the absence of DDR2 in mice exacerbates injury, leading to increased liver fibrosis. The data also underscore the importance of ECM receptors in the interaction between HSCs, macrophages, and the ECM in the generation of a fibrotic response.
Materials and Methods
Animals
DDR2 knock-out mice (DDR2−/−) were generated as described.18 Heterozygous DDR2 knock-out/LacZ knock-in mice were kindly donated by Regeneron Pharmaceuticals (Tarrytown, NY).
Model of Liver Injury
To induce liver injury, 6- to 8-week-old male DDR2+/+ and DDR2−/− mice were administered biweekly doses of 0.5 μL of CCl4 per gram of body weight by gavage diluted in olive oil.12 Control animals received olive oil. As in our prior studies,12 we considered “acute” injured livers those submitted to a single gavage dose of 0.5 μL of CCl4 per gram. They were analyzed 3 days after intoxication.
Immunohistochemical Detection of DDR2
Liver sections (10 μm thick) fixed in cold acetone were processed for H&E stain or reacted with 1:100 dilution of polyclonal antibody (pAb) R2-JM, raised against the juxtacrine domain of mouse DDR2 (amino acids 429−557), followed by the appropriate fluorescent secondary antibody.
For the detection of β-galactosidase activity, livers were fixed in 0.4% (v/v) glutaraldehyde, 2 mmol/L MgCl2, and 5 mmol/L EGTA in a sodium phosphate buffer (77 mmol/L NaHPO4/22 mmol/L NaH2PO4), washed three times for 15 minutes in a solution containing 2 mmol/L MgCl2, 0.01% sodium deoxycholate, and 0.02% Nonidet P-40 in sodium phosphate buffer, and stained for 2 to 4 hours at 30°C in the washing solution containing 1 mg/mL X-gal, 5 mmol/L potassium ferricyanide, and 5 mmol/L potassium ferrocyanide. Livers then were rinsed in phosphate-buffered saline (PBS) and cleared in a benzyl benzoate:benzyl-alcohol (2:1) solution.
Immunohistochemistry and Image Analysis
Picrosirius red staining was performed as previously described.19 Liver sections (10 μm thick) fixed in cold acetone were processed with 1:50 dilution of anti-human α-smooth muscle actin (α-SMA) monoclonal antibody (Sigma Chemical Co, St. Louis, MO), rabbit anti-mouse desmin polyclonal antibody (1:200; Dako, Glostrup, Denmark), followed by the appropriate fluorescent secondary antibodies. Irrelevant appropriate immunoglobulins were used as negative controls. Images were quantified using the AnalySIS V3.2 program (Olympus Software Imagine Solutions GMBH, Munster, Germany). Results were expressed as a percentage of specifically colored tissue area relative to the whole analyzed area.
Histologic Analysis of Macrophages
Liver sections (10 μm thick) fixed in cold acetone were immunostained with F4/80 antibody (1:100), followed by the appropriate fluorescent secondary antibody. Stained cells were counted in 10 randomly selected high-power fields. In some studies, scar-associated macrophages were counted. A positive cell was included only if it was within or apposed to the scar.20
DDR2 Immunodetection
Immunoprecipitation/Western blot analysis for DDR2 was performed as previously described12 using pAb R2-JM. In brief, primary rat stellate cell cultures were lysed in radio immunoprecipitation assay buffer containing protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN), 2 mmol/L sodium orthovanadate, and 10 mmol/L sodium fluoride. Lysate (250 μg) was incubated with R2-JM polyclonal anti-DDR2 Ab for 1 hour at 4°C. Protein A-Sepharose beads (Sigma Chemical Co) were added for 1 hour at 4°C, then precipitated by centrifugation, and washed three times with radio immunoprecipitation assay buffer. The protein A–Sepharose–bound proteins were extracted by 10-minute incubation in Laemmli buffer at 80°C. Protein aliquots (80 to 300 μg) were separated through 8% SDS-PAGE electrophoresis and blotted onto a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). DDR2 was detected using the R2-JM anti-DDR2 Ab (1:1000) and horseradish-peroxidase–conjugated protein A (1:20,000). Phosphorylated DDR2 was detected using the antiphosphotyrosine monoclonal Ab 4G10 (1:20,000; Upstate Biotechnology, Inc, Lake Placid, NY) and horseradish-peroxidase–conjugated goat anti-mouse IgG (1:20,000). The bands were visualized using the Super Signal Femto Substrate kit (Pierce Chemical Co, Rockford, IL).
Gelatinase and Collagenase Activities
Flash-frozen liver samples were homogenized in 100 mmol/L Tris, 200 mmol/L NaCl, 0.1% Triton X-100 (pH 7.4). The homogenates were centrifuged at 10,000 × g for 20 minutes at 4°C. Gelatinolytic and collagenolytic activity in the supernatant of tissue homogenates and in supernatants from cell cultures were determined using biotinylated gelatinase or collagenase substrates and detected at OD 450 nm following the manufacturer's instructions (Chemicon International). To activate latent MMPs, conditioned media were incubated for 2 hours at 37°C with 2.5 mmol/L APMA (p-aminophenylmercuric acetate; Sigma Chemical Co.).21
Isolation and Primary Culture of HSCs and Liver Macrophages from DDR2+/+ and DDR2−/− Mice
Quiescent hepatic sinusoidal cells from untreated mice and in vivo activated ones from mice treated for 10 days with CCl4 were isolated as described.22 In brief, enzymatic perfusion was followed by isopycnic gradient centrifugation of dispersed liver cells placed on top of a triple layer of Percoll solutions (50%, 31%, and 24%). HSCs were collected from the 24%/31% Percoll solution interphase and macrophages from the 31%/50% interphase. All cells were centrifuged to remove cell debris and resuspended in serum-free RPMI-1640 media at 1.5 × 106 cells/mL. HSCs were shed onto uncoated plastic wells and nonadhered cells, collected after 15 minutes. As a result, HSC cultures were 99% macrophage-free. Then, HSCs immediately were submitted to RNA extraction or were plated on culture wells coated with a 0.5-mm layer of collagen type I (Becton Dickinson). Media was changed and cells were maintained in RPMI-1640 media plus 5% fetal calf serum (FCS) for up to 48 hours. Plate efficiency was 81 ± 20. For migration experiments, HSCs were placed on the upper compartment of a migration well.
We used HSCs from untreated mice as quiescent HSCs. They contained high amounts of large vitamin A droplets and of the succinate receptor (GPR91) mRNA, two markers of quiescent HSCs.23 As previously reported,23 we used HSCs isolated from livers treated for 10 days with CCl4 as the source of in vivo activated HSCs. Finally, some quiescent HSCs were activated in vitro by culture on plastic surface in RPMI-1640 media plus 5% FCS for 7 days, detached with ice-cold EDTA solution in PBS, and plated onto migration wells for migration experiments. Both in vivo and in vitro activated HSCs contained small vitamin A droplets while expressing α-SMA, desmin, and very low amounts of GPR91 mRNA (data not shown). They did not display signs of apoptosis (data not shown). Western blot revealed that DDR2 expression and phosphorylation were detectable in quiescent DDR2+/+ HSCs after 16 hours of culture on collagen type I, and strongly expressed by the in vivo activated DDR2+/+ HSCs (data not shown). Conditioned media was obtained on the second day of culture after 16 hours under serum-free conditions.
Macrophages from untreated and fibrotic livers were cultured for 16 to 24 hours at 1.5 × 105 cells/mL of RPMI-1640 media plus 5% FCS. On average, 86% of the cells expressed F4/80. Conditioned media was obtained on the second day of culture after 16 hours under serum-free conditions.
Cell Growth Assays
Cell proliferation was measured using the nontoxic alamarBlue assay (BioSource International, Camarillo, CA), according to the manufacturer's instructions. In brief, freshly isolated HSCs were cultured on top of a thin matrix of collagen type I in RPMI-1640 plus 1% FCS.12 AlamarBlue was added, and absorbance at 595 nm and 575 nm was measured every 8 hours using a Perkin Elmer HTS700-Plus plate reader (Applied Biosystems, Foster City, CA). Results were expressed as a percentage of reduced alamarBlue.
Cell Migration Through Type I Collagen
Cell migration assays were performed as described previously12 with minor modifications. Culture inserts with 8-μm pores coated with 0.001% collagen type I (Becton Dickinson) were placed on top of 24-well plates. HSCs suspended in RPMI-1640 supplemented with 0.5% FCS were placed on the culture insert. Macrophages were isolated from DDR2−/− or DDR2+/+ fibrotic livers, and their conditioned media was used as chemoattractants in the lower compartment of DDR2−/− or DDR2+/+ HSC migration chambers. In other experiments, RPMI-1640 supplemented with 10% FCS was used as a chemoattractant. Cells were allowed to migrate for 20 hours. Nonmigrated cells were removed with a cotton swab. Cells that migrated and adhered to the underside of the membrane were fixed in 100% methanol, stained with 0.2% crystal violet, and counted in 10 random, high-power fields per membrane.
Quantitative RT-PCR
Total RNA from whole liver lysates or freshly isolated cells was extracted using RNeasy columns (Qiagen GmbH, Hilden, Germany). RNA quantity and quality were analyzed with a capillary 2100 Bio-analyzer (Agilent Technologies, Palo Alto, CA). Quantitative RT-PCR was performed using the commercially available primer sets SYBR Green PCR Core Reagent kit and the Gene Amp 5700 sequence detection system (all from Applied Biosystems).
Statistics
Results refer to mean ± SEM. Comparisons among groups were performed with the Student's t-test, and P values less than 0.5 were considered significant. Each experiment was performed in duplicate or triplicate and repeated at least two times with independent samples derived from at least two mouse livers or two independent primary cultures.
Results
Chronic CCl4 Injury Increases Fibrogenic and ECM Remodeling Responses and Alters Cell Recruitment to the Fibrotic Scars in DDR2-Deficient Livers
Based on the role of DDR2 in ECM signaling, we sought to assess the impact of DDR2 deficiency on the hepatic response to sustained liver injury. DDR2+/+ and DDR2−/− mice were injected chronically with CCl4 for up to 30 days. Control animals received olive oil. By quantitative RT-PCR analysis of whole liver lysates, collagen α1(I) mRNA expression was equal between DDR2+/+ and DDR2−/− mice that were oil-treated and at day 4, after a single acute CCl4, and slightly but not significantly increased after a 10-day CCl4 administration in DDR2−/− livers compared to DDR2+/+ fibrotic livers. CCl4 treatment for an additional 20 days rendered the relative amount of collagen type I mRNA in DDR2−/− fibrotic livers approximately twofold higher than in DDR2+/+ mice (Figure 1A). Morphometric quantitation of Picrosirius red staining confirmed an equal accumulation of fibrillar collagen in the fibrotic tissue of all mice after 10 days of CCl4 administration, whereas it was 2.5-fold greater in DDR2−/− mice at day 30 (Figure 1, B–D). In contrast, there was no difference in the extent of injury. Specifically, at both days 3 and 30 of CCl4 administration, hepatocyte damage was similar in both DDR2+/+ and DDR2−/− livers based on similar levels of centrilobular necrosis, and serum alanine aminotransferase (270 and 258 mL/L), aspartate aminotransferase (170 and 160 IU/L), and albumin levels (20 and 24 g/dL).
Figure 1.
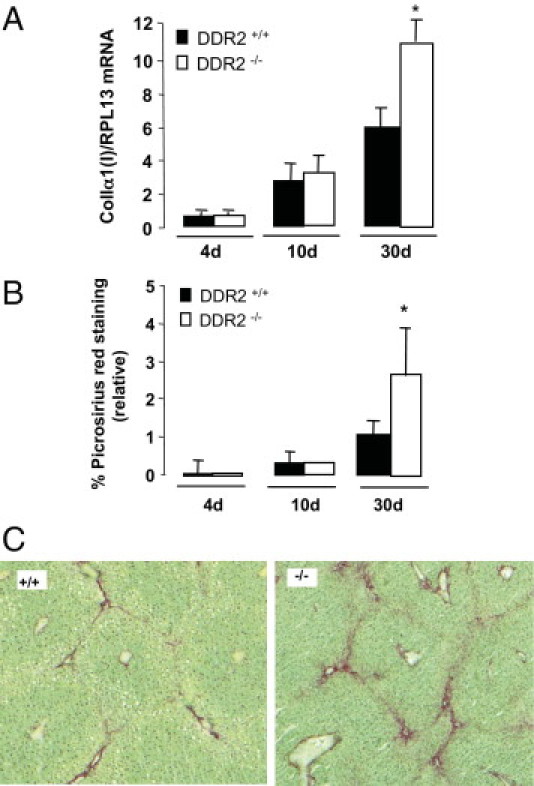
Collagen expression in DDR2−/− and DDR2+/+ livers after chronic CCl4. A: Real-time quantitative analyses of collagen type 1(I) mRNA after 10 and 30 days of treatment with CCl4. Results are expressed as a ratio of collagen type I mRNA versus RPL13 mRNA. B: Computer-assisted semiquantitation of picrosirius red–stained livers after 10 and 30 days of treatment with CCl4. Results are expressed as the percentage of region of interest. *P < 0.05, significantly different compared with DDR2+/+ group. C: Picrosirius red staining of collagen deposits in DDR2+/+ and DDR2−/− livers treated for 30 days with CCl4.
ECM remodeling is another key feature of liver fibrosis. Gelatinolytic and collagenolytic (G/C) activities were below detection levels in untreated and oil-treated livers (data not shown). Liver tissue extracts after acute injury contained low G/C activities (data not shown) that reached measurable levels after 10 days of treatment, a time point at which DDR2−/− extracts contained 50% less activity than DDR2+/+ (Figure 2A). As expected, further treatment with CCl4 increased G/C activity in all of the livers. G/C activity in DDR2+/+ mice was augmented by 60%. Interestingly, the increase in the gelatinolytic activity in the DDR2−/− livers was 2.8-fold higher than in the DDR2+/+ livers, whereas the increase in collagenolytic activity was not significantly different (Figure 2B).
Figure 2.
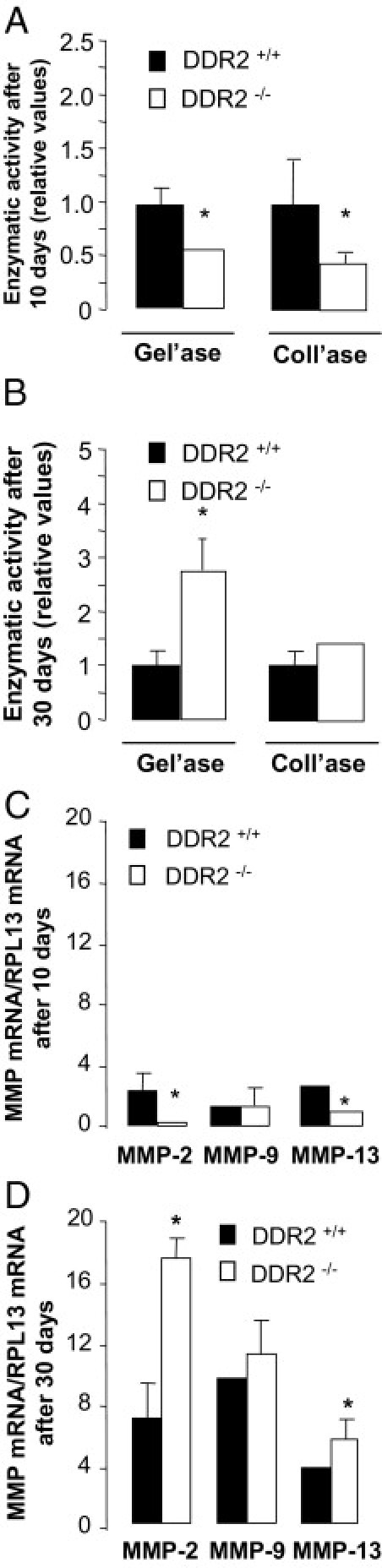
Gelatinase and collagenase activities in livers of DDR2−/− and DDR2+/+ mice after CCl4 administration. Relative gelatinase (A) and collagenase (B) activities in tissue extracts from DDR2−/− and DDR2+/+ livers after 10 or 30 days of treatment with CCl4. For each time point, enzymatic activity in DDR2+/+ livers was considered 1. Real-time quantitative analyses on MMP-2, MMP-9, and MMP-13 mRNA induction in DDR2−/− and DDR2+/+ livers after 10 days (C) and 30 days (D) of treatment with CCl4. Results are expressed as a ratio versus RPL13 mRNA. *P < 0.05, significantly different compared with DDR2+/+ group.
MMPs are the main enzymes responsible for G/C activities in the fibrotic liver.24 MMP-2 and MMP-9 are the main mouse liver gelatinases, and MMP-13 is the main collagenase. Mouse DDR2 signaling is associated with MMP-2 and MMP-10 activities, whereas MMP-9 activity is DDR2-independent.25, 26 Figure 2C documents significantly lower levels of MMP-2 and MMP-13 mRNAs, and equal levels of MMP-9 in fibrotic DDR2−/− livers after a 10-day treatment, compared with DDR2+/+ livers. As observed for G/C activities, chronic treatment induced MMP mRNA expression. However, DDR2−/− livers expressed 2.1-fold greater MMP-2 mRNA and 30% more MMP-13 mRNA, which correlated with their augmented collagenolytic activity (Figure 2D). These results indicate that during persistent injury, DDR2 expression inversely correlates with hepatic gelatinase, probably resulting from reduced MMP-2 activity.
All untreated and control livers contained similar levels of desmin/α-SMA–stained cells. As previously described,27 desmin/α-SMA–expressing spindle-shaped cells increased significantly in sinusoidal segments around centrilobular veins after chronic CCl4 treatment. This occurred regardless of DDR2 gene expression. However, we observed a higher density of desmin and α-SMA–expressing cells at bridging fibrotic tissue in DDR2−/− mice that became significantly different at day 10, and reached a threefold difference at day 30 (Figure 3, A and B). These data correlate inversely with DDR2 expression in recruitment of activated HSCs to fibrotic scars of livers after chronic CCl4.
Figure 3.
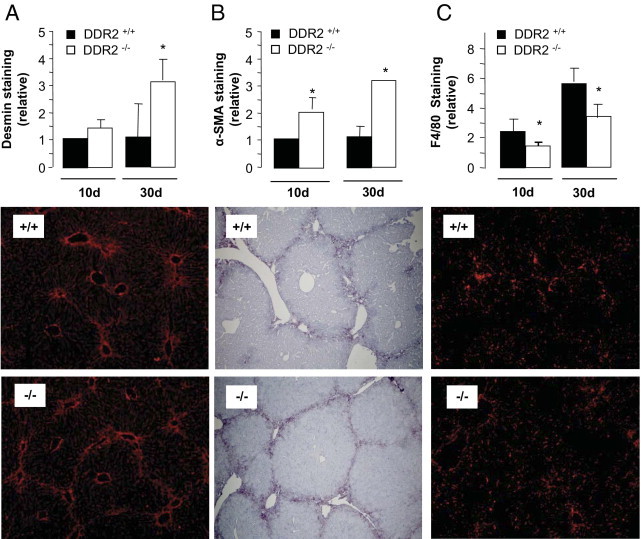
Immunohistochemical analysis of HSCs and macrophage recruitment after CCl4 administration. Liver sections from DDR2−/− and DDR2+/+ mice treated with CCl4 for 10 or 30 days were stained with anti-desmin, anti–α-SMA, or anti-F4/80 antibodies. A and B: Computer-assisted semiquantitation of stained cells within the fibrotic scar in microphotographed labeled sections. Data are expressed as the percentage of region of interest, which was restricted to scarring tissue C: Semiquantitation of anti-F4/80–stained macrophages associated with the scar. Only F4/80-positive cells located within or opposed the scar tracts were counted. *P < 0.05, significantly different compared with the DDR2+/+ group. Lower panels:DDR2+/+ group. Lower panels: Microphotographs of liver sections from DDR2+/+ and DDR2-/- mice treated with CCL4 for 30 days and stained with anti-desmin (red; A), anti–α-SMA (blue; B) and anti-F4/80 (red; C) antibodies.
Immunostaining of fibrotic livers revealed increased numbers of F4/80 macrophages as fibrosis developed, both within the sinusoids and opposite the collagenous septa surrounding the periportal regions.20, 28, 29, 30 This effect was observed irrespective of the DDR2 status (data not shown). Despite this, Figure 3C shows that recruitment of F4/80 macrophages to the scar tissue of DDR2−/− livers was 40% lower by day 10 and remained so at day 30. Both untreated and control livers contained similar levels of F4/80-stained sinusoids regardless of DDR2 expression, whereas acute treatment showed 18% reduced staining in DDR2−/− tissue (data not shown).
These results indicated that chronic CCl4 intoxication induced a more vigorous fibrogenic response associated with altered cell recruitment in DDR2−/− mice. Although we previously described DDR2 mRNA and protein in isolated rat HSCs 48 hours after an acute dose of CCl4 or after culture activation,12 the contribution of DDR2 to chronic liver fibrosis was not established. Therefore, we next analyzed in situ hepatic DDR2 expression after CCl4 administration.
DDR2 Expression Is Enhanced in HSCs of Livers Treated Chronically with CCl4
Immunohistochemical analysis using a polyclonal antibody against murine DDR2 (see Materials and Methods) displayed minimal DDR2 protein expression in control livers (Figure 4A). Ten days of CCl4 administration stimulated DDR2 expression in spindle-shaped cells within fibrotic scars (Figure 4B), and fibrotic lesions after 30 days contained a greater density of DDR2-expressing cells (Figure 4C). To further clarify hepatic expression of DDR2 in situ, heterozygous DDR2 knock-out/LacZ knock-in mice were treated for 30 days with CCl4, and β-galactosidase expression was analyzed as a surrogate marker for DDR2. As shown in Figure 4D, β-galactosidase was barely detectable in untreated livers, whereas it appeared in fibroblast-like cells of injured livers (Figure 4E), mainly located in the fibrotic bands, as revealed by Masson staining (Figure 4F).
Figure 4.
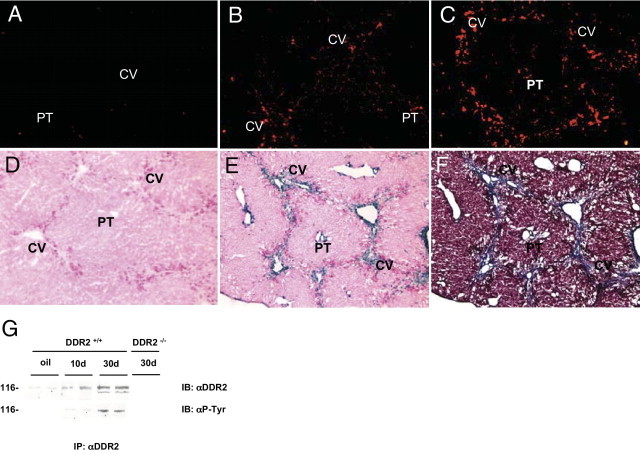
DDR2 is induced in stellate cells during chronic CCl4 injury. Immunohistochemical detection of DDR2 expression in mouse liver. DDR2 expression (red) was analyzed in untreated livers (A) after 10 days (B) or after 30 days (C) of CCl4 treatment. D and E: β-galactosidase activity (blue) in DDR2 knock-out/LacZ knock-in mice untreated (D) or treated with CCl4 for 30 days (E). F: Masson staining of DDR2 knock-out/LacZ knock-in mice treated with CCl4 for 30 days. G: Western blot of DDR2 expression and phosphorylation in DDR2−/− and DDR2+/+ stellate cells freshly isolated from untreated livers, or treated with CCl4 for 10 and 30 days. CV, central vein; IB, immunoblot; IP, immunoprecipitation; PT, portal tract.
Immunoprecipitation followed by Western blot analysis of DDR2 expression in freshly isolated HSCs showed enhanced expression after CCl4 treatment of a 116-kDa protein corresponding to DDR2.12 Antiphosphotyrosine antibody revealed a significant increase of phosphorylated DDR2 in HSCs from chronically treated livers compared with HSCs from untreated mice (Figure 4G).
These data pinpoint HSCs as the major DDR2-expressing cells in the fibrotic mouse liver after chronic injury with CCl4, which correlates with their role as the main cell type responsible for pericentral fibrosis after persistent CCl4 administration. HSC transdifferentiation from a quiescent cell to a migratory and fibrogenic phenotype reflects active changes in gene expression. Thus, our next goal was to analyze mRNA expression of DDR2-dependent genes in HSCs after chronic CCl4.
To do so, we built on our previous studies of gene expression signatures obtained by comparative DNA microarray analysis of freshly isolated quiescent DDR2+/+ and DDR2−/− HSCs (I. Badiola, E. Olaso, O. Crende, F.L. Friedman, F. Vidal-Vanaclocha, unpublished data).
Amplified DDR2-Dependent Expression Levels of Immunomodulatory and Fibrogenic Genes in HSCs from Chronic CCl4-Injured DDR2−/− Livers
First, we selected several DDR2-dependent genes that are both differentially expressed in quiescent DDR2+/+ HSCs and DDR2−/− HSCs (I. Badiola, E. Olaso, O. Crende, F.L. Friedman, F. Vidal-Vanaclocha, unpublished data), and also have been associated strongly with fibrogenic and immunomodulatory responses to chronic liver injury. After their validation by quantitative RT-PCR analysis, we further compared their expression in activated HSCs from chronically injured DDR2+/+ and DDR2−/− livers.
Basal osteopontin and transforming growth factor (TGF)-β mRNA levels were significantly higher in isolated DDR2−/− HSCs (Table 1). Moreover, the expected up-regulation of osteopontin and TGF-β mRNA levels in DDR2+/+ HSCs after chronic CCl4 was increased twofold in HSCs from the CCl4-treated DDR2−/− livers. Furthermore, basal mRNA expression of the immunosuppressive cytokine IL-10 was doubled in quiescent DDR2−/− HSCs, whereas chronic CCl4 exacerbated an IL-10 mRNA decrease in DDR2−/− HSCs. Quantitative RT-PCR analysis validated reduced expression of three key genes that regulate fibrogenesis. Collagen type I, MMP-2, and MMP-13 mRNA expression were reduced significantly in quiescent DDR2−/− HSCs. More importantly, their mRNA expression remained reduced in HSCs from chronic fibrotic DDR2−/− livers. Thus, quantitative RT-PCR analysis confirmed that DDR2 deficiency remarkably altered HSC expression of important genes whose immunomodulatory and fibrogenic activities can modulate fibrosis. It also indicates that mRNA expression driven by DDR2 signaling that characterizes normal liver function in quiescent HSCs is exacerbated in activated HSCs after chronic CCl4 administration.
Table 1.
Real-Time PCR Quantification of mRNA Levels in HSCs Isolated from Control and CCl4-Treated DDR2−/− and DDR2+/+ Livers
| mRNA | Oil |
CCl4 |
||
|---|---|---|---|---|
| +/+ | −/− | +/+ | −/− | |
| Osteopontin | 1.0 ± 0.025 | 5.7 ± 0.003⁎ | 8.1 ± 0.001⁎ | 15.4 ± 0.012⁎† |
| TGF-β | 1.0 ± 0.003 | 4.8 ± 0.030⁎ | 2.7 ± 0.025⁎ | 12.5 ± 0.020⁎† |
| IL-10 | 1.0 ± 0.050 | 1.9 ± 0.014⁎ | 0.8 ± 0.004 | 0.4 ± 0.003⁎† |
| Collagen I α1 | 1.0 ± 0.040 | 0.5 ± 0.125 | 3.5 ± 0.213⁎ | 2.1 ± 0.198⁎† |
| MMP-2 | 1.0 ± 0.210 | 0.7 ± 0.050⁎ | 10.6 ± 2.050⁎ | 3.2 ± 0.512⁎† |
| MMP-13 | 1.0 ± 0.060 | 0.8 ± 0.090 | 3.1 ± 0.048 | 1.5 ± 0.290⁎† |
N = 16 mice from 2 independent treatments, 2 mice per group. Relative mRNA expression by HSCs from control (oil-treated) DDR2+/+ livers compared to RPL13 mRNA expression: osteopontin: 1.29 ± 0.01; TGF-β: 0.18 ± 0.01; IL-10: 0.71 ± 0.004; collagen I α1: 1.0 ± 0.024; MMP-2: 0.04 ± 0.008; MMP-13: 0.01 ± 0.000.
P < 0.05 compared to HSCs from control DDR2+/+ livers.
P < 0.05 compared to HSCs from DDR2+/+ livers after CCl4 administration.
Next, to further analyze our in vivo data on altered ECM remodeling and HSC recruitment, we isolated HSCs from chronically injured DDR2−/− and DDR2+/+ livers, and analyzed in vitro their migration and proliferation rates, and their ECM degrading activities.
Enhanced in Vitro Migration and Proliferation and Reduced Matrix-Degrading Activities in HSCs from Chronically Injured DDR2−/− Livers
Amplification of HSCs within fibrotic areas is the net result of migration and proliferation. Therefore, we next assessed the impact of DDR2 on migration capacity of isolated HSCs using a Boyden chamber migration assay. The number of quiescent DDR2−/− HSCs that migrated across the collagen-coated membrane in response to a FCS gradient was half that of quiescent DDR2+/+ cells, whereas the number of migrated DDR2−/− HSCs from fibrotic livers was twice that of the in vivo activated DDR2+/+ cells (Figure 5A). We used alamarBlue, a nontoxic dye, to measure proliferation of activated HSCs (Figure 5B). The proliferation rate was 70% lower in quiescent DDR2−/− HSCs than in quiescent DDR2+/+ones, whereas it was 50% more in HSCs from fibrotic DDR2−/− mice than from fibrotic DDR2+/+mice. No signs of apoptosis or cell detachment were observed under any of the assayed conditions (data not shown). Quiescent DDR2−/− HSCs activated by in vitro culture (see Materials and Methods) migrated and proliferated significantly less than quiescent DDR2+/+ HSCs cultured in the same conditions (data not shown).
Figure 5.
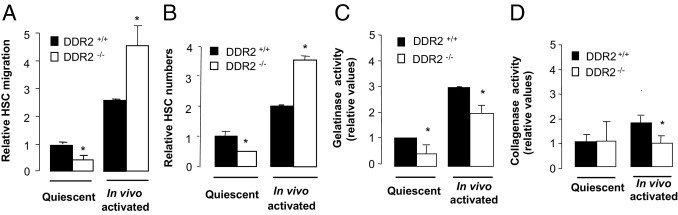
Enhanced chemotactic migration and proliferation and reduced ECM remodeling in DDR2−/− HSCs after chronic liver injury. Quiescent and activated DDR2−/− and DDR2+/+ HSCs were isolated from control and chronic fibrotic livers, respectively. A: HSCs were allowed to migrate for 16 hours through collagen type I–coated porous upper wells of migration chambers in response to a fetal calf serum gradient. B: HSC proliferation rates using the alamarBlue assay. Colorimetric-based assay of gelatinase (C) and collagenase (D) activities in HSCs isolated from DDR2−/− and DDR2+/+ fibrotic livers after 16 hours culture on top of thin collagen type I–coated wells. *P < 0.05, significantly different compared with DDR2+/+ group.
These results indicate that under basal conditions, DDR2 signaling induces migration and proliferation of quiescent HSCs, as shown by the reduced migration and proliferation of quiescent DDR2−/− HSCs. In this regard, overexpression of nonsignaling DDR2 reduced migration of HSC-T6 cells,12 a cell line derived from normal rat liver HSCs.31 On the other hand, in vitro data on HSCs from fibrotic DDR2−/− livers correlate with histologic data showing larger numbers of activated HSCs recruited to the fibrotic scars after chronic CCl4 (Figure 2). These apparent contradictory responses of DDR2−/− HSCs suggest that a specific DDR2-dependent conditioning of the liver occurs that in turn affects HSC responses after chronic injury. These responses underscore the importance of DDR2 in orchestrating activation signals sent by the profibrogenic milieu generated during chronic liver injury.
Next, we challenged HSCs with collagen type I, the preferred ligand of DDR2 (see Materials and Methods), and their cellular supernatants were assayed for G/C activities. G/C activities were confined to the medium, and none were found in cell lysates (not shown). These activities accounted for approximately 50% of those present in the corresponding APMA-activated conditioned media, indicating that 50% of G/Cs were in their active form, regardless of the DDR2 status of the cells. Supernatants from quiescent DDR2−/− HSCs contained approximately 54% of gelatinase activity, and supernatants from activated DDR2−/− HSCs contained significantly (∼30%) less (Figure 5C). Collagenase activity in response to collagen type I was similar in all quiescent cells but 45% lower in activated DDR2−/− HSCs (Figure 5D). These data, in concert with the results on quantitative RT-PCR (Table 1), indicate that DDR2 induces MMP-2/MMP-13 expression and associated G/C activity in HSCs under both normal conditions and after chronic CCl4 administration. It also indicates that other cellular sources of DDR2-dependent MMP activity exist in the fibrotic livers.
Thus, we next explored how DDR2 may contribute to the profound changes in the cellular milieu characteristic of chronic fibrosis. Although we found no evidence of DDR2 expression by macrophages (data not shown), increasing data indicate a direct role of macrophages (Kupffer cells and monocyte-derived macrophages) in liver fibrosis by release of MMPs and fibrosis-related cytokines,30 and through HSC recruitment to the fibrotic tissue.32 Furthermore, addition of Kupffer cell–conditioned medium during culture activation of HSCs leads to a closer recapitulation of in vivo gene expression that resembles changes seen after chronic CCl4.23
Thus, to analyze the role of DDR2 in the chronic responses of the fibrogenic liver microenvironment, we next analyzed the chemotactic and ECM-degrading activities of macrophages from livers after chronic CCl4. We correlated these findings with quantitative RT-PCR analysis for a set of critical chemotactic and profibrogenic cytokine mRNAs secreted by macrophages during hepatic fibrosis that are altered in whole DDR2−/− liver extracts.
Paracrine DDR2-Mediated Modulation of Macrophages Mediates the Enhanced Migration of HSCs in Chronically Injured DDR2−/− Livers
Next, we studied HSC migration in response to soluble factors derived from liver macrophages. Quiescent HSCs and macrophages were isolated from oil-treated livers, and activated HSCs and macrophages were isolated from CCl4-treated livers. HSCs were placed in the upper chamber of a migration device, and supernatants from macrophage cultures were added to the lower compartment. Under this condition, quiescent DDR2−/− HSCs migrated approximately twofold more than quiescent DDR2+/+ HSCs (Figure 6A). Maximal migration was displayed by activated DDR2−/− HSCs cultured with media from macrophages isolated from DDR2−/− livers. The promigratory effect of macrophages from CCl4-treated DDR2−/− livers was stronger toward DDR2−/− HSCs than toward DDR2+/+ HSCs. This migratory enhancement was not observed using media conditioned by macrophages from untreated or oil-treated mice (data not shown), which suggests an inhibitory paracrine effect of DDR2 in the secretion of chemotactic factors for HSCs by macrophages from fibrotic livers.
Figure 6.
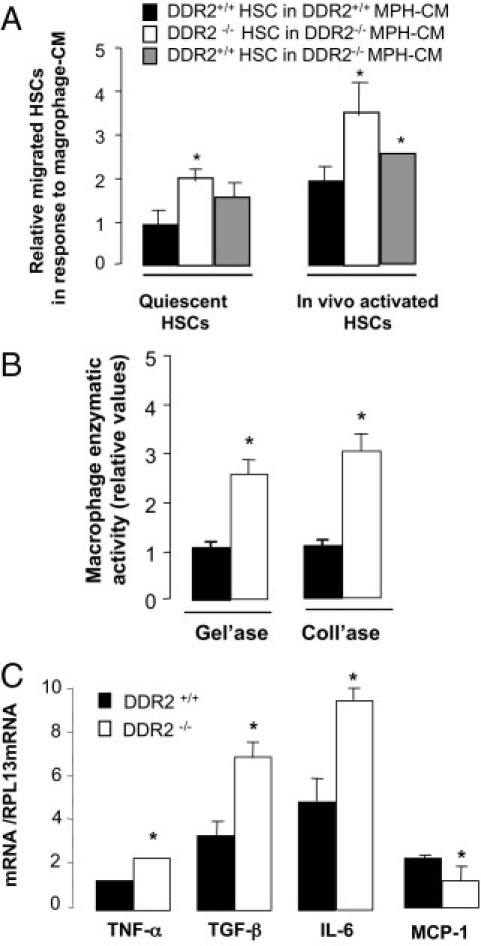
Profibrotic role of macrophages from chronically injured DDR2−/− livers. A: Quiescent HSCs from oil-treated and activated HSCs from CCl4-intoxicated livers were allowed to migrate for 16 hours through collagen type I–coated porous upper wells of migration chambers in response to media conditioned by macrophages from fibrotic livers (MPH-CM). B: Gelatinase and collagenase activities in supernatants from macrophages isolated from fibrotic livers and cultured for 16 hours. C: Differential mRNA expression pattern of fibrosis-related genes in macrophages isolated from fibrotic livers. (n = 16 mice from 2 independent treatments, 2 mice per group). Relative mRNA expression in macrophages from control DDR2+/+ livers compared to RPL13 mRNA expression: TGF-β: 0.06 ± 0.007; tumor necrosis factor-α (TNF-α): 1.61 ± 0.254; IL-6: 0.08 ± 0.002; monocyte chemoattractant protein-1 (MCP-1): 0.31 ± 0.034. *P < 0.05 relative to DDR2+/+ HSC in DDR2+/+ MPH-CM (A) or control DDR2+/+ livers (B and C).
Altered MMP Activity and Cytokine mRNA Expression in Macrophages from Chronically Injured DDR2−/− Livers
Next, macrophages were isolated from fibrotic livers and analyzed for MMP activity and expression of genes implicated in the paracrine modulation of HSC migration and fibrogenic responses. Figure 6B shows that supernatants of macrophages from fibrotic DDR2−/− livers contained 3.3-fold higher gelatinolytic activity and twofold higher collagenolytic activity than cells from DDR2+/+ livers. These results suggest a combined DDR2-dependent role for HSCs and macrophages in ECM remodeling during chronic liver fibrosis.
Scar-associated macrophages during injury are associated with a high expression of fibrogenic TGF-β1 in the scars and high levels of liver tumor necrosis factor-α and IL-6 mRNAs.20, 30 Figure 6C indicates that macrophages from fibrotic DDR2−/− livers displayed a greater induction of TGF-β, tumor necrosis factor-α, and IL-6 mRNAs and a reduced expression of chemotactic monocyte chemoattractant protein-1 mRNA. Quiescent macrophages from untreated DDR2−/− showed no significant differences in gene expression compared with macrophages from untreated DDR2+/+ livers, indicating that the effect of DDR2 removal on macrophages occurs only after persistent liver injury. These results are in correlation with gene expression analysis of whole liver extracts (see Supplemental Table S1 at http://ajp.amjpathol.org).
Discussion
In this work, we show enhanced liver fibrosis after CCl4 administration in DDR2-deficient mice, suggesting that in normal liver, DDR2 suppresses fibrosis in chronic injury. Mechanistically, DDR2 deficiency attenuates the function of quiescent HSCs, as shown by their enhanced basal mRNA expression of fibrogenic and inflammatory factors, and their impaired migratory, proliferative, and gelatinolytic activities. CCl4 administration within this altered, DDR2-deficient sinusoidal milieu affects secretion by macrophages of fibrogenic and chemotactic factors. Ultimately, chronic liver fibrosis in DDR2-deficient mice yields larger collagenous scar tissue that is populated by greater numbers of activated HSCs.
DDR2 expression has been associated with inflammatory and fibrogenic reactions such as wound healing, arthritis, fibrosis, and cancer. We previously reported enhanced DDR2 mRNA expression in HSCs after acute CCl4 administration.12 In this work, we show that DDR2 expression increases after chronic CCl4 treatment, and that HSCs that initiated their activation program in response to liver injury are the main cellular source of DDR2.
Regulation of gene expression by DDR2 has been described primarily in vitro using cells overexpressing DDR212, 33, 34, 35 and studies have been restricted to ECM-related genes. Using primary cells isolated from DDR2-deficient mice, we recently described DDR2-dependent basal gene expression in murine HSCs and skin fibroblasts.26 In this work, we analyzed for the first time the regulatory role of DDR2 in the expression of genes related to immunomodulation and fibrogenesis in HSCs activated by persistent injury. To support the possible role of DDR2 as a suppressor of chronic fibrosis, quantitative RT-PCR showed that expression of TGF-β1, a pluripotent immunomodulatory and fibrogenic factor that largely regulates HSC transition to myofibroblast-like cells,36 of immunosuppressive cytokine IL-10 and of chemokine-like protein osteopontin37 are partially DDR2-dependent in HSCs. The relevance of DDR2 signaling in chronic injury is reinforced by the augmented DDR2-dependent altered gene expression in HSCs from fibrotic livers, a situation in which intense DDR2 phosphorylation is taking place.
We also found that DDR2 modulation of collagen type I, MMP-2, and MMP-13 occurs in HSCs irrespective of their activation status. Surprisingly, although collagen type I directly promoted DDR2 expression and augmented MMP activity in quiescent HSCs, chronic liver injury in DDR2-deficient mice led to larger collagen deposits, MMP activity, and HSC migration. Thus, the positive loop created between ECM composition, DDR2 signaling, and ECM remodeling in HSCs under normal liver function is altered in chronic liver injury.
Experimental models of liver fibrosis highlight the importance of macrophages in perpetuating an inflammatory phase, resulting in the massive release of proinflammatory cytokines including tumor necrosis factor-α, IL-6, TGF-β, and chemokines, as well as activation of HSCs.32 Hepatic macrophages do not express DDR2, but results indicate that DDR2 gene deletion in HSCs alters macrophage responses to chronic injury. It is tempting to speculate that changes in the liver microenvironment generated by chronic DDR2 signaling in activated HSCs influence macrophage profibrogenic activities. For example, HSC expression of DDR2-dependent cytokines monocyte chemoattractant protein-1 and osteopontin modulate macrophage recruitment and macrophage release of MMPs38 and TGF-β production.37
Our results also may indicate that enhanced fibrosis after chronic liver injury in the absence of DDR2 in HSCs result from the disruption of the fine balance between secretion of pro-activation factors by both activated macrophages and activated HSCs, to enhance recruitment of activated HSCs to the fibrotic scar, and secretion of antimigratory factors for macrophages by scar-recruited activated HSCs.
In summary, our work underscores the role of altered receptor-ECM interactions in HSCs within a fibrogenic microenvironment. Our findings support a model in which DDR2 signaling in HSCs down-regulates gene expression of fibrosis-related cytokines and up-regulates ECM-related genes such as fibrillar collagen type I, basal membrane degrading MMP-2, and collagenolytic MMP-13. Chronic injury induces an HSC activation program that includes enhanced DDR2 expression and signaling, leading to downstream modulation of DDR2-dependent genes. Furthermore, the altered fibrogenic sinusoidal milieu resulting from tonic DDR2 signaling leads to paracrine inhibition of profibrogenic activities by scar-associated macrophages.
Acknowledgments
We thank Joana Marquez, Iratxe Basaldua, and Aritz Lopategi for skillful expertise, Hsin-Chie Lin (Regeneron Pharmaceuticals) for preliminary in vivo and in vitro experiments, and Fernando Vidal-Vanaclocha for his helpful comments.
Footnotes
Supported by the International Association for Cancer Research, Spanish Ministry of Health, and Basque Department of Health (E.O.); and NIH grants DK56621 and 1P20AA017067-01 (S.L.F.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.09.002.
Supplementary data
References
- 1.Friedman S.L. Liver fibrosis—from bench to bedside. J Hepatol. 2003;38:S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R., Brenner D.A. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabele E., Brenner D.A., Rippe R.A. Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci. 2003;8:D69–D77. doi: 10.2741/887. [DOI] [PubMed] [Google Scholar]
- 4.Naito M., Hasegawa G., Ebe Y., Yamamoto T. Differentiation and function of Kupffer cells. Med Electron Microsc. 2004;37:16–28. doi: 10.1007/s00795-003-0228-x. [DOI] [PubMed] [Google Scholar]
- 5.Thurman R.G. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 6.Canbay A., Friedman S., Gores G.J. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 7.Milani S., Herbst H., Schuppan D., Kim K.Y., Riecken E.O., Stein H. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology. 1990;98:175–184. doi: 10.1016/0016-5085(90)91307-r. [DOI] [PubMed] [Google Scholar]
- 8.Viñas O., Bataller R., Sancho-Bru P., Ginès P., Berenguer C., Enrich C., Nicolás J.M., Ercilla G., Gallart T., Vives J., Arroyo V., Rodés J. Human hepatic stellate cells show features of antigen-presenting cells and stimulate lymphocyte proliferation. Hepatology. 2003;38:919–929. doi: 10.1053/jhep.2003.50392. [DOI] [PubMed] [Google Scholar]
- 9.Marra F. Hepatic stellate cells and the regulation of liver inflammation. J Hepatol. 1999;31:1120–1130. doi: 10.1016/s0168-8278(99)80327-4. [DOI] [PubMed] [Google Scholar]
- 10.Maher J.J. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 2001;21:417–426. doi: 10.1055/s-2001-17555. [DOI] [PubMed] [Google Scholar]
- 11.Barczyk M., Carracedo S., Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olaso E., Ikeda K., Eng F.J., Xu L., Wang L.H., Lin H.C., Friedman S.L. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108:1369–1378. doi: 10.1172/JCI12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shrivastava A., Radziejewski C., Campbell E., Kovac L., McGlynn M., Ryan T.E., Davis S., Goldfarb M.P., Glass D.J., Lemke G., Yancopoulos G.D. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 14.Konitsiotis A.D., Raynal N., Bihan D., Hohenester E., Farndale R.W., Leitinger B. Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. Biol Chem. 2008;283:6861–6868. doi: 10.1074/jbc.M709290200. [DOI] [PubMed] [Google Scholar]
- 15.Xu L., Servais J., Polur I., Kim D., Lee P.L., Chung K., Li Y. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010;62:2736–2744. doi: 10.1002/art.27582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao T.K., Kimura Y., Kenny T.P., Branchi A., Gishi R.G., Van de Water J., Kung H.J., Friedman S.L., Gershwin M.E. Elevated expression of tyrosine kinase DDR2 in primary biliary cirrhosis. Autoimmunity. 2002;35:521–529. doi: 10.1080/0891693021000057784. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X.H., Yan M., Liu L., Wu T.J., Ma L.L., Wang L.X. Expression of discoidin domain receptors (DDR2) in alcoholic liver fibrosis in rats. Arch Med Res. 2010;41:586–592. doi: 10.1016/j.arcmed.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda K., Wang L.H., Torres R., Zhao H., Olaso E., Eng F.J., Labrador P., Klein R., Lovett D., Yancopoulos G.D., Friedman S.L., Lin H.C. Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J Biol Chem. 2002;277:19206–19212. doi: 10.1074/jbc.M201078200. [DOI] [PubMed] [Google Scholar]
- 19.Junqueira L.C., Bignolas G., Brentani R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 20.Duffield J.S., Forbes S.J., Constandinou C.M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., Iredale J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellers A., Cartwright E., Murphy G., Reynolds J.J. Evidence that latent collagenases are enzyme-inhibitor complexes. Biochem J. 1997;163:303–307. doi: 10.1042/bj1630303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen B., Arteta B., Smedsrød B. The physiological scavenger receptor function of hepatic sinusoidal endothelial and Kupffer cells is independent of scavenger receptor class A type I and II. Mol Cell Biochem. 2002;240:1–8. doi: 10.1023/a:1020660303855. [DOI] [PubMed] [Google Scholar]
- 23.De Minicis S., Seki E., Uchinami H., Klube J., Zhang Y., Brenner D.A., Schwabe R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132:1937–1946. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 24.Arthur M.J. Fibrogenesis II: metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G245–G249. doi: 10.1152/ajpgi.2000.279.2.G245. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz P.A., Jarai G. Collagen I induces discoidin domain receptor (DDR) 1 expression through DDR2 and a JAK2-ERK1/2-mediated mechanism in primary human lung fibroblasts. J Biol Chem. 2011;286:12912–12923. doi: 10.1074/jbc.M110.143693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olaso E., Lin H.C., Wang L.H., Friedman S.L. Impaired dermal wound healing in discoidin domain receptor 2-deficient mice associated with defective extracellular matrix remodeling. Fibrogenesis Tissue Repair. 2011;4:5. doi: 10.1186/1755-1536-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rockey D.C., Boyles J.K., Gabbiani G., Friedman S.L. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol. 1992;24:193–203. [PubMed] [Google Scholar]
- 28.Ferré N., Martínez-Clemente M., López-Parra M., González-Périz A., Horrillo R., Planagumà A., Camps J., Joven J., Tres A., Guardiola F., Bataller R., Arroyo V., Clària J. Increased susceptibility to exacerbated liver injury in hypercholesterolemic ApoE-deficient mice: potential involvement of oxysterols. Am J Physiol Gastrointest Liver Physiol. 2009;296:G553–G562. doi: 10.1152/ajpgi.00547.2007. [DOI] [PubMed] [Google Scholar]
- 29.Fallowfield J.A., Mizuno M., Kendall T.J., Constandinou C.M., Benyon R.C., Duffield J.S., Iredale J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 30.Luckey S.W., Petersen D.R. Activation of Kupffer cells during the course of carbon tetrachloride-induced liver injury and fibrosis in rats. Exp Mol Pathol. 2001;71:226–240. doi: 10.1006/exmp.2001.2399. [DOI] [PubMed] [Google Scholar]
- 31.Xu L., Hui A.Y., Albanis E., Arthur M.J., O'Byrne S.M., Blaner W.S., Mukherjee P., Friedman S.L., Eng F.J. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wynn T.A., Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faraci E., Eckber M., Gerstmayerc B., Bosioc A., Vogel W.F. An extracellular matrix-specific microarray allowed the identification of target genes downstream of discoidin domain receptors. Matrix Biol. 2003;22:373–381. doi: 10.1016/s0945-053x(03)00053-2. [DOI] [PubMed] [Google Scholar]
- 34.Su J., Yu J., Ren T., Zhang W., Zhang Y., Liu X., Sun T., Lu H., Miyazawa K., Yao L. Discoidin domain receptor 2 is associated with the increased expression of matrix metalloproteinase-13 in synovial fibroblasts of rheumatoid arthritis. Mol Cell Biochem. 2009;330:141–152. doi: 10.1007/s11010-009-0127-0. [DOI] [PubMed] [Google Scholar]
- 35.Ferri N., Carragher N.O., Raines E.M. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling: potential implications in atherosclerosis and lymphangioleiomyomatosis. Am J Pathol. 2004;164:1575–1585. doi: 10.1016/S0002-9440(10)63716-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gressner A.M., Weiskirchen R., Breitkopf K., Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7 doi: 10.2741/A812. d793-d780. [DOI] [PubMed] [Google Scholar]
- 37.Mori R., Shaw T.J., Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205:43–51. doi: 10.1084/jem.20071412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawashima R., Mochida S., Matsui A., YouLuTuZ Y., Ishikawa K., Toshima K., Yamanobe F., Inao M., Ikeda H., Ohno A., Nagoshi S., Uede T., Fujiwara K. Expression of osteopontin in Kupffer cells and hepatic macrophages and stellate cells in rat liver after carbon tetrachloride intoxication: a possible factor for macrophage migration into hepatic necrotic areas. Biochem Biophys Res Commun. 1999;256:527–531. doi: 10.1006/bbrc.1999.0372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.