

Abstract
Hypersensitivity pneumonitis (HP), an inflammatory lung disease, develops after repeated exposure to inhaled particulate antigen and is characterized by a vigorous T helper type 1-mediated immune response, resulting in the release of IL-12 and interferon (IFN)-γ. These T helper type 1 cytokines may participate in the pathogenesis of HP. Stachybotrys chartarum (SC) is a dimorphic fungus implicated in a number of respiratory illnesses, including HP. Here, we have developed a murine model of SC-induced HP that reproduces pathology observed in human HP and hypothesized that toll receptor-like 9 (TLR9)-mediated dendritic cell responses are required for the generation of granulomatous inflammation induced by inhaled SC. Mice sensitized and challenged with 106 SC spores develop granulomatous inflammation with multinucleate giant cells, accompanied by increased accumulation of neutrophils and CD4+ and CD8+ T cells. SC sensitization and challenge resulted in robust pulmonary expression of tumor necrosis factor-α, IL-12, and IFN-γ. SC-mediated granulomatous inflammation required IFN-γ and was TLR9 dependent, because TLR9−/− mice displayed reduced peribronchial inflammation, decreased accumulation and/or activation of polymorphonuclear (PMN) and CD4+ and CD8+ T cells, and reduced lung expression of type 1 cytokines and chemokines. T-cell production of IFN-γ was IL-12 dependent. Our studies suggest that TLR9 is critical for dendritic cell-mediated development of a type 1 granulomatous inflammation in the lung in response to SC.
Hypersensitivity pneumonitis (HP) is an inflammatory lung disease that develops after repeated exposure to inhaled particulate antigen.1 Exposure to these antigens occurs most frequently in certain occupations or hobbies or within the home environment. Accumulating evidence suggests that molds may represent an underappreciated cause of HP. For instance, molds are increasingly being recognized as a cause of HP after exposure to contaminated humidifiers, heating ducts, and, more recently, flooded old buildings. Stachybotrys chartarum (SC) is a fungus that causes “black mold disease” and “sick building syndrome” and has been speculated as a main offender in promoting HP.
Histopathology in HP is characterized by dense accumulation of macrophages, lymphocytes, epithelioid cells, and fibroblasts around indigestible particles or antigens.2 Hypersensitivity-type granulomas are believed to be induced by the interaction of activated T cells, macrophages, and dendritic cells (DCs) in an antigen-specific manner.3 DCs are potent antigen-presenting cells in the lungs and play a main regulatory role in T-cell activation and differentiation. T cells are necessary for the development and progression of HP.4, 5 For instance, athymic nude mice that lack T cells are protected from the development of HP in response to inhaled antigenic challenge. In rodent models of HP, there is a shift in T-cell population to CD8+ predominance by 24 to 48 hours after challenge with antigen.5, 6, 7 In humans with HP, there is an increase in the number of CD8+ T and CD8+ natural killer T cells in bronchoalveolar lavage fluid or lung interstitium or both.8, 9, 10, 11 Mechanisms that promote CD8+ T-cell accumulation/activation have not been thoroughly defined.
The generation of granulomatous inflammation in HP depends on the expression of T helper type 1 (Th1) cytokines, including tumor necrosis factor (TNF)-α, IL-12, and interferon (IFN)-γ.12, 13 In a murine model of HP due to the thermophillic fungus Saccharopolyspora rectivirgula, genetic deletion or antibody neutralization of IFN-γ resulted in a decrease in TNF-α and IL-12 production, lung permeability, granuloma formation, and subsequent fibrotic response, compared with wild-type (WT) mice.14 Moreover, mice that are Th1 biased are more susceptible to the development of HP in response to antigen, whereas Th2-biased mice are generally resistant.15 Conidia from SC can function as antigens to promote allergic or hypersensitivity responses. In the limited animal studies performed to date, the intranasal (i.n.) installation of SC spores to nonsensitized mice resulted in an influx of mononuclear cells and neutrophils, which is associated with the induction of pro-inflammatory cytokines (IL-1, IL-6, and TNF-α) and chemokines [macrophage inflammatory protein (MIP)-1α and monocyte chemoattractant protein 1].16, 17 In presensitized animals, i.n. challenge with SC resulted in a granulomatous inflammatory cell infiltrate in lung peribronchial, perivascular, and alveolar spaces and an increase in the lung expression of IL-1, TNF-α, and MIP-1α.18, 19 Despite the substantial clinical effect of SC, little is known about the specific mechanisms responsible for SC-mediated lung disease.
Toll-like receptors (TLRs) are a family of type I transmembrane receptors that respond to pathogen-associated molecular patterns expressed by a diverse group of infectious microorganisms, resulting in activation of the host's immune system.20, 21, 22, 23 The role of TLRs in the generation of HP is incompletely defined. Given the composition of fungal cell wall components,24, 25 the most relevant TLRs are TLR4, which binds to and is activated by lipopolysaccharide and fungal mannans, and TLR2, which recognizes fungal β-glycans and zymosan.26, 27 TLR9 is of particular interest, because this TLR recognizes fungal DNA, and this TLR has been shown to drive type 1 responses to both microbial and nonmicrobial antigens.28 We have shown that TLR9 is required for optimal expression of activating type 1 cytokines and chemokines in response to both intracellular and extracellular lung bacterial pathogens.29, 30 TLR9 may also contribute to granulomatous inflammation. For example, TLR9−/− mice displayed a marked reduction of mononuclear cell accumulation, granuloma formation within the liver, and reductions in both IFN-γ and TNF-α production in response to heat-killed Propionobacterium acnes, compared with WT mice.31, 32 The cellular components and pathogen recognition receptors involved in responding to intrapulmonary SC spores have not been defined and are the focus of this study.
Materials and Methods
Reagents
Murine recombinant cytokines were purchased from R&D Systems (Minneapolis, MN). Polyclonal antimurine cytokine antisera used in enzyme-linked immunosorbent assay (ELISA) or neutralization experiments were produced by immunization of rabbits with either recombinant murine cytokines in multiple intradermal sites with complete Freund's adjuvant. ELISA for quantization of murine IFN-γ was purchased from PBL Biomedical Laboratories (Piscataway, NJ).
Animals
Specific pathogen-free BALB/c mice and IFN-γ−/− mice on a BALB/c genetic background (matched for age and sex) were purchased from The Jackson Laboratory (Bar Harbor, ME). TLR9−/− mice (BALB/c background) were bred in house. All mouse strains were housed in specific pathogen-free conditions within the animal care facility (Unit for Laboratory Animal Medicine) until the day of sacrifice.
Administration of SC Conidia
Intratracheal
Mice were injected with ketamine and xylazine by the i.p. route. After adequate anesthesia, mice were restrained in the supine position. The anterior aspect of the neck was exposed, and an incision was made to visualize the trachea. A 26-gauge needle was advanced endotracheally, and, through this, 30 μL of normal saline containing SC conidia was injected. The animals were then allowed to recover, and the wound was closed with surgical staples. No adverse effects were encountered with this procedure.
Intranasal
Animals were lightly anesthetized with ketamine and xylazine. After adequate anesthesia, 10 to 20 μL of a solution containing SC conidia was placed in the nares of mice until the solution was inhaled.
Intraperitoneal
Mice were injected i.p. with 50 μL of a solution containing SC conidia.
Removal of Various Organs at the Time of Necropsy
Mice were euthanized at various intervals after SC sensitization and challenge by inhalation of carbon dioxide and exsanguinated; organs (lungs, spleens) were removed.
Macrophage Isolation
At various times after i.t. inoculation, mice were euthanized in a rapid and painless fashion while deeply anesthetized. The trachea was cannulated with a plastic tube; the lungs were lavaged with a 3-mL aliquot of calcium, magnesium-free PBS, and alveolar macrophages were purified with the adherence purification method.
Total Lung Leukocyte Preparation
Lungs were removed from euthanized animals, and leukocytes were prepared as previously described. Briefly, lungs were minced with scissors to a fine slurry in 15 mL/lung digestion buffer [RPMI/5% fetal calf serum/1 mg/mL collagenase (Boehringer Mannheim Biochemical, Germany)/30 μg/mL DNase (Sigma-Aldrich, St. Louis, MO)]. Lung slurries were enzymatically digested for 30 minutes at 37°C. The total lung cell suspension was pelleted, resuspended, and spun through a 20% Percoll gradient to enrich for leukocytes before further analyses.
Multiparameter Flow Cytometric Analyses
Total lung leukocytes were isolated as described above. Lymphocyte subsets were analyzed by first gating on CD45+ “lymphocyte-sized” leukocytes and then examined for lymphocyte-associated markers. Antibodies (BD Pharmingen, San Diego, CA, and Caltag, UK) used for phenotyping include T-cell markers (anti-CD4, anti-CD8, anti-CD69), B cells (anti-CD19), pan-natural killer cells (anti-DX5), CD11c, major histocompatibility complex type II (MHCII), and Gr-1 (DC). Cells were collected on a FACSCalibur cytometer (Becton Dickinson, San Jose, CA) with the use of Cellquest software version 7 (Becton Dickinson). Analyses of data were performed using the Cellquest software package.
Cytokine ELISA
Lung homogenate or DC supernatant fluids were collected, and cytokine levels were determined by a sandwich ELISA method as previously described.
Generation of Bone Marrow–Derived DCs
Bone marrow was harvested from WT and mutant mice and seeded in tissue culture flasks in RPMI 1640–based complete media with 10 ng/mL murine recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF). Cells were fed after 3 days, and loosely adherent cells were collected after 6 to 7 days and incubated with anti-CD11c antibody coupled to magnetic beads (Miltenyi Biotec, Auburn, CA). Cells were purified with positive selection for CD11c+ cells by running the cell suspension through a magnetic column. CD11c+ DCs were plated overnight and resuspended in fresh media the next day.
Isolation of Lung DCs
Leukocytes from lung digests were purified with positive selection for CD11c+ cells by running the cell suspension through a magnetic column. CD11c+ cells were plated on plastic for 1 hour to remove residual lung macrophages, then nonadherent cells were plated for further analysis. Final cells were >90% DCs, as determined by flow cytometry (CD11chi, MHCIIhi, Gr-1low).
mRNA Extraction and Real-Time (TaqMan) Quantitative PCR
Total RNA from cells was isolated per manufacturer's protocol for the RNAeasy Mini kit (Qiagen, Valencia, CA). RNA amounts were determined by spectrometric analysis at 260 nm. All primers were designed with Primer Express software version 2 (Applied Biosystems, Foster City, CA). Levels of mRNA were determined by real-time quantitative RT-PCR analysis with the use of an ABI PRISM 7000 Sequence Detection System (ABI/Perkin Elmer Co, Foster City, CA).
For inflammatory score grading, H&E staining performed on lung sections was harvested from mice at day 2 after SC challenge. The total number of peribronchial inflammatory cells was quantitated per 40× high-power fields in 10 random fields as follows: 0 to 5 cells were graded as 0, 5 to 10 cells were graded as 1, 10 to 20 cells were graded as 2, 20 to 30 cells were graded as 3, and >40 cells were graded as 4. The value from all of the 10 fields is represented as mean ± SEM.
Statistical Analyses
Ratio scale data were evaluated by analysis of variance with Newman-Keuls follow-up. Statistical significance was determined with the unpaired, two-tailed Student's t-test and nonparametric Mann-Whitney U test. Calculations were performed with InStat for Macintosh version 6 (GraphPad Software, San Diego, CA). In all cases, P < 0.05 was considered significant.
Results
Effect of Sensitization and Challenge with SC Conidia on Lung Histopathology
We have developed a murine model of HP with the use of the fungus SC, which has been associated with a variety of respiratory diseases, including sick building syndrome and black mold disease. This is a particularly attractive model, because SC is a clinically relevant cause of lung disease, and the model nicely mimics the repeated exposure to antigen that would occur in the natural setting. In this model, we sensitized BALB/c mice (WT) with i.p. injections of 106 SC spores on days −14 and −7 and then administer 106 spores i.n. on days −5 and −3. At day 0, we challenged sensitized mice with 106 fungal spores i.t., then harvested the lungs at day 2 and day 5. We used BALB/c mice for our experiments, because these mice are Th1 biased when challenged with various fungi, and studies suggest a more robust type 1 granulomatous response in Th1-biased mice.15 The i.t. challenge with SC spores in sensitized mice resulted in a marked accumulation of neutrophils and mononuclear cells within the interstitial compartments and a predominantly lymphocytic infiltration within the peribronchial regions (Figure 1), maximal at day 2 after i.t. SC (Figure 1C). In addition, giant cells and loose epitheloid granulomas were observed within both the interstitial and peribronchial areas. Partial resolution or i.t. saline (Figure 1A) of granulomatous inflammation was noted by day 5 (Figure 1D). Nonsensitized mice with i.t.-administered SC spores did not develop appreciable inflammation or evidence of granulomas (Figure 1B). We also challenged mice with defective TLR4 signaling (TLR4lps-d, BALB/c genetic background) with SC conidia and found a similar degree of granulomatous inflammation as observed in WT mice, indicating that granulomatous inflammation in this model did not depend on TLR4 signaling, excluding a possible role for lipopolysaccharide contamination (data not shown).
Figure 1.
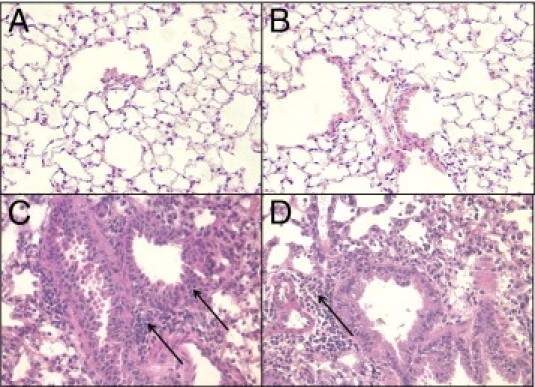
Lung histopathology after SC challenge. H&E staining of lung sections obtained from mice after saline (A) or nonsensitized (B) or sensitized mice challenged with SC at day 2 (C) or day 5 (D). Accumulation of neutrophils and mononuclear cells is shown within the interstitial compartments, and a predominantly lymphocytic infiltration within the peribronchial regions as indicated by thin arrows, as well as multinucleated giant cells (thicker arrow) and loose epitheloid granulomas within both the interstitial and peribronchial areas in SC-challenged mice. Four to five mice were used per group, and the experiments were repeated twice. Original magnification, ×40. N = 4 to 5; mean (SEM of two experiments).
SC Sensitization and Challenge Promote Lung PMN- and T-Cell Accumulation
We next examined lung inflammatory cell influx in mice after SC sensitization and subsequent i.t. challenge. We observed a robust accumulation of inflammatory cells in lung digests of WT animals 2 days after i.t. SC challenge, representing a >10-fold increase over baseline number of cells (Figure 2A), with a decrease in number of inflammatory cells by day 5. There was an approximately fourfold increase in the number of neutrophils at day 2 after SC (Figure 2B). No change in number of lung macrophages was observed after SC sensitization/challenge (data not shown). SC sensitization and challenge also resulted in an increase in the total number of CD4+ T cells (Figure 2C) and especially CD8+ T cells (Figure 2D), maximal at 2 days after i.t. SC challenge (2.8-fold and 6.4-fold increase over baseline, respectively; P < 0.05).
Figure 2.
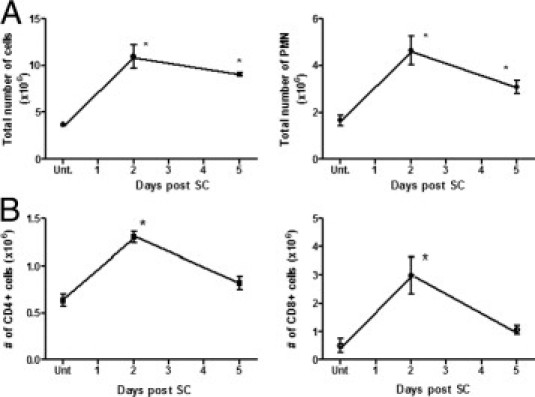
A: Accumulation of inflammatory cells after SC challenge. WT mice were sensitized and challenged with SC as described, lungs were harvested at day 2 and day 5 after challenge, lung collagenase digests were performed, total number of leukocytes were counted with a hemocytometer, cytospins were performed, and total PMN cell were quantitated. B: Flow cytometry was performed, and total CD4+ and CD8+ T cells were quantitated. N = 4 to 5; mean ± SEM of two experiments. *P < 0.05 compared with untreated controls.
SC Sensitization and Challenge Result in Type 1 Cytokine Production, and Granulomatous Inflammation Is Reduced in IFN-γ−/− Mice
To further assess the mechanisms underlying cellular changes observed in mice after SC sensitization and challenge, we profiled type 1 cytokine expression in whole lungs 2 and 5 days after challenge. SC-challenged mice displayed a substantial induction of the type 1 cytokines IFN-γ, IL-12, and TNF-α (Figure 3A), maximal at day 2 after i.t. SC administration.
Figure 3.
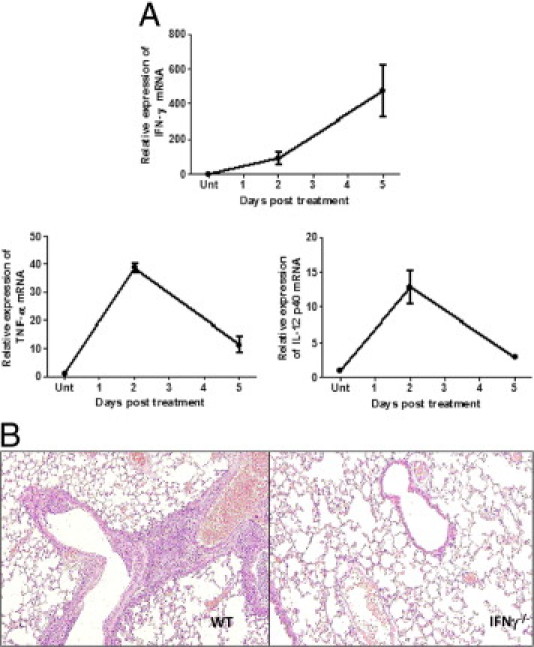
A: Type 1 cytokine expression in lung and lung histopathology in IFNγ−/− mice after SC sensitization and challenge. WT mice were sensitized and challenged with SC as described, and lungs were harvested at day 2 and day 5. Relative expression of IFNγ, TNF-α, and IL-12 p40 was measured by real-time PCR. Error bars indicate mean (SEM of two experiments). B: H&E staining was performed on lung sections harvested from WT and IFN-γ−/− mice at day 2 after SC challenge. N = 5 in each experiment, repeated twice.
Previous studies have shown that IFN-γ is essential for the generation of the granulomatous inflammatory response seen in HP. To examine the role of IFN-γ in SC-induced HP, WT and IFN-γ−/− mice were sensitized and challenged with SC, then lungs were harvested at day 2. As previously observed, WT mice displayed histologic evidence of peribronchiolar mononuclear aggregates and loosely formed granulomas; these were graded on the basis of the inflammation score described in Materials and Methods (Table 1). In contrast, IFN-γ−/− mice were protected after SC challenge, with minimal to no evidence of inflammation at days 2 (Figure 3B) and 5 (histology not shown), further supporting a necessary role for IFN-γ in SC-induced HP.
Table 1.
Inflammation Score and Granulomas in Lung Sections Harvested from WT and IFN-γ−/−Mice at Day 2 after SC Challenge
| Inflammation score/10 fields | No. of granulomas/10 fields | |
|---|---|---|
| Wild type | 3.75 ± 0.23 | 1.91 ± 0.7 |
| IFNγ−/− | 0.55 ± 0.11 | 0 ± 0 |
H&E staining performed on lung sections harvested from WT and IFNγ−/− mice at day 2 after SC challenge and total number of inflammatory cells and granulomas quantitated per magnification of ×40 high-power fields. Inflammatory cells were graded as described in Materials and Methods.
TLR9−/− Mice Have Reduced Granulomatous Inflammation after Sensitization and Challenge with SC Conidia
TLR9 is known to play a role in the regulation of Th1 responses.29, 33, 34 To investigate the contribution of TLR9 to SC-induced granulomatous inflammation, we sensitized and challenged WT and TLR9-deficient mice with i.p. injections of 106 SC spores on days −14 and −7, and then administer 106 spores i.n. on days −5 and −3. At day 0, sensitized mice were administered 106 fungal spores, and lungs were harvested on day 2. As observed previously, WT mice developed peribronchial and interstitial inflammatory infiltrates, multinucleated giant cells, and loose epitheloid granulomas (Figure 4A). By comparison, TLR9−/− mice had a significantly blunted inflammatory response, including a marked decrease in inflammatory cells, giant cells, and granuloma formation (Figure 4B). To quantify the difference between the WT and TLR9−/− mice, inflammation scores were performed as described in Materials and Methods, as well as the number of granulomas quantified, shown as Table 2.
Figure 4.
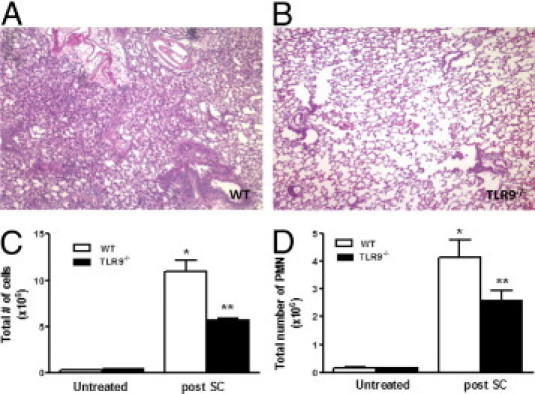
Lung histopathology and inflammatory cell accumulation in WT and TLR9−/− mice after SC challenge. WT and TLR9−/− mice were sensitized and challenged with SC, and lungs were harvested at day 2. H&E staining was performed on lung sections as shown in A and B. C: Lung collagenase digests were performed, and total leukocytes were quantitated in different groups with a hemocytometer. D: Total PMN cells were quantified on cytospins. N = 4 to 5 in each group; experiments were performed twice. *P < 0.05 compared with untreated controls; **P < 0.05 compared with SC challenged WT mice.
Table 2.
Inflammation Score and Granulomas in Lung Sections Harvested from WT and TLR9−/− Mice at Day 2 after SC Challenge
| Inflammation score/10 fields | No. of granulomas/10 fields | |
|---|---|---|
| Wild type | 3.97 ± 0.11 | 1.8 ± 0.37 |
| TLR9−/− | 1.43 ± 0.13 | 0 ± 0 |
H&E staining performed on lung sections harvested from WT and TLR9−/− mice at day 2 after SC challenge and total number of inflammatory cells and granulomas quantitated per magnification of ×40 high-power field. Inflammatory cells were graded as described in Materials and Methods.
To quantify changes in the number of inflammatory cells, WT and TLR9−/− mice were sensitized and challenged as before, lungs were harvested at day 2 after challenge, and collagenase digests were performed. As observed previously, WT mice had a robust increase in the number of total cells (Figure 4C) and PMN cells at day 2 after challenge (Figure 4D). In comparison, TLR9-deficient mice had fewer numbers of both total cells and PMN cells (50% and 35% decrease, respectively).
TLR9-Deficient Mice Have Decreased Accumulation and Activation of T Cells and DCs Compared with that in WT Mice after SC Sensitization and Challenge
Next, the number and activational state of T-cell populations present in the lungs of WT and TLR9−/− mice were quantitated at baseline and after SC administration by flow cytometry. As shown in Figure 5A, SC sensitization and challenge resulted in an increase in the total number of CD4+ T cells and activated CD4+ T cells (as indicated by expression of the activational marker CD69; P < 0.05) in WT mice (Figure 5B). TLR9−/− mice had a similar increase in total CD4+ T cells, but the number of activated CD4+ T cells was significantly reduced in SC-challenged TLR9−/− mice (P < 0.05). We observed an even more impressive increase in the total and activated CD8+ T cells (Figure 5, C and D, respectively) in WT mice 2 days after i.t. SC challenge (6.4-fold and ninefold increase over baseline, respectively). Compared with WT animals, there was a 35% decrease in total CD8+ T cells (P = 0.05) and a nearly 70% reduction in CD69+ CD8+ T cells in TLR9−/− mice after sensitization and challenge with SC conidia (P < 0.05). Furthermore, a considerable increase was observed in the number of natural killer cells after SC challenge which was significantly reduced in TLR9−/− mice (data not shown). Collectively, these studies suggest that TLR9 mediates T-cell accumulation and activation during the evolution of SC-induced HP.
Figure 5.
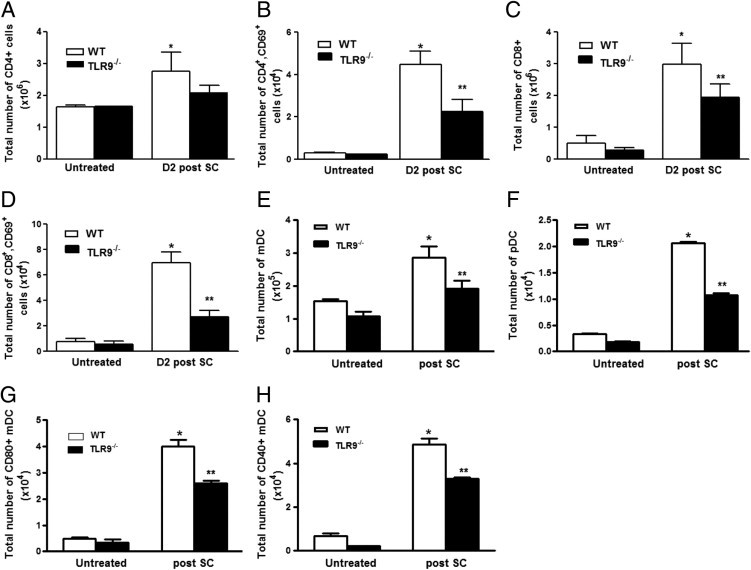
Accumulation and activation of T cells and DCs in WT and TLR9−/− mice after SC challenge. WT and TLR9−/− mice were sensitized and challenged with SC, and lungs were harvested at day 2. Lung collagenase digests were performed, and total number of CD4+ T cells (A), activated CD4+ T cells (B), total number of CD8+ T cells (C), activated CD8+ T cells (D), total number of DCs [both myeoloid dendritic cell (mDC) (E) and plasmacytoid dendritic cell (pDC) (F)], as well as activated mDCs indicated by expression of costimulatory marker CD80 (G) and CD40 (H) were measured by flow cytometry. N = 6 to 8 in each group; experiments were performed twice. Results are expressed as mean ± SEM. *P < 0.05 compared with untreated controls; **P < 0.05 compared with WT mice after SC.
DCs are potent antigen-presenting cells in the lung and are necessary for T-cell activation and polarization. To assess DC accumulation in lungs harvested from SC-challenged WT and TLR9−/−mice, T cells, B cells, and autofluorescent macrophages were eliminated by forward and side scatter characteristics, and the remaining cells were assessed for expression of MHCII, CD11c, and Gr-1. We found no difference in the number of conventional DC (MHCII+,CD11c+, GR-1−) in the lungs of WT and TLR9−/− mice in the unsensitized state (Figure 5E). However, the sensitization and challenge with SC resulted in a 2.5-fold increase in the number of conventional DCs (Figure 5F) (MHCII+, CD11c+, GR-1low) and a fourfold increase in plasmacytoid DCs (Figure 5G) (GR-1+) in lungs of WT mice 48 hours after challenge, whereas a more modest increase was observed in the total number and percentage of these cells in spore-challenged TLR9−/− mice (P < 0.05). Furthermore, with the use of four-color flow cytometry, we observed impairment in the activation/maturation of conventional DCs as indicated by reduced expression of the costimulatory molecules of CD40 and CD80 in the lungs of infected TLR9−/− mice 48 hours after i.t. SC administration compared with infected WT animals (Figure 5H) (P < 0.01).
TLR9−/− Mice Have Reduced Type 1 Cytokine and Chemokine Production after Sensitization and Challenge with SC Spores
We next assessed the expression of TNF-α, type I cytokines IL-12 and IFN-γ, and chemokines in the lungs of WT and TLR9-deficient mice challenged with SC. In WT animals, sensitization and subsequent challenge with SC induced a vigorous time-dependent expression of these cytokines, with TNF-α and IL-12 p40 mRNA levels peaking at day 2, and IFN-γ mRNA levels continuing to rise out to day 5 after i.t. SC spore administration (Figure 6A). In comparison to WT mice, mRNA expression of TNF-α, IL-12 p40, and IFN-γ was substantially diminished in the lungs of TLR9-deficient mice after challenge with SC. Similar to the mRNA data, WT mice had a substantial induction of TNF-α, IL-12, and IFN-γ protein at day 2 after sensitization and challenge, which was significantly decreased in TLR9−/− mice (Figure 6B). Given that impaired neutrophil and activated T-cell influxes were observed in TLR9−/− mice after SC, we also assessed neutrophil and effector T cell–specific chemokine production. Compared with WT animals, TLR9−/− mice had significantly lower levels of MIP-2 and keratinocyte-derived chemokine at day 2 (54% and 77% reduction, respectively; P < 0.01), which was maintained at day 5. In addition, the maximum induction of IFN-inducible protein (IP)-10 was reduced by nearly 50% (P < 0.01).
Figure 6.
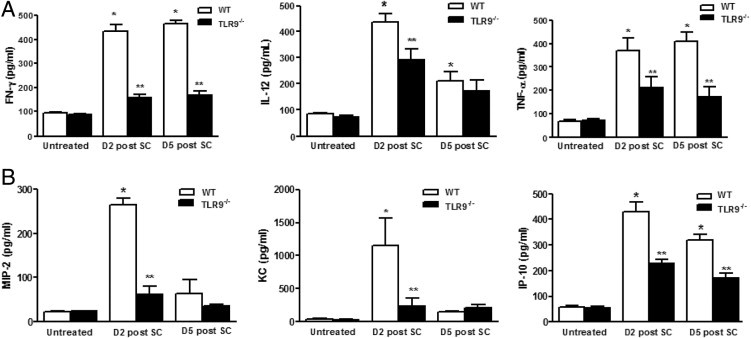
Cytokine and chemokine levels in lungs of WT and TLR9−/− after SC sensitization and challenge. WT and TLR9−/− mice were sensitized and challenged with either saline or SC, and lungs were harvested at day 2 and day 5. Cytokine levels of IFN-γ, TNF-α, IL-12 (A) and chemokine levels of keratinocyte-derived chemokine (KC), MIP-2, and IFN-inducible protein 10 (IP-10) (B) were measured by ELISA. N = 6 to 8 in each group; experiments were performed twice. Results are expressed as mean ± SEM. *P < 0.05 compared with untreated controls; **P < 0.05 compared with WT mice after SC sensitization and challenge.
DCs from TLR9−/− Mice Display Impaired SC-Induced IL-12 Production and Reduced Ability to Activate T Cells
DCs are required for the development of antigen-specific type 1 cytokine responses, particularly IFN-γ production by T cells.35 To determine whether the diminished granulomatous inflammatory response observed in the TLR9−/− mice was secondary to altered DC function, WT and TLR9−/− mice were sensitized (i.p. and i.n.) as described previously, and bone marrow was harvested on day 0. Bone marrow cells were cultured for 6 days in GM-CSF, DC-purified by CD11c+ magnetic bead separation on day 6, and stimulated with SC spores in a ratio of 1:10. Spontaneous ex vivo production of IL-12 was measured by ELISA, and IL-12 p40 mRNA expression was determined by real-time PCR. As shown in Figure 7A, incubation of sensitized WT DCs with SC resulted in considerable induction of IL-12 p40 mRNA and IL-12 protein (Figure 7B), which was substantially mitigated in lung DCs from TLR9−/− mice (P < 0.05). To assess the contribution of IL-12 production from DCs to expression of IFN-γ from T cells, bone marrow was isolated from sensitized WT and TLR9−/− mice on day 0, then DCs were maturated for 6 days in GM-CSF. DCs were then stimulated with SC and cocultured with splenic T cells harvested from naive WT mice for 18 hours in the presence and absence of IL-12 antibody. T cells cocultured with WT DCs produced large quantities of IFN-γ. The induction of IFN-γ depended on IL-12 production by DCs, because IFN-γ expression was totally abolished in the presence of IL-12 antibody (Figure 7C). Interestingly, T cells when cocultured with sensitized TLR9−/− DCs produced 60% less IFN-γ compared with incubation with WT DCs (P < 0.01). To confirm a similar defect in IL-12 production in lung DCs, DCs were harvested from the lung on day 0 after sensitization, then stimulated with SC, and cell supernatant IL-12 levels were determined after 18 hours in culture. As observed in bone marrow–derived DCs, the production of IL-12 from TLR9−/− lung DCs was considerably reduced (P < 0.05) compared with their WT counterparts (Figure 7D). Collectively, these findings suggest that the reduction of IFN-γ responses to SC administration in TLR9-deficient mice is probably attributable to impaired antigen-induced production of IL-12 by DCs.
Figure 7.
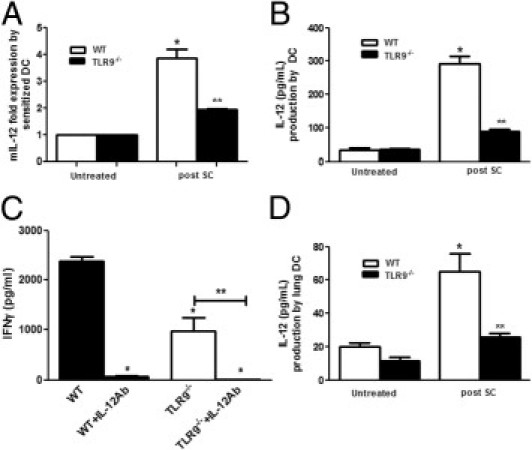
Production of IL-12 by sensitized bone marrow DCs and lung DCs harvested from WT and TLR9−/− and antigen presentation by WT and TLR9−/− DCs. WT and TLR9−/− mice were sensitized by SC, and bone marrow cells were harvested at day 0, cells were cultured with GM-CSF, and DCs were purified by CD11c+ magnetic beads at day 6 and stimulated with SC spores (1:10). IL-12 protein levels were measured by ELISA in supernatant fluids collected at 18 hours after stimulation (B) and IL-12 p40 mRNA expression was measured by real-time PCR (A). In a separate experiment, sensitized bone marrow DCs from WT and TLR9−/− mice were cocultured with naive WT splenic T cells for 18 hours, and their antigen-presenting function was measured by T-cell IFN-γ production in the presence or absence of IL-12 antibody by ELISA (C). Lung DCs were harvested from sensitized WT and TLR9−/− mice as described previously and stimulated with SC, and cell supernatant levels of IL-12 were measured by ELISA (D). N = 4 to 5 in each group; each experiment was repeated twice. Results represent mean ± SEM. *P < 0.05 compared with untreated controls; **P < 0.05 compared with WT mice after SC or *P <0.01 compared with IL-12 antibody (Ab) treatment.
Discussion
In this study, we show for the first time the critical role of TLR9 in generation of the Th1 response in a mouse model of SC-induced HP. Several pattern recognition receptors have previously been implicated in the immunopathogenesis of HP, including TLR2 and TLR6, as well as the downstream adaptor protein MyD88 in a SR mouse model of farmer's lung disease.36, 37 Although TLR9 has been shown to be important for granuloma formation and IFN-γ production during Gram-positive infection (Propionibacterium acnes)38 or in response to mycobacterial antigen,39 the role of TLR9 in HP was previously unknown.
We found reduced accumulation of neutrophils and activated CD4+ and CD8+ T cells in TLR9−/− mice during SC sensitization/challenge. Neutrophils have previously been shown to contribute to IFN-γ production during the acute phase of Saccharopolyspora rectivirgula–induced HP, and the neutrophilic response in this model was MyD88 dependent.37 We observed a significant decrease in production of neutrophil chemokines MIP-2 and keratinocyte-derived chemokine in TLR9−/− mice after SC challenge. Similarly, TLR9-deficient mice also had a substantial defect in the production of IFN-inducible protein 10, a potent chemoattractant of effector CD4+ and CD8+ T cells.40 Effector T cells, especially CD8 T cells, are main cellular sources of IFN-γ that drive the cell-mediated, delayed-type hypersensitivity immune response in HP.41 Our results indicate that granulomatous inflammation in our murine model of SC-induced HP is IFN-γ dependent, and the defect in PMN-, effector T-, and natural killer–cell accumulation in TLR9−/− mice could all contribute meaningfully to reduce IFN-γ production found in these animals.
In addition to defects in IFN-γ production, decreases in IL-12 expression in TLR9−/− mice during SC-induced HP were also observed. Although IL-12 is made by multiple lung immune cells, we found impaired production of IL-12 by sensitized DCs isolated from TLR9−/− mice. DCs drive Th1 immune responses via the production of IL-12.42 The role of DCs in the pathophysiology of HP has not been clearly defined. Girard and colleagues43 have demonstrated a significant increase in the number of mature CD11c+ cells in lungs of SR-challenged mice simultaneously infected with Sendai virus, suggesting the role of DCs in the generation of HP. Moreover, exposure to mycobacterial antigen has been shown to induce accumulation of lung DCs, which was reduced in TLR9-deficient mice.44 We found decreased DC recruitment and blunted DC activation in TLR9−/− mice in vivo. Moreover, DC-derived IL-12 was required for SC-induced IFN-γ production be splenic T cells, and both bone marrow–derived DCs and lung DCs from TLR9−/− mice showed defects in antigen-induced IL-12 production. These observations suggest that protection against granulomatous inflammation in TLR9−/− mice is due, at least in part, to alterations in DC function.
We have previously shown that lung macrophages from TLR9−/− mice default to a alternatively activated (M2), rather than classically activated (M1) phenotype during active bacterial or fungal pulmonary infection.29, 30, 45 The M2 phenotype is characterized by reduced production of inflammatory genes, such as TNF-α and iNOS, and increased expression of arginase, and is induced by cytokines expressed during type 2 responses (eg, IL-4, IL-13).46 Similar to observations made in infection models, we observed evidence of reduced classical activation of lung macrophages (as manifest by decreased TNF-α expression and nitric oxide production) in sensitized TLR9−/− mice 48 hours after i.t. SC challenge (see Supplemental Figure S1 at http://ajp.amjpathol.org). This defect is probably linked to reduced expression of IFN-γ, and this macrophage phenotype may contribute to impaired production of TNF-α and possibly neutrophil-specific chemokines.
In summary, our studies have identified SC as a potential cause of HP, an effect that depends on Th1 cytokines. Moreover, TLR9 is a critical component of the granulomatous response to SC, and TLR9-mediated DC responses are necessary for antigen-specific T-cell responses in this model. TLR9 signaling may serve as a therapeutic target in the treatment of patients with this devastating disease.
Footnotes
Supported by National Institutes of Health grants K08HL094762, R01 HL097564, and R01 HL25243.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.08.019.
Supplementary data
NO production and TNF-α expression by alveolar macrophages. WT and TLR9−/− mice were sensitized and challenged with SC as described, and bronchoalveolar lavage was performed at day 2. Alveolar macrophages were purified by adherence purification. Spontaneous ex vivo production of NaNO2 was measured by Greiss reaction (A), and TNF-α expression was measured by real-time PCR (B). N = 5 in each group; results represent mean ± SEM. *P < 0.01 compared with WT mice.
References
- 1.Girard M., Israel-Assayag E., Cormier Y. Pathogenesis of hypersensitivity pneumonitis. Curr Opin Allergy Clin Immunol. 2004;4:93–98. doi: 10.1097/00130832-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Suga M., Yamasaki H., Nakagawa K., Kohrogi H., Ando M. Mechanisms accounting for granulomatous responses in hypersensitivity pneumonitis. Sarcoidosis Vasc Diffuse Lung Dis. 1997;14:131–138. [PubMed] [Google Scholar]
- 3.Denis M. Proinflammatory cytokines in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1995;151:164–169. doi: 10.1164/ajrccm.151.1.7812548. [DOI] [PubMed] [Google Scholar]
- 4.Denis M., Cormier Y., Laviolette M., Ghadirian E. T cells in hypersensitivity pneumonitis: effects of in vivo depletion of T cells in a mouse model. Am J Respir Cell Mol Biol. 1992;6:183–189. doi: 10.1165/ajrcmb/6.2.183. [DOI] [PubMed] [Google Scholar]
- 5.Israel-Assayag E., Fournier M., Cormier Y. Blockade of T cell costimulation by CTLA4-Ig inhibits lung inflammation in murine hypersensitivity pneumonitis. J Immunol. 1999;163:6794–6799. [PubMed] [Google Scholar]
- 6.Schuyler M., Gott K., Cherne A., Edwards B. Th1 CD4+ cells adoptively transfer experimental hypersensitivity pneumonitis. Cell Immunol. 1997;177:169–175. doi: 10.1006/cimm.1997.1107. [DOI] [PubMed] [Google Scholar]
- 7.Richerson H.B., Coon J.D., Lubaroff D. Selective early increases of bronchoalveolar CD8+ lymphocytes in a LEW rat model of hypersensitivity pneumonitis. J Allergy Clin Immunol. 1995;96:113–121. doi: 10.1016/s0091-6749(95)70040-4. [DOI] [PubMed] [Google Scholar]
- 8.Walker C., Bauer W., Braun R.K., Menz G., Braun P., Schwarz F., Hansel T.T., Villiger B. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med. 1994;150:1038–1048. doi: 10.1164/ajrccm.150.4.7921434. [DOI] [PubMed] [Google Scholar]
- 9.Kim H.J., Kim H.Y., Kim B.K., Kim S., Chung D.H. Engagement of glucocorticoid-induced TNF receptor costimulates NKT cell activation in vitro and in vivo. J Immunol. 2006;176:3507–3515. doi: 10.4049/jimmunol.176.6.3507. [DOI] [PubMed] [Google Scholar]
- 10.Barrera L., Mendoza F., Zuniga J., Estrada A., Zamora A.C., Melendro E.I., Ramirez R., Pardo A., Selman M. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177:44–55. doi: 10.1164/rccm.200701-093OC. [DOI] [PubMed] [Google Scholar]
- 11.Semenzato G., Agostini C., Zambello R., Trentin L., Chilosi M., Pizzolo G., Marcer G., Cipriani A. Lung T cells in hypersensitivity pneumonitis: phenotypic and functional analyses. J Immunol. 1986;137:1164–1172. [PubMed] [Google Scholar]
- 12.Schuyler M., Gott K., Cherne A. Mediators of hypersensitivity pneumonitis. J Lab Clin Med. 2000;136:29–38. doi: 10.1067/mlc.2000.107694. [DOI] [PubMed] [Google Scholar]
- 13.Yi E.S. Hypersensitivity pneumonitis. Crit Rev Clin Lab Sci. 2002;39:581–629. doi: 10.1080/10408360290795583. [DOI] [PubMed] [Google Scholar]
- 14.Vogelmeier C. [Interferon-gamma is necessary for the expression of hypersensitivity alveolitis] German. Pneumologie. 1997;51:1065. [PubMed] [Google Scholar]
- 15.Jimenez-Alvarez L, Zuniga J, Gaxiola M, Checa M, Becerril C, Mendoza F, Pardo A, Selman M: Inflammatory response and dynamics of lung T cell subsets in Th1, Th2 biased and Th2 deficient mice during the development of hypersensitivity pneumonitis, Exp Mol Pathol, 88:407–415 [DOI] [PubMed]
- 16.Leino M.S., Alenius H.T., Fyhrquist-Vanni N., Wolff H.J., Reijula K.E., Hintikka E.L., Salkinoja-Salonen M.S., Haahtela T., Makela M.J. Intranasal exposure to Stachybotrys chartarum enhances airway inflammation in allergic mice. Am J Respir Crit Care Med. 2006;173:512–518. doi: 10.1164/rccm.200503-466OC. [DOI] [PubMed] [Google Scholar]
- 17.Yike I., Rand T.G., Dearborn D.G. Acute inflammatory responses to Stachybotrys chartarum in the lungs of infant rats: time course and possible mechanisms. Toxicol Sci. 2005;84:408–417. doi: 10.1093/toxsci/kfi080. [DOI] [PubMed] [Google Scholar]
- 18.Flemming J., Hudson B., Rand T.G. Comparison of inflammatory and cytotoxic lung responses in mice after intratracheal exposure to spores of two different Stachybotrys chartarum strains. Toxicol Sci. 2004;78:267–275. doi: 10.1093/toxsci/kfh064. [DOI] [PubMed] [Google Scholar]
- 19.Pestka J.J., Yike I., Dearborn D.G., Ward M.D., Harkema J.R. Stachybotrys chartarum, trichothecene mycotoxins, and damp building-related illness: new insights into a public health enigma. Toxicol Sci. 2008;104:4–26. doi: 10.1093/toxsci/kfm284. [DOI] [PubMed] [Google Scholar]
- 20.Akira S., Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 21.Gao J.J., Diesl V., Wittmann T., Morrison D.C., Ryan J.L., Vogel S.N., Follettie M.T. Regulation of gene expression in mouse macrophages stimulated with bacterial CpG-DNA and lipopolysaccharide. J Leukoc Biol. 2002;72:1234–1245. [PubMed] [Google Scholar]
- 22.An H., Yu Y., Zhang M., Xu H., Qi R., Yan X., Liu S., Wang W., Guo Z., Guo J., Qin Z., Cao X. Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2: TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002;106:38–45. doi: 10.1046/j.1365-2567.2002.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aderem A., Ulevitch R.J. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 24.Karunasena E., Cooley J.D., Straus D., Straus D.C. Protein translation inhibition by Stachybotrys chartarum conidia with and without the mycotoxin containing polysaccharide matrix. Mycopathologia. 2004;158:87–97. doi: 10.1023/b:myco.0000038438.48072.02. [DOI] [PubMed] [Google Scholar]
- 25.Seo S.C., Reponen T., Levin L., Borchelt T., Grinshpun S.A. Aerosolization of particulate (1–>3)-beta-D-glucan from moldy materials. Appl Environ Microbiol. 2008;74:585–593. doi: 10.1128/AEM.01791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozinsky A., Underhill D.M., Fontenot J.D., Hajjar A.M., Smith K.D., Wilson C.B., Schroeder L., Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Netea M.G., Gow N.A., Munro C.A., Bates S., Collins C., Ferwerda G., Hobson R.P., Bertram G., Hughes H.B., Jansen T., Jacobs L., Buurman E.T., Gijzen K., Williams D.L., Torensma R., McKinnon A., MacCallum D.M., Odds F.C., Van der Meer J.W., Brown A.J., Kullberg B.J. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116:1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasperkovitz P.V., Cardenas M.L., Vyas J.M. TLR9 is actively recruited to Aspergillus fumigatus phagosomes and requires the N-terminal proteolytic cleavage domain for proper intracellular trafficking. J Immunol. 2010;185:7614–7622. doi: 10.4049/jimmunol.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhan U., Lukacs N.W., Osterholzer J.J., Newstead M.W., Zeng X., Moore T.A., McMillan T.R., Krieg A.M., Akira S., Standiford T.J. TLR9 is required for protective innate immunity in Gram-negative bacterial pneumonia: role of dendritic cells. J Immunol. 2007;179:3937–3946. doi: 10.4049/jimmunol.179.6.3937. [DOI] [PubMed] [Google Scholar]
- 30.Bhan U., Trujillo G., Lyn-Kew K., Newstead M.W., Zeng X., Hogaboam C.M., Krieg A.M., Standiford T.J. Toll-like receptor 9 regulates the lung macrophage phenotype and host immunity in murine pneumonia caused by Legionella pneumophila. Infect Immun. 2008;76:2895–2904. doi: 10.1128/IAI.01489-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalis C., Gumenscheimer M., Freudenberg N., Tchaptchet S., Fejer G., Heit A., Akira S., Galanos C., Freudenberg M.A. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes. J Immunol. 2005;174:4295–4300. doi: 10.4049/jimmunol.174.7.4295. [DOI] [PubMed] [Google Scholar]
- 32.Velayudham A., Hritz I., Dolganiuc A., Mandrekar P., Kurt-Jones E., Szabo G. Critical role of toll-like receptors and the common TLR adaptor: MyD88, in induction of granulomas and liver injury. J Hepatol. 2006;45:813–824. doi: 10.1016/j.jhep.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Bafica A., Scanga C.A., Feng C.G., Leifer C., Cheever A., Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards L., Williams A.E., Krieg A.M., Rae A.J., Snelgrove R.J., Hussell T. Stimulation via Toll-like receptor 9 reduces Cryptococcus neoformans-induced pulmonary inflammation in an IL-12-dependent manner. Eur J Immunol. 2005;35:273–281. doi: 10.1002/eji.200425640. [DOI] [PubMed] [Google Scholar]
- 35.Lieberman L.A., Hunter C.A. Regulatory pathways involved in the infection-induced production of IFN-gamma by NK cells. Microbes Infect. 2002;4:1531–1538. doi: 10.1016/s1286-4579(02)00036-9. [DOI] [PubMed] [Google Scholar]
- 36.Fong DJ, Hogaboam CM, Matsuno Y, Akira S, Uematsu S, Joshi AD: Toll-like receptor 6 drives interleukin-17A expression during experimental hypersensitivity pneumonitis. Immunology, 130:125–136 [DOI] [PMC free article] [PubMed]
- 37.Nance S.C., Yi A.K., Re F.C., Fitzpatrick E.A. MyD88 is necessary for neutrophil recruitment in hypersensitivity pneumonitis. J Leukoc Biol. 2008;83:1207–1217. doi: 10.1189/jlb.0607391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tchaptchet S., Kirberg J., Freudenberg N., Schamel W.W., Galanos C., Freudenberg M.A. Innate, antigen-independent role for T cells in the activation of the immune system by Propionibacterium acnes. Eur J Immunol. 2010;40:2506–2516. doi: 10.1002/eji.200939860. [DOI] [PubMed] [Google Scholar]
- 39.Ito T., Schaller M., Hogaboam C.M., Standiford T.J., Chensue S.W., Kunkel S.L. TLR9 activation is a key event for the maintenance of a mycobacterial antigen-elicited pulmonary granulomatous response. Eur J Immunol. 2007;37:2847–2855. doi: 10.1002/eji.200737603. [DOI] [PubMed] [Google Scholar]
- 40.Zeng X., Moore T.A., Newstead M.W., Deng J.C., Lukacs N.W., Standiford T.J. IP-10 mediates selective mononuclear cell accumulation and activation in response to intrapulmonary transgenic expression and during adenovirus-induced pulmonary inflammation. J Interferon Cytokine Res. 2005;25:103–112. doi: 10.1089/jir.2005.25.103. [DOI] [PubMed] [Google Scholar]
- 41.Hunninghake G.W., Garrett K.C., Richerson H.B., Fantone J.C., Ward P.A., Rennard S.I., Bitterman P.B., Crystal R.G. Pathogenesis of the granulomatous lung diseases. Am Rev Respir Dis. 1984;130:476–496. doi: 10.1164/arrd.1984.130.3.476. [DOI] [PubMed] [Google Scholar]
- 42.Godfrey D.I., Kennedy J., Gately M.K., Hakimi J., Hubbard B.R., Zlotnik A. IL-12 influences intrathymic T cell development. J Immunol. 1994;152:2729–2735. [PubMed] [Google Scholar]
- 43.Girard M., Israel-Assayag E., Cormier Y. Mature CD11c(+) cells are enhanced in hypersensitivity pneumonitis. Eur Respir J. 2009;34:749–756. doi: 10.1183/09031936.00140908. [DOI] [PubMed] [Google Scholar]
- 44.Ito T., Schaller M., Hogaboam C.M., Standiford T.J., Sandor M., Lukacs N.W., Chensue S.W., Kunkel S.L. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J Clin Invest. 2009;119:33–46. doi: 10.1172/JCI35647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y., Wang F., Bhan U., Huffnagle G.B., Toews G.B., Standiford T.J., Olszewski M.A. TLR9 signaling is required for generation of the adaptive immune protection in Cryptococcus neoformans-infected lungs. Am J Pathol. 2010;177:754–765. doi: 10.2353/ajpath.2010.091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Modolell M., Corraliza I.M., Link F., Soler G., Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NO production and TNF-α expression by alveolar macrophages. WT and TLR9−/− mice were sensitized and challenged with SC as described, and bronchoalveolar lavage was performed at day 2. Alveolar macrophages were purified by adherence purification. Spontaneous ex vivo production of NaNO2 was measured by Greiss reaction (A), and TNF-α expression was measured by real-time PCR (B). N = 5 in each group; results represent mean ± SEM. *P < 0.01 compared with WT mice.