

Abstract
The p53 family member p73 is essential for brain development, but its precise role and scope remain unclear. Global p73 deficiency determines an overt and highly penetrant brain phenotype marked by cortical hypoplasia with ensuing hydrocephalus and hippocampal dysgenesis. The ΔNp73 isoform is known to function as a prosurvival factor of mature postmitotic neurons. In this study, we define a novel essential role of p73 in the regulation of the neural stem cell compartment. In both embryonic and adult neurogenesis, p73 has a critical role in maintaining an adequate neurogenic pool by promoting self-renewal and proliferation and inhibiting premature senescence of neural stem and early progenitor cells. Thus, products of the p73 gene locus are essential maintenance factors in the central nervous system, whose broad action stretches across the entire differentiation arch from stem cells to mature postmitotic neurons.
Keywords: p73, p73 isoforms, neural stem cells, embryonic neurogenesis, adult neurogenesis
p73 is a structural homolog of p53 and p63. The p73 locus encodes two main classes of transcription factors called TAp73 and ΔNp73, that either contain or lack the N-terminal transactivation domain.1 p73 has a significant but poorly understood role in brain development. The overwhelming phenotype of mice deficient in all p73 isoforms (global p73−/− mice) centers on the central nervous system (CNS) and is characterized by cortical loss causing ex vacuo hydrocephalus and dysgenesis and hypoplasia of the hippocampus and caudal cortex.2,3
The current view holds that the prominent cortical loss and hydrocephalus of global p73−/− mice are solely a consequence of unopposed apoptosis of mature postmitotic neurons occurring in the weeks and months after birth due to the absence of ΔNp73. Thus, in vivo loss of mature cortical neurons in p73−/− mice was reported to start after birth at day P1–3 and be significant at day P14–16.4 In this model, as part of the normal ‘pruning’ process in CNS formation, developmental neuronal death is mediated by p53 and the proapoptotic c-Jun N-terminal kinase cascade, whereas ΔNp73 has an important counterbalancing role, promoting the survival of mature neurons.4 Indeed, direct functional studies showed that ΔNp73 can function as a prosurvival factor of differentiated neurons.4,5 In neuronal cultures derived from sympathetic cervical ganglia, over-expression of ΔNp73 was found to inhibit apoptosis induced by nerve growth factor withdrawal or p53 overexpression.5,6 The prosurvival role of ΔNp73 for differentiated mature neurons was recently confirmed in vivo by a ΔNp73 isoform-specific knockout (KO) model.7 Homozygous ΔNp73 KO mice show increased cell death of mature neurons, albeit only in select regions including the preoptic area, vomeranasal neurons, GnRH-positive cells and Cajal–Retzius neurons. In addition, the choroid plexus appears atrophic. However, overall ΔNp73 KO mice exhibit a surprisingly subtle and discrete CNS phenotype. They are healthy, lack hydrocephalus, do not show massive cortical loss and display no overt neurological abnormalities.7 A recently reported second independent ΔNp73 KO mouse shows a similarly subtle phenotype with late signs of neuronal loss and neuro-degeneration after 10 months of age, but again no cortical thinning or overt hydrocephalus.8 However, the fact that the severe CNS phenotype of global p73−/− mice is not phenocopied by ΔNp73−/− mice (nor by TAp73−/− mice, see below) strongly suggests that the p73 locus must control additional important aspects of brain development such as neurogenesis that could explain cortical hypoplasia and hydrocephalus seen in the absence of all p73 isoforms.
Neurogenesis is the process by which newborn neurons are created from neural stem cells (NSC). In mouse development it starts around day E11 and is completed at birth, but continues in two select regions of the adult CNS, the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) and the ventricular/subventricular zone (VZ/SVZ) of the forebrain. Neural progenitors in the embryonic CNS consist of multipotent self-renewing stem cells as well as precursors with a more limited capacity for renewal and differentiation.9 Proper formation of cortex and hippocampus depends on maintaining sufficient numbers of neural progenitors over the entire course of neurogenesis.
Of note, another important phenotype reported for global p73−/− brains and absent in ΔNp73−/− brains is a severe impairment of adult neurogenic zones, the hippocampus and olfactory bulbs. p73−/− mice exhibit reduction in hippocampal size and severe hippocampal dysgenesis characterized by either complete absence or truncation of the lower blade of the DG (the hippocampal neurogenic zone) and abnormal wave-like gyrations of the cornu ammonis (CA) layers of the Ammon’s horn.2,3 This is largely due to the absence of TAp73 isoforms, as was revealed by TAp73 isoform-specific KO mice. TAp73−/− brains selectively phenocopy the malformation of the DG lower blade present in p73−/− mice, but show none of the other CNS malformations.10 Moreover, global p73−/− mice also exhibit abnormally small olfactory bulbs (that receive neuroblasts from the SVZ and the rostral migratory stream).4,11 Such findings are further indications that p73 could have important functions in neurogenesis. So far this has not been investigated.
The limitations of the isoform-specific p73 mice render the global p73−/− model highly relevant to better understand the full actions of p73 in CNS development and homeostasis. In sum, deletion of either ΔNp73 or TAp73 isoforms does not cause the massive cortical loss and ventricular collapse seen in p73−/− mice,7,8,10 suggesting a role for p73 in neurogenesis and a cooperation or redundancy between the isoforms during this process. In this study, we report a hitherto unknown function of p73 as an essential regulator of neural stem and progenitor cells required for embryonic and adult neurogenesis in the CNS.
Results
Cortical hypoplasia, hydrocephalus and hippocampal dysgenesis in p73−/− mice are already present at birth and correlate with prenatal proliferative defects in SVZ
It has been the general experience that 80% of global p73−/− mice die within 4–6 weeks because of the chronic infections, gastrointestinal hemorrhage and intracranial bleeding (e.g., Yang et al.3; Pozniak et al.4). However, as recently published,12 this poor survival rate of p73−/− mice is mainly due to starvation caused by their failure to compete with wild-type (WT) and p73 +/− littermates. Identifying p73−/− pups right after birth and moving them to foster mothers essentially normalizes their weight and rescues longevity into late adulthood (> 18 months) in 75% of p73−/− mice. Infections, although present, are mild and do not cause death. This discovery allowed us to generate a large colony of p73−/− mice (n>55) and characterize their brain defects in more details.
The current model holds that the cortical loss and ex vacuo hydrocephalus of adult p73−/− mice are a consequence of uncontrolled postnatal apoptosis of mature cortical neurons due to the lack of the ΔNp73 isoform, an essential anti-apoptotic survival factor of postmitotic neurons.4,5,7 Corticogenesis in mice terminates around day E18 and is complete at birth (P0). Of note, upon closer inspection a substantial fraction of p73−/− mice (38%; 6 out of 16 animals) already exhibited moderate cortical thinning and hydrocephalus at birth (Figure 1a). The caudal cortex of these mutant P0 mice was 23% thinner on average than in WT littermates (Figure 1a). In addition, the thickness of the main neurogenic area, the subventricular zone (SVZ), was substantially reduced in hydrocephalic P0 p73−/− mice compared with WT littermates (Figure 1a bottom). Thus, this early mutant phenotype cannot solely be explained by unopposed apoptosis of mature cortical neurons but instead suggests an additional impairment of the pre-natal corticogenesis phase. Moreover, 100% of p73−/− newborns exhibited hippocampal dysgenesis with severe truncation or absence of the lower blade of the DG, which harbors the neurogenic subgranular zone (SGZ, Figure 1a top). Together, these data suggested that p73, in addition to its protective role in postnatal mature neurons, plays a critical role in the neural stem and progenitor compartment.
Figure 1.
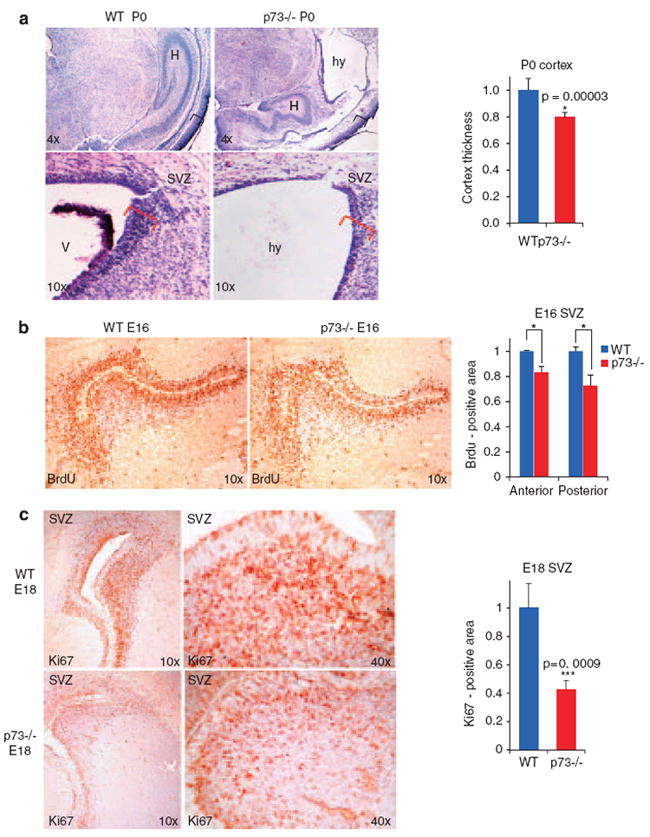
Cortical hypoplasia, hydrocephalus and hippocampal dysgenesis in p73−/− mice are already present at birth and correlate with prenatal proliferative defects in subventricular zone (SVZ) neurogenesis. (a) Top: a significant fraction of p73−/− mice (6 of 16 examined) show cortical hypoplasia and hydrocephalus already at birth (P0). This phenotype is progressive and soon affects 100% of animals (P30). Hippocampal dysgenesis is 100% penetrant already at birth. Brackets indicate cortical thickness, quantitated on the right from n=3 mice per genotype. WT thickness is set as 1. Bottom: at birth (P0), the SVZ of hydrocephalic p73−/− mice is prematurely exhausted, indicated by a thinning of this zone (red brackets). Representative hematoxylin-eosin sections. H, hippocampus; hy hydrocephalus; V, lateral ventricle. Error bars, S.E.M. (b) E16 p73−/− VZ/SVZ show reduced number of proliferating progenitor cells compared with WT VZ/SVZ as revealed by in vivo BrdU labeling. Results of quantitative morphometry are shown on the right. The proliferative loss ranged from 17 to 28% along the anterior–posterior axis (anterior, P=0.0392; posterior, P=0.0254). WT levels are set as 1. Error bars, S.E.M. Same stereotaxic plane. (c) E18 p73−/− brains show reduced precursor proliferation in the SVZ and ventricular zone of the telencephalon, detected by Ki67 immunostaining. The degree varies; an extreme case shows 60% decrease in SVZ/VZ proliferation, quantitated on the right. WT levels are set as 1. Error bars, S.E.M. Same stereotaxic plane, but the p73−/− section shows some degree of malformation of the basal ganglia
Consistent with previous reports, by 1 month of age (P30), 100% of rescued mutant mice showed marked loss of cortical mass and hydrocephalus indicative of a rapidly progressive process. In fact, 20% of P30 mice exhibited cortical reduction to 65% of normal thickness with extreme hydrocephalus, causing their early death (Supplementary Figure S1a). The hippocampal and cortical phenotype of surviving P30 mutant mice (Supplementary Figure S1b) was underlined by defective neurohistologic architecture as revealed by Tuj1/β-III tubulin staining, an axonal marker (Supplementary Figure S1c). Thus, in p73−/− brains, the hippocampal CA layers of Ammon’s horn exhibited aberrant axon orientation (Supplementary Figure S1c, left panels). As previously reported,2 the caudal p73−/− cortex showed abnormal lamination, and mutant axons appeared much shorter than WT axons (Supplementary Figure S1c, right panels). This phenotype, albeit less prominently, was also present in old p73−/− mice, indicating that their long-term survival is likely due to less severe brain disorganization (data not shown). An additional 5% of mutant mice died in the ensuing 8 months from their brain defects. Cortical reduction in all long-term survivors (75% of the colony) was milder and, although functionally compromising (see below), still compatible with life-sufficient activities under protected housing conditions.
Collectively, the above data are best explained by hypothesizing that abnormal neurogenesis also contributes to the profoundly disturbed brain development in p73−/− mice. To test this notion, we first examined the VZ/SVZ of E16 mice. Indeed, although at this stage the overall thickness of this zone in p73−/− brains appeared normal (data not shown), p73−/− VZ/SVZ showed a clear reduction in the number of proliferating progenitor cells, as revealed by in vivo BrdU labeling (Figure 1b, Supplementary Figure S1d). Quantitative morphometry showed a significant reduction in the number of BrdU-positive proliferating cells in p73−/− VZ/SVZ areas compared with corresponding WT areas. The proliferative loss ranged from 17% to 28% along the anterior–posterior axis (Figure 1b). In further support, Ki67 immunostaining revealed a continuous decrease in proliferating cells in p73−/− VZ/SVZ by day E18. An extreme case with 60% reduction in Ki67-positive cells is shown in Figure 1c (magnified in Supplementary Figure S1e).
p73 is an essential regulator of NSC survival and self-renewal
As the above results suggest that p73−/− mice possess fewer neural stem/precursor cells, we next examined the functional properties of WT and p73−/− NSC in vitro. Stem cells derived from mouse CNS can be maintained in an undifferentiated proliferative state in the classic neurosphere assay (NSA).13 The three-dimensional neurospheres consist primarily of Nestin-positive NSC and progenitor cells.14 Although the NSA might not completely reflect the in vivo developmental progression, it is a very useful informative tool that enables long-term propagation and quantitation of NSC and allows the study of their self-renewal. To this end, we assessed self-renewal by the total number of neurospheres that serially regenerated from105 dissociated single cells at the end of each passage plated at clonal density of 20 cells/μl. We focused on the early (E14) and late (P0) stages of neurogenesis and analyzed neurospheres generated from either whole brain or isolated SVZ and SGZ zones, as described in Materials and Methods. Of note, WT NSC cultures expressed high levels of TAp73 throughout development, whereas ΔNp73 levels were 27-fold lower, as determined by quantitative RT (qRT)-PCR (Figure 2a, Supplementary Figure S2a). Thus, TAp73 isoforms seem to be the dominant factors in the TA: ΔN ratio of NSC. This ratio is maintained at subsequent passages in culture (Supplementary Figure S2b). At early neurogenesis (E14), the number of neurospheres generated from WT and p73−/− whole brains were identical at passage 1 (Figure 2b). However, continuous passaging revealed a profound defect in self-renewal/survival of mutant NSC. The frequency of E14 p73−/− NSC capable of regenerating new neurospheres from single cells decreased progressively and was significantly reduced by passage 6. By passage 10, p73−/− cultures were completely exhausted of NSC and died out completely at passage 11, whereas the corresponding WT cultures were still fully viable at that time (Figure 2b).
Figure 2.
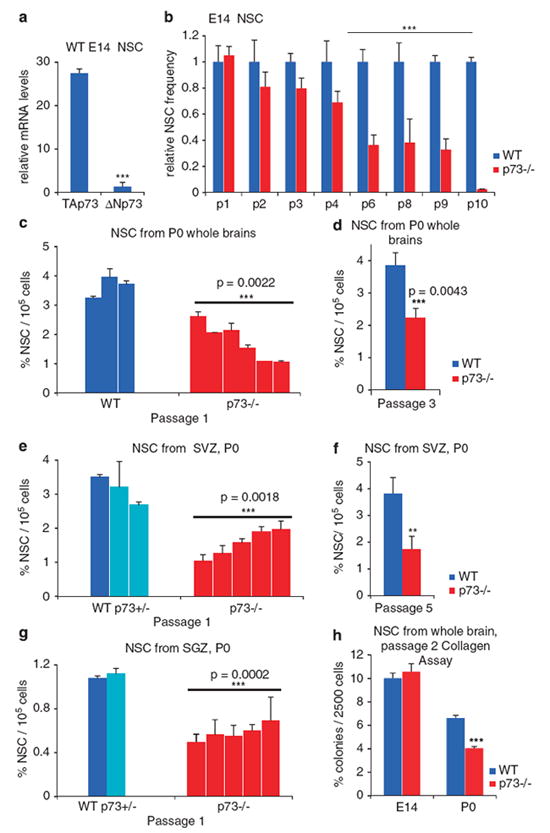
p73 is an essential regulator of neural stem cell survival and self-renewal. (a) Neurospheres derived from WT brains at E14 and P0 express both TAp73 and ΔNp73 isoforms. However, the levels of TAp73 are about 27-fold higher than those of ΔNp73, as determined by qRT-PCR. Values from whole brains at E14 are shown. See also Supplementary Figure 2. (b–g) p73−/− neural stem cells (NSC) at E14 and P0 exhibit self-renewal and maintenance defects. (b) Neurosphere assays derived from E14 forebrains. By passage 6, mutant cultures are severely impaired. p73−/− relative to WT neurosphere numbers from triplicate plates after each passage are indicated (n=3 each). (c–g) Neurosphere assays derived from P0 whole brains (c and d), the subventricular zone (SVZ; e and f) and hippocampus sub-granular zone (SGZ; g). Cultures derived from individual p73−/− pups from a representative litter of five independent litters with comparable results are shown (c, e and g). At P0, p73−/− cultures have severely impaired self-renewal, already evident at passage 1 (c, e and g) which persists with passaging (d, passage 3; and f, passage 5). Overall, the self-renewal capacity of p73−/− neural stem cells is reduced by 47% in whole brain (d), 56% in SVZ (f) and 48% in SGZ, compared with corresponding WT cultures. (h) Neural colony-forming cell assay in collagen. Passage 2 dissociated neurospheres derived from E14 and P0 whole brain WT and p73−/− mice were plated at clonal density (2500 cells/1.5 ml) in collagen. Number and size of newly formed colonies were evaluated after 21 days. A total of four mice/age/genotype were evaluated in triplicate in two independent experiments. Similar to the neuropshere assays, WT and p73−/− E14 cells produced a similar number of colonies, whereas p73−/− P0 showed ~ 50% decrease in the total number of colonies (P<0.0005)
In contrast to E14 cultures, in which self-renewal defects appeared gradually with passaging, newborn (P0) mutant neurospheres from whole brains exhibited self-renewal defects from the outset, at passage 1. p73 +/− cultures did not differ significantly from WT cultures. Figure 2c shows P0 NSC of individual mice from a representative litter to illustrate the general tendency in the p73−/− genotype as well as a certain phenotypic variability among animals. Identical results were obtained from multiple litters. p73−/− P0 cultures contained on average only 1–2% self-renewing stem cells, compared with 4% in WT P0 cultures, the latter being consistent with the literature.13 Therefore, individual p73−/− P0 cultures retained only 25–50% of the stemness potential of corresponding WT P0 cultures. This seriously diminished potential was then stably maintained in subsequent passages (Figure 2d). To determine which of the neurogenic zone(s) at P0 is mostly affected, we generated specific neurosphere cultures from SVZ and hippocampal SGZ from newborn brains. Of note, both SVZ and SGZ P0 cultures from p73−/− brains showed significant loss of self-renewal capacity at passage 1 (Figures 2e and g). Similar to whole brains, this diminished potential of about half the WT rate was then stably maintained over the next 5 passages, the endpoint of our experiment (Figure 2f).
As an alternative means of calculating the frequency of NSC in the neurosphere cultures, we also performed a clonality assay in collagen based on the neural colony-forming cell (NCFC) assay.15 The NCFC assay is more stringent than the NSA in its evaluation of the long-term proliferative and self-renewal capabilities of cells15 by preventing inherent fusion of neurospheres. In this assay, 2500 cells, derived from passage 2 dissociated neurospheres, were plated in 1.5 ml collagen mixed with medium and EGF and colonies were scored with respect to number and size 21 days later. This type of experiment was performed twice in triplicate for both E14 and P0 whole brain NSC. The results obtained in this clonal assay mirrored the results obtained in the neurosphere assays shown in Figures 2b–g. Thus, although WT and p73−/− NSC generated colonies with similar frequencies at E14, the sphere-forming cells from p73−/− NSC were only half than those of WT at P0 (Figure 2h). These results confirmed and validated the NSAs described above and proved that the cell density used in the NSAs was sufficiently low to generate clonal neurospheres.
In sum, we conclude that p73 has a major role in self-renewal and maintenance of NSC during most of embryonic neurogenesis, ranging from E14 to birth. The magnitude of this p73 effect is substantial, as its loss reduces the neural stem and progenitor compartment by 50%.
p73 is an essential regulator of NSC proliferation
The pronounced defect in p73−/− NSC self-renewal and maintenance was accompanied by a strong decrease in neurosphere size, indicating an additional impairment in the proliferation of progenitors (Figure 3a). Neurospheres derived from E14 brains exhibited large differences in size starting from passage 6 on. The average size of p73−/− neurospheres was 35% smaller than the corresponding WT neurospheres (table, Figure 3b). Moreover, when their distribution was assessed, WT neurospheres accumulated primarily in the upper size range (> 100 μm), whereas p73−/− neurospheres accumulate more in the lower range so that <20% reached >200 μm in diameter compared with 40% in WT (Figure 3b). At this stage, the frequency of p73−/− NSC was also markedly reduced compared with WT (see Figure 2b). Further passaging intensified the defects observed in p73−/− cultures both in number and size, and the percentage of big neurospheres continued to decline (data not shown). In confirmation, when assessed under the stringent conditions of the collagen clonality assay, E14 p73−/− NSC generated fewer colonies >1 mm compared with WT NSC already at passage 2 (Supplementary Figure S3a).
Figure 3.
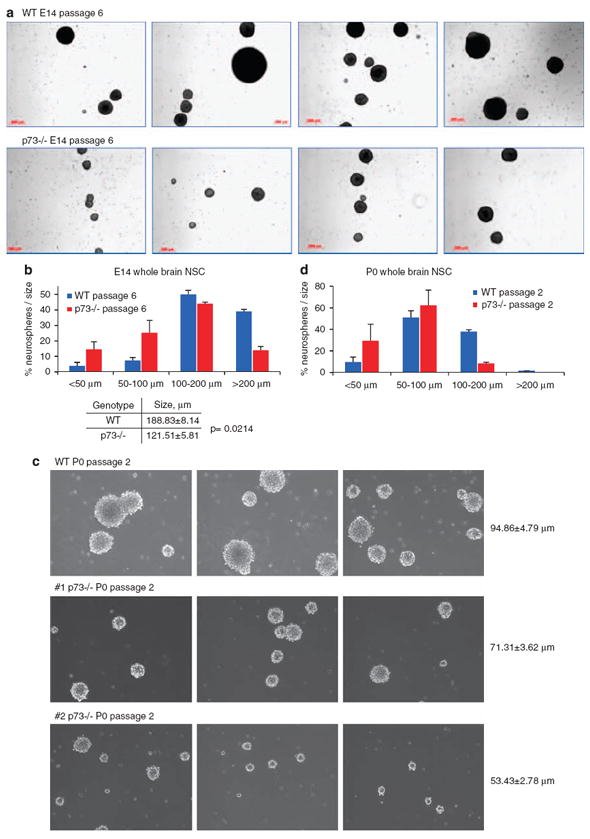
p73 is an essential regulator of neural stem cell proliferation. (a–d) Neurospheres from E14 and P0 p73−/− mice grow slower and are much smaller compared with WT neurospheres. (a) E14 neurospheres at passage 6. Representative images from WT and p73−/− littermates cultures are shown. (b) Size distribution of E14 neurospheres at passage 6 shown in (a). WT neurospheres tend to accumulate in the upper range, whereas a substantial number of p73−/− neurospheres accumulate in the lower range. At this stage, the frequency of their neural stem cells is also markedly reduced compared with WT (See Figure 2b). A total of >500 neurospheres were counted per genotype. (c) P0 neurospheres at passage 2. Most of the mutant cultures grow slower and form smaller spheres already at passage 2, compared with corresponding WT cultures. Some mutant variation exists. One WT and two mutant mice are shown as representative examples. (d) Size distribution of the P0 neurosphere shown in (c). WT neurospheres tend to accumulate in the upper range, whereas a substantial number of mutant neurospheres accumulate in the lower range. A total of >500 neurospheres were counted per genotype
At P0, the effect of p73 loss on the size of NSC cultures was even more dramatic and already visible by passage 2–3. Representative images from a P0 litter of one WT and two p73−/− whole brains are shown in Figure 3c. Thus, size reduction ranged from 25 to 50% in p73−/− cultures compared with WT cultures. Size distribution at P0 was even more lopsided than at E14 (Figure 3d). Similar proliferative defects were observed in P0 cultures from p73−/− SVZ and SGZ (Supplementary Figure S3b and data not shown). Of note, the difference in neurosphere size was not because of the smaller cell size, as dissociated single cells of both genotypes had the same diameter, as determined by fluorescence-activated cell sorting (FACS) analysis (Supplementary Figure S3c). This confirms that the smaller neurosphere size in p73−/− cultures is the result of fewer cells per neurosphere. Together, these indicate that p73-deficient NSC are not only deficient in self-renewal but also in proliferation and generation of progeny.
p73−/− NSC exhibit impaired S-phase and increased senescence, associated with deregulation of the Sox and Notch signaling pathways of stem maintenance
To further explore the proliferation defects revealed by neurosphere size reduction (Figure 3), we measured BrdU incorporation into neurospheres under self-renewal conditions. To this end, P0-derived neurospheres at passage 2 were plated as 105 dissociated cells, allowed to grow for 2 days to reform spheres and then pulsed for 20 min with BrdU. The number of BrdU-positive cells was assessed by immunofluorescence. p73−/− NSC cultures showed a 36% decrease in the percentage of cells in S-phase compared with WT controls (Figure 4a). Similar results were obtained in E14-derived cultures (data not shown). On the other hand, apoptosis did not contribute to the growth retardation of p73−/− cultures at E14 and P0 (Figure 4b and data not shown). In further support of impaired S-phase, p73−/− NSC cultures exhibited reduced levels of cyclin D1 (indicative of G1/S transition) and cyclin E (indicative of S-phase progression) at E14 and P0, compared with corresponding WT cultures, whereas cyclin B1 levels remained unchanged (Figures 4g and i and data not shown).
Figure 4.
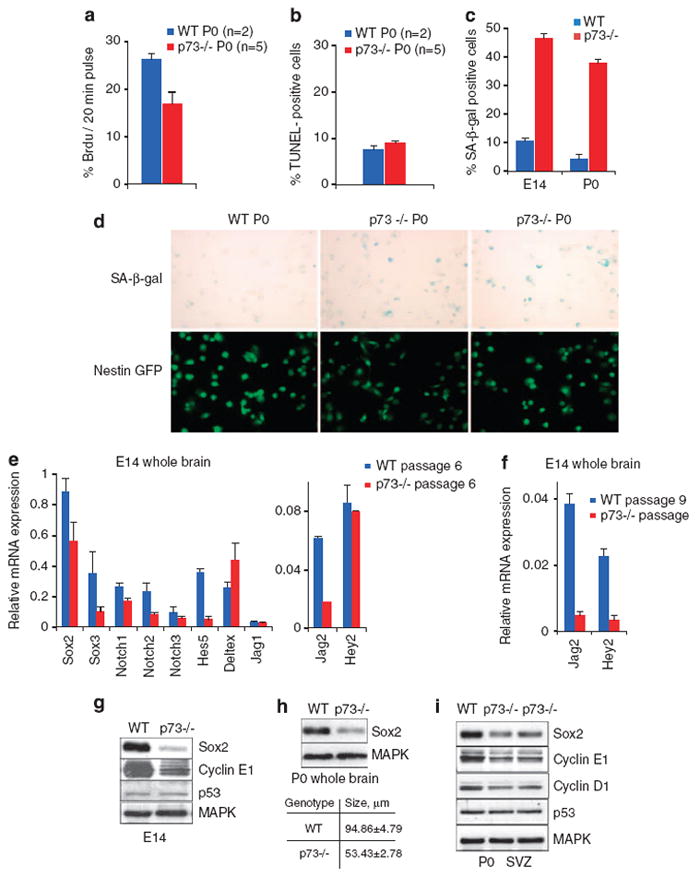
p73−/− neural stem cells exhibit impaired S-phase and increased senescence, associated with deregulation of the Sox and Notch signaling pathways of stem maintenance. (a) BrdU incorporation in p73−/− neurospheres is decreased. NSC at passage 2 from P0 whole brains of two WT and five mutant mice were plated in duplicates at equal cell numbers and allowed to form spheres for 48 h as in Figure 3, followed by a 20 min pulse with BrdU. Spheres were analyzed by immunofluorescence for BrdU incorporation and >1000 were counted per genotype. Results are representative of two independent experiments. (b) Apoptosis does not have a role in the growth impairment of p73−/− neurospheres. TUNEL-positive cells per neurosphere derived from P0 brains. (c and d) p73−/− stem/progenitor cells show increased senescence. (c) Quantification of senescent SA-β-gal-positive stem/progenitor cells in WT and p73−/− neurospheres obtained from E14 (passage 6) and P0 (passage 3) whole brains (n=4 per genotype). A minimum of 1000 cells per animal were counted. Similar results were seen in P0 SVZ and SGZ. (d) NSC from P0 whole brains of one WT Nestin–GFP and two representative p73−/− Nestin–GFP mice. SA-β-gal staining (top) of Nestin–GFP-positive cells (bottom). SA-β-gal staining was performed on freshly dissociated passage 3 neurospheres. Similar results were obtained with SA-β-gal staining of undissociated neurospheres (data not shown). (e and f) Relative mRNA levels of endogenous Sox2, Sox3, members of the Notch pathway (Notch1, Notch2, Notch3, Jag1, Jag2, Deltex, Hes5 and Hey2). qRT-PCR assays from E14 neurospheres at passage 6 (e) and passage 9 (f). All values were normalized to GAPDH. All error bars, S.E.M. (g–i) Sox2 protein is downregulated in p73−/− neurospheres. Immunoblot analysis of E14 NSC at passage 9 (g), P0 whole brain at passage 2 (h) and P0 SVZ at passage 5 (i), probed for NSC factor Sox2 and cell cycle regulators cyclin E1, cyclin D1 and p53. Equal amounts of total protein were loaded, MAPK is the loading control. (h) The degree of Sox2 downregulation in mutant mice (top) correlates with their average neurosphere size (bottom)
Previous studies have shown that NSC have developed strict regulatory mechanisms to prevent premature senescence and prolong their proliferative capacity.16 Therefore, we scored p73−/− neurosphere cultures for senescence by staining with senescence-associated β-galactosidase (SA-β-gal). To this end, we crossed Nestin–GFP transgenics with p73 +/− mice to generate WT Nestin–GFP and p73−/− Nestin–GFP transgenic mice. Nestin is an intermediate filament expressed in radial glial cells and is the most widely used marker for neural stem and early progenitor cells. Crossing into a Nestin–GFP background had no impact on the p73−/− brain phenotype, but allowed us to selectively trace the properties and quality of NSC in vitro and in vivo. Neurosphere cultures from E14 and P0 WT and p73−/− Nestin–GFP whole brains were generated. There was no difference in the intensity of Nestin–GFP between the genotypes (FACS analysis, Supplementary Figure S4a and b). Moreover, passaging the NSC as neurospheres enriched for the proportion of Nestin–GFP-positive cells from 65% at passage 0 to 85–90% at passage 2–3 in both genotypes (Supplementary Figure S4a and b). SA-β-gal staining was performed on freshly dissociated neurospheres that were allowed to attach on poly-d-lysine coated slides for 30 min. Of note, littermate cultures show that 48% of E14 and 38% of P0 Nestin–GFP-positive p73−/− cells stain strongly positive for SA-β-gal. In contrast, WT cultures were essentially negative (10 and 4%, respectively; Figure 4c and d). Identical results were obtained with undissociated P0 neurospheres and with dissociated P0 neurospheres derived from SVZ and SGZ (data not shown). Thus, in addition to promoting S-phase and proliferation, p73 is also critical in preventing premature senescence of neural stem and progenitor cells.
To obtain insight into the mechanisms elicited by p73 in maintaining the NSC compartment, we investigated the impact of p73 loss on several canonical neurodevelopmental pathways. The Sox B1 subfamily of HMG-box transcription factors (Sox1–3) is expressed by precursors in the embryonic nervous system, where these factors maintain neural progenitors in an undifferentiated state while suppressing neuronal differentiation.17 For example, Sox2 is a critical regulator of neural stem and precursor cells, as established by hypomorphic or brain-specific Sox2-deficient mice.18,19 Moreover, Sox2 controls NSC maintenance in postnatal hippocampus in vivo and neurosphere cultures in vitro via cell-autonomous Sonic Hedgehog-mediated18 and non-autonomous20 pathways. Similarly, the Notch signaling pathway has fundamental roles during maintenance, proliferation and differentiation of NSC in the developing brain.21
Indeed, as indicated in Figure 4e, qRT-PCR analysis in E14 neurospheres at passage 6 revealed lower levels of Sox2 mRNA expression in p73−/− neurospheres compared with littermate WT neurospheres. Moreover, Sox3 expression, another family member involved in NSC maintenance17 was downregulated. Furthermore, the Notch signaling pathway was profoundly altered at all levels of regulation. Overall, expression of Notch receptors was reduced in p73−/− NSC. Specifically, Notch2, involved in SVZ stem maintenance,22 was most severely affected. Moreover, levels of various Notch ligands (the activatory Jag2 and the inhibitory Deltex), as well as Notch effectors (Hes5 and Hey2) were dysregulated by loss of p73 (Figure 4e). In support, the activity of the Notch pathway further diminished with continued passaging (Figure 4f, passage 9).
Moreover, a similar profile of dysregulated Sox2, Sox3, Notch1, Notch3 and Hes5 was obtained in Nestin–GFP-positive FACS-sorted cells from p73−/− P0 neurospheres (Supplementary Figure S4c).
To confirm these results, we performed immunoblot analyses of the growth-impaired p73−/− NSC cultures from E14 whole brain (passage 9, Figure 4g), P0 whole brain (passage 3, Figure 4h) and P0 SVZ (passage 5, Figure 4i). Importantly, Sox2 protein levels were significantly down-regulated in all p73−/− neurosphere cultures from whole brain and SVZ obtained from individual mice at E14 and P0 (Figures 4g–i), whereas the percentage of Sox2-positive cells in NSC from both genotypes were identical (~75%, Supplementary Figure S4d). Thus, the Sox2 content per mutant stem/progenitor cell is lower. The lower Sox2 levels are also reflected by the smaller neurosphere size of the mutant cultures from which the protein lysates were obtained (table, Figure 4h). On the other hand, neurospheres of both genotypes contained the same proportion of Nestin-positive cells, as endogenous Nestin mRNA levels showed no difference compared with WT cultures (Supplementary Figure S4e) and Nestin–GFP distribution by FACS analysis was identical (Supplementary Figures S4a and b). Together, these data suggest that Nestin-positive p73−/− neural stem and progenitor cells exhibit lower activities of the Sox and Notch pathways and raise the possibility that they are downstream effectors of p73 function.
To gain insight into the mechanism of p73 loss-induced senescence in NSC, we investigated a possible role of p53. We had previously found that p73 loss results in impaired proliferation and premature senescence of differentiated mouse embryo fibroblasts because of the compensatory activation of p53.23 However, neither total p53 nor activated p53Ser15 protein was upregulated compared with the corresponding WT cultures (Figures 4g and i and data not shown). In agreement, levels of p53 mRNA and p21 protein also remained unchanged (Supplementary Figure S4f and data not shown). The inhibition of endogenous telomerase activity resulting in telomere shortening was shown to lead to replicative senescence.24,25 Moreover, telomeres shorten with age in NSC of SVZ and telomerase-deficient mice exhibit reduced neurogenesis with impaired neuronal differentiation and neuritogenesis.26 It is known that TERT levels correlate with telomere length.26 Thus, we determined the level of mTERT mRNA in Nestin–GFP-positive FACS-sorted cells. In support of the observed premature senescence, the results indicate about a 6-fold reduction of mTERT RNA levels in p73−/− NSC derived from P0 mice compared with WT controls (Supplementary Figure S4g).
In sum, these results indicate that inactivation of p73 causes a general disturbance in NSC maintenance factors which leads to premature exhaustion and depletion of neural precursors through S-phase impairment and senescence.
Impaired generation of mature neurons from p73−/− NSCs
Next, to examine the differentiation potential of p73−/− neural progenitors, we cultured passage 1 or 2 neurospheres generated from E14 and P0 whole brains and from P0 SVZ and SGZ for 7–9 days in conditions allowing tri-lineage differentiation (see Materials and Methods). Dissociated p73−/− NSC from E14 and P0 were able to differentiate into neurons (Tuj1/β-III tubulin positive), astrocytes (GFAP positive) and oligodendrocytes (CNPase positive), demonstrating that p73−/− NSC are in fact multipotent (Figure 5a). However, defects were present in the number and quality of neurons (Figures 5b–e) and oligodendrocytes (Supplementary Figure S5a and b). Astrocyte properties were unaltered.
Figure 5.
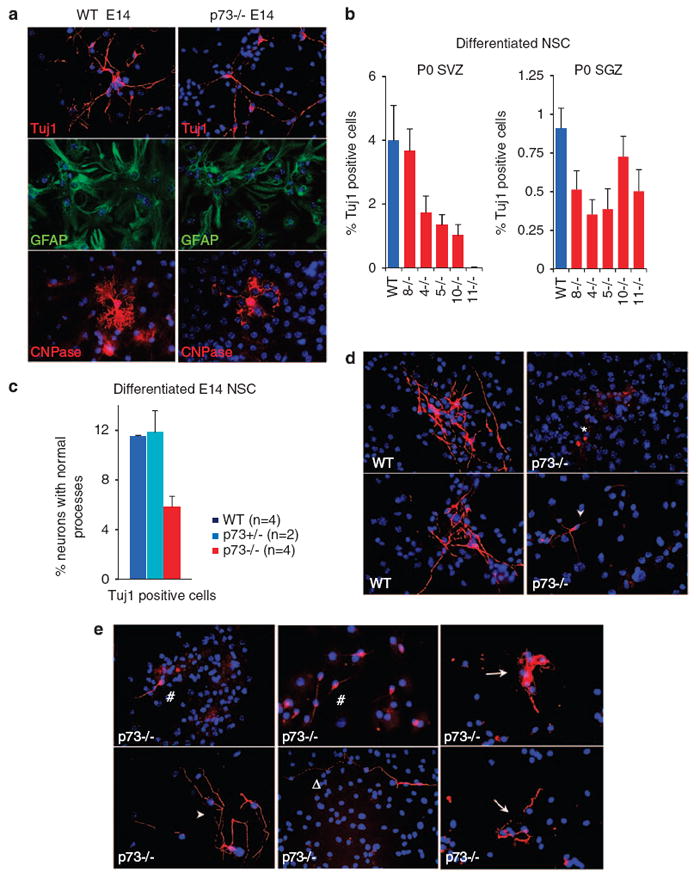
Impaired generation of mature neurons from p73−/− neural stem cells. (a) p73−/− NSC are multipotent. In vitro differentiation assay of dissociated cells from E14 passage 1 neurospheres. At day 7, Tuj1, GFAP and CNPase-positive cells marking neurons, astrocytes and oligodendrocytes, respectively, were assessed by immunofluorescence for cell number and morphology. Mutant precursors are multilineage competent but show defects in neurons and oligodendrocytes. Astrocytes seem normal. Hoechst counterstain, × 40 magnification. (b) p73−/− NSC generate fewer neurons. In vitro differentiation assays of dissociated cells from P0 passage 1 neurospheres derived from SVZ (left) and SGZ (right). At day 7, all Tuj1-positive cells irrespective of aberrant morphology were scored. All positive cells exceeding 1500 in total in randomly selected fields were counted per animal and plotted as percentage of total cells. The average of three WT mice (blue bar) and five individual p73−/− mice (red bars) from the same litter are shown. Data from two independent experiments, each carried out in duplicate. Error bars, S.E.M. (c) p73−/− NSC generate neurons with aberrant processes. At day 7, only Tuj1-positive cells with WT-like robust neuronal processes were scored. All cells in randomly selected fields, exceeding 2500 cells in total, were counted per genotype. Data from two independent experiments, each performed in duplicate. (d and e) Representative images show the poor quality of many of the generated p73−/− neurons. This includes multiple defects in process formation that are either completely absent (*), short (#), contorted (arrow), exhibit abnormal arborization (triangle) and/or poor interconnectivity with neighboring neurons (arrow head)
Given the massive neuronal loss in p73−/− cortex, we focused on the number and quality of neurons yielded by p73−/− NSC. Concerning neuron numbers, at E14 passage 1 both WT and p73−/− NSC generated the same total number of Tuj1-positive cells (irrespective of morphology; data not shown). However, at later passages the p73−/− NSC fell behind in the number of neurons produced, indicating an inability to maintain and/or produce neuronal precursors and an impaired potential to differentiate into neurons (Supplementary Figure S5c). Of note, at P0 the total number of Tuj1-positive cells was already reduced at passage 1 (Figures 5b). Similar results were obtained in E14 and P0 WT and p73−/− Nestin–GFP cultures.
Concerning neuron quality, a significant number of Tuj1-positive cells derived from p73−/− E14 NSC passage 1 had major defects in neurite outgrowth compared with WT neurons (Figure 5c). In WT and p73 +/− cultures most neurons show mature morphology with long Tuj1-positive processes exhibiting extensive arborization and physical connections with other neurons (Figures 5a and d). In contrast, in mutant cultures a large proportion of Tuj1-positive cells seemed to be underdeveloped and of poor quality and showed multiple defects in process formation (Figures 5d and e). Similar morphologic defects were seen in P0 p73−/− NSC derived from whole brain, SVZ and SGZ, as revealed by Tuj1 and Map2a,b neuronal markers (Supplementary Figure 5d and data not shown). In sum, this indicates that p73 is not only required for the maintenance and self-renewing capacity of NSC, but also for precursor proliferation and differentiation into neuronal and oligodendrocytic progeny.
p73 is also required in adult hippocampal neurogenesis
Our data so far indicate that p73 has an important role in embryonic neurogenesis. However, neurogenesis continues in two select regions of the adult CNS, the SGZ in the DG of the hippocampus and the SVZ of the forebrain. As already discussed, global p73−/− and TAp73−/− mice show a profound hippocampal dysgenesis, marked by unusual folding of the CA layers, an extended upper blade but absence/truncation of the lower blade of the DG.3,10 Thus, we hypothesized that p73 also has a role in adult neurogenesis. The SGZ differentiation cascade is comprised of discrete steps identifiable by a combination of markers and morphology.27 The primordial NSC of the DG coexpress Nestin together with GFAP and have a distinct radial glial morphology. Through asymmetric divisions these NSC generate early progenitors that continue to express Nestin but lose radial glial morphology and lose GFAP.28,29 These so-called amplifying neural progenitors undergo several rounds of symmetric divisions and subsequently exit the cell cycle to become postmitotic neuroblasts that are positive for Tuj1/βIII-tubulin. Further maturation yields immature neurons and finally mature granule neurons which functionally integrate into the existing granule layer.27
To specifically trace NSC in adult brain in vivo, we again crossed p73 +/− mice with Nestin–GFP transgenics.30 The resulting adult p73−/− Nestin–GFP mice were neuroanatomically identical to non-transgenic p73−/− mice, and faithfully mimicked their hippocampal dysgenesis. This in vivo traceable model allowed us to detect additional defects in the p73−/− neurogenic DG compartment. Representative images and quantitative results obtained from 6-week-old DG of WT and p73−/− Nestin–GFP mice are shown in Figure 6. The SGZ of the p73−/− upper blade seems to contain fewer Nestin-positive progenitors than the corresponding WT upper blade (Figure 6a, green cells). Likewise, the preserved portion of the truncated p73−/− lower blade also exhibits dramatically lower numbers of Nestin-positive progenitors compared with WT lower blade (Figure 6a and c, green cells) and the corresponding granule layer is missing.
Figure 6.
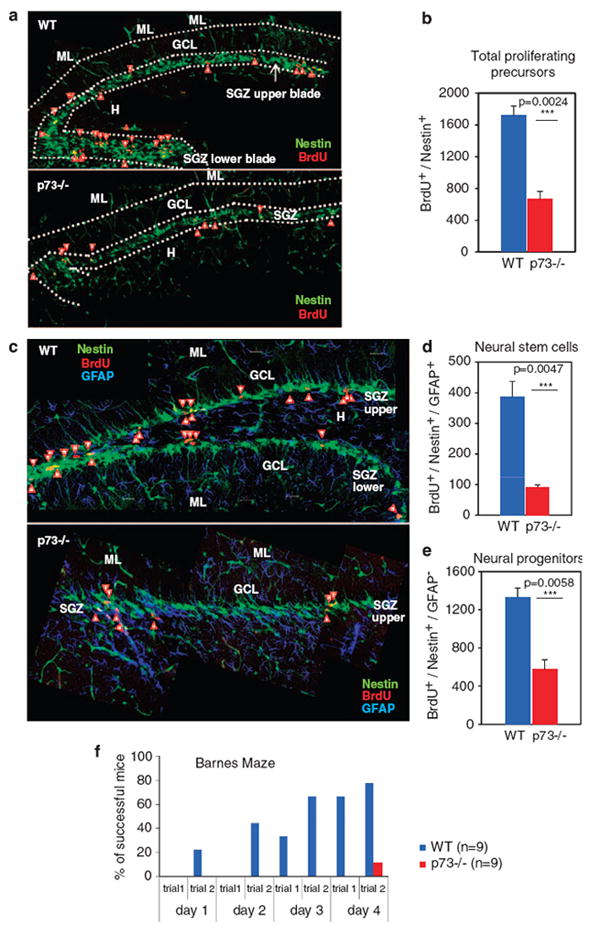
p73 is also required in adult neurogenesis. Loss of p73 expression severely impairs proliferation of hippocampal progenitors in vivo. p73−/− Nestin–GFP mice (6-week old) are compared with WT-Nestin–GFP mice. (a) Total proliferative cells in the neurogenic sub-granular zones (SGZs) of the upper and lower blades of the dentate gyrus (DG). BrdU+/Nestin+ double-positive cells comprising neural stem and progenitor cells are scored (arrowheads). p73−/− mice show a reduced number of double-positive cells. Normally each blade contains a molecular layer (ML), granule cell layer (GCL) and sub-granular layer (SGZ). H, hilus. (b). Quantitation of the total number of proliferating cells per DG (n=5 mice per genotype). (c–e) p73 loss affects proliferation in both the neural stem and the progenitor compartments of the DG. (c) Double immunofluorescence for BrdU (in red) and GFAP (in blue) in the SGZ (green) of p73−/− Nestin–GFP versus WT-Nestin–GFP mice. Arrowheads point to BrdU+/Nestin +/GFAP+ triple-positive cells. (d and e) Quantitation of the number of triple-positive stem cells (d) and double-positive but GFAP-negative (e) progenitor cells per DG (n=3 mice per genotype). (f) Barnes maze test of WT and p73−/− mice (n=9 per genotype) indicates a profound lack of spatial memory and learning in mutant mice, and is associated with severely impaired adult hippocampal neurogenesis
This reduced number of dentate stem and progenitor cells in adult p73−/− mice is unlikely to be simply a consequence of impaired embryonic neurogenesis, but rather suggests a proliferative defect in this neurogenic zone that is ongoing and persistent in the adult. In the adult brain, only stem cells and their early descendants divide actively and can be labeled by BrdU in vivo.31 Thus, we determined the actively proliferating dentate NSC population. To this end, we labeled these cells by BrdU administration and scored the total number of BrdU-positive cells within the Nestin–GFP-positive population in the DG of 6-week-old WT and p73−/− Nestin–GFP mice. Standardized serial sections covering the entire DG were then counted. Indeed, the number of BrdU+/Nestin + double-positive cells in p73−/− Nestin–GFP mice was decreased by 61% (Figure 6a, cells indicated by red arrowheads; quantification of 5 mice each in Figure 6b). The mean number of BrdU+/Nestin+ cells per p73−/− DG was 666±97.5 compared with 1720±119 cells per WT DG (P=0.0024; Figure 6b). Next, we subdivided the Nestin-positive population into neural stem and progenitor cells by costaining BrdU-positive cells for GFAP. This allowed us to separately determine the number of proliferating neural stem (triple positive BrdU+/Nestin+/GFAP+) and progenitor cells (double positive BrdU+/Nestin+/GFAP−). In p73−/− DG, proliferating NSC were reduced by 75% (mean value 386±51 cells per WT DG compared with 92±7 cells per p73−/− DG; P=0.0047; Figures 6c and d). Likewise, proliferating GFAP-negative progenitor cells were decreased by 57% (mean value 1334±95 cells per WT DG compared with 574±104 cells per p73−/− DG; P=0.0058; Figure 6e). Collectively, these data indicate that p73 is essential for proliferation of all dentate Nestin+ progenitor populations, with a particular emphasis on the earliest stem cell population. To determine if defects in subsequent differentiation steps also have a role in DG dysgenesis, we performed neuron birthdating experiments. To this end, 6-week-old WT and p73−/− Nestin–GFP were injected with BrdU, killed 2 weeks later and the brain sections were analyzed for BrdU and NeuN (a marker for mature neurons) colocalization. The results showed a decrease by 35% in the number of BrdU+/NeuN+ cells in p73−/− DG compared with corresponding WT sections (Supplementary Figure S6a). However, p73−/− mice start with fewer proliferating NSC (over 60% decrease; Figure 6b). When the numbers of BrdU+/NeuN+ cells after 2 weeks were normalized against single BrdU+ cells present at the start, the results emphasize that the defect of p73−/− DG lies in the proliferation of NSC, whereas further differentiation, at least quantitatively, occurs normally. Moreover, p73−/− SGZ showed a complete absence of single BrdU+ cells, whereas WT SGZ retained a low number (3.4%) of labeled stem/progenitor cells which did not leave the compartment (Supplementary Figure S6a, arrow head). This suggests impaired symmetrical division of this niche in p73−/− DG.
Apoptosis as a probable cause for adult stem and precursor loss in p73−/− SGZ could be ruled out since brain sections from 6-week-old aged-matched WT and p73−/− Nestin–GFP animals showed no difference in in situ TUNEL and cleaved-caspase 3 stainings (Supplementary Figure S6b and c). To further exclude neurodegeneration that might possibly contribute to the observed adult hippocampal abnormalities, we performed Bielschowsky’s silver staining and phospho-Tau immunohistochemistry to look for plaques and neuro-fibibrillary tangles in sections of 6-week-old whole brains including hippocampus. We were not able to detect signs of neurodegeneration in p73−/− brains (Supplementary Figure S6d and data not shown).
The importance of the hippocampus in learning and memory formation is well recognized.32 To determine the behavioral consequences of the hippocampal abnormalities in adult mice, we performed classic Barnes maze cognitive tests in age-matched p73−/− and WT mice (n=9 each). As shown in Figure 6f, all WT mice showed robust spatial learning and new memory formation. In stark contrast, only a single p73−/− animal was eventually successful, but only very late in the test (second trial on the fourth day) and only after an erratic parcours, which seemed to be more accidental than learned. Moreover, p73−/− mice performed poorly in reflex and neuromuscular function tests, sensorimotor and anxiety tests, collectively reflecting their severe brain phenotype (Supplementary Figure S6e).
Discussion
In this study, we report a new role for p73 in neural stem and progenitor cell maintenance that is essential for proper embryonic and adult neurogenesis. Heretofore, the known action of this p53 family member in the CNS was limited to the anti-apoptotic prosurvival role of ΔNp73 in postmitotic mature neurons of the postnatal brain. Our data offer an additional layer of understanding to explain the severe CNS phenotype of p73−/− mice, marked by cortical hypoplasia, ex vacuo hydrocephalus and hippocampal dysgenesis.
Importantly, in vivo studies of neurogenic zones showed a reduced proliferative pool in E16 and E18 VZ/SVZ and a reduction in the total SVZ area at birth compared with WT littermates. In agreement, in vitro studies of E14 neurospheres revealed an essential role for p73 in maintenance of the self-renewal potential of neural stem/progenitor cells, which at that early stage appeared gradually during serial passaging. As a consequence, p73−/− mice have a significantly depleted stem cell compartment at the time of birth (P0). This is reflected by the fact that P0 mutant NSC cultures have reduced self-renewal potential already at passage 1. The pronounced defect in p73−/− NSC self-renewal and maintenance was accompanied by a strong decrease in neurosphere size. We uncovered that this is due to an additional impairment in proliferation associated with premature senescence in neural stem and progenitor cells. Consequently, the reduced population of neural precursors led to a reduced differentiation potential, reflected by lower numbers and poor quality of differentiated products in two of the three basic cell lineages, neurons and oligo-dendrocytes. In support, previous studies had already shown that overexpressed TAp73 greatly increased the spontaneous differentiation of cultured oligodendrocyte precursor cells.33
Moreover, a large proportion of p73−/− neurons exhibited abnormal neurite projections, suggesting that p73 might also be actively involved in the neuronal differentiation process per se. In support, E14 p73−/− neurospheres at passage 1 produced normal numbers of Tuj1-expressing cells, yet half of these cells failed to mature into proper neurons. Thus, in addition to protecting mature postmitotic neurons,4 our studies reveal that p73 is also critically important for maintaining an adequate stem cell/progenitor pool that enables the generation of a sufficient number of cortical neurons. We propose that cortical thinning in p73−/− mice occurs in part as a consequence of aberrant NSC through interrelated mechanisms characterized by impaired self-renewal, premature exit from the self-renewing compartment through senescence, and impaired production and quality of differentiated derivatives (neurons and oligodendrocytes). The fact that NSC express high levels of TAp73 compared with ΔNp73 suggests that TAp73 tilts the balance toward a proliferative state in stem/progenitor cells, whereas ΔNp73 has a supportive role in this cell type. A similar function was identified for its homolog TAp63 in skin stem cells.34
Furthermore, our studies show that this novel stem cell function of p73 is not limited to embryonic neurogenesis but extends to the maintenance of adult neurogenesis. This was confirmed in the hippocampal DG through in vivo quantification of proliferating neural stem and progenitor cells. In vivo BrdU labeling of p73−/− mice demonstrated an impaired proliferation of both stem cells and amplifying progenitor cells, which correlated with lower total numbers of Nestin-positive cells in the SGZs of the upper and remaining lower blades.
At the molecular level, we found a direct correlation between the reduced proliferative capacity of stem/progenitor cells and the levels of Sox2 protein and mRNA. Moreover, Sox3, Notch1 and Notch2, Jag2, Hes5 and Deltex appear dependent on direct or indirect regulation by p73, as they are dysregulated at the transcriptional level in p73−/− neurospheres. Thus, p73 loss induces a disturbance in the canonical Sox and Notch NSC signaling pathways, which presumably underlies the impaired self-renewal capacity and proliferation of p73−/− stem cells. We propose that the extent to which these factors are affected determines the outcome and severity of the neural phenotype in p73−/− mice. Most important for our studies, Sox2 hypomorhic mice and CNS-specific conditional Sox2 KO mice closely phenocopy p73−/− mice. These Sox2 mutant mice show reduction in cortical mass, hydrocephalus, progressive loss of the lower DG blade and a gradual depletion of stem cells from cultured neurospheres.18,35 In addition, neurospheres derived from hypomorphic Sox2 mutants are unable to properly differentiate into neurons.19 Moreover, deletion of Sox2 transcriptional targets, such as sonic hedgehog (Shh) and loss of genes that control Shh signaling, such as Kif3a and Smoothened (Smo), also produce comparable CNS phenotypes. All are marked by enlarged telencephalic ventricles and a reduction in brain size because of the reduced proliferation in the SVZ compartment. They also exhibit postnatal arrest of hippocampal development, the latter caused by a failure of GFAP-positive radial glial NSC to develop in the DG after embryonic development.36 Future studies should address if Shh signaling is also impaired in p73−/− brains.
Overall, our data establishes that the products of the p73 gene locus are essential maintenance factors in the nervous system whose broad action stretches across the entire differentiation arch from stem cells to mature postmitotic neurons. Despite the apparent predominance of the TAp73 isoform in NSC (our data), the lesson learned from both isoform-specific KO mice7,8,10 with their surprisingly mild CNS phenotypes is that TAp73 and ΔNp73 isoforms are redundant and can compensate for each other on a cell and organ level. Thus, both p73 isoforms are indispensable for brain homeostasis. On the other hand, p53 has only a limited proapoptotic role in CNS development, reflected by the fact that up to 23% of female p53−/− embryos die in utero because of the exencephaly resulting from overproduction of neuronal tissue.37,38 Thus, p73 emerges as a unique member of the p53 family with a central role in CNS development and maintenance.
Materials and Methods
Animals
This study was approved by Stony Brook University Animal Care Committee, and use was in accordance with institutional guidelines. p73 +/− mice3 were maintained on a mixed 129Sv:Balbc 75 : 25 background. Transgenic Nestin–GFP hemizygous mice30 were crossed with p73 +/− mice to generate F2 p73−/−; Nestin–GFP and p73 +/+; Nestin–GFP mice for analysis.
Neurosphere assays
Whole brains or carefully dissected SVZ/SGZ were harvested from E14 and P0 WT and p73−/− mice as indicated, dissociated into single cells by trituration, filtered and plated in standardized condition at 105 cells/60 mm dishes/5 ml NeuroCult NSC Basal Medium (Mouse) with proliferation supplements and EGF (all from StemCell Technologies Inc., Vancouver, BC, Canada) following the manufacturer’s instructions. Neurospheres were allowed to form for 5 days (E14) or 7 days (P0). This was considered passage 0. To avoid variation related to brain processing, we only started to quantify neurospheres (NSC) generated after the end of passage 0. Neurospheres were passaged every 5 days. Passage 0 neurospheres were dissociated with accutase, filtered and replated in triplicates at 105cells/60 mm dishes/5 ml medium and regeneration of new neurospheres was monitored. At the end of each passage, the number and size of neurospheres and the total number of cells was assessed. Subsequently, 105 dissociated cells were replated in triplicates for the next passage. Results shown are representative of multiple independent litters. For BrdU labeling, P0-derived neurospheres at passage 2 were plated as 105 dissociated cells, allowed to grow for 2 days to reform neurospheres and then pulsed for 20 min with 10 μM BrdU (BD BioSciences, San Jose, CA, USA, Cat No. 550891). TUNEL staining was performed with the In Situ cell death detection kit (TMR Red, Roche, Indianapolis, IN, USA).
Immunoblot analysis
Neurospheres were lysed in RIPA buffer containing protease inhibitors (Roche) and processed for western blotting. Antibodies are in Supplementary Data.
SA-β-galactosidase staining
SA-β-Galactosidase staining was performed as previously described.39 Details are found in Supplementary Data.
Differentiation assays
Cells from freshly dissociated neurospheres from E14 and P0 were seeded on poly-d-lysine/laminin-coated glass slides (BD BioCoat, San Jose, CA, USA, Cat No. 354688) in NeuroCult NSC Basal Medium with differentiation supplements (StemCells Technologies Inc.) following the manufacturer’s instructions. Differentiated cells were immunostained and quantitated as indicated.
Neuroanatomy
For histology, perfusion-fixed brain sections were stained with hematoxylin-eosin, silver stain or antibodies. Details are in Supplementary Data.
Immunohistochemistry
Paraffin-embedded sections (3 μm) were immunostained as previously described.40 Details and list of antibodies are in Supplementary Data.
In vivo BrdU labeling of fetal brains
Pregnant mouse females were intraperitoneally injected with BrdU (150 mg/kg of body weight, 1 injection, BD BioSciences) dissolved in PBS at embryonic day E16. The embryos were dissected after 2 h, and their PFA-fixed brains were paraffin-embedded followed by coronal sectioning.
In vivo BrdU labeling in adult mice
WT Nestin–GFP and p73−/− Nestin–GFP transgenic mice (6-week old) were intraperitoneally injected with BrdU (150 mg/kg per injection; three injections at 4 h intervals, BD BioSciences) and killed 24 h later for short-term labeling and 2 weeks later for neuronal birthdating experiments. Mice were deeply anesthetized by i.v. avertin solution (2.5%) and transcardially perfused with PBS followed by 4% paraformaldehyde in PBS. Brains were dissected out and stored in 4% PFA/PBS at 4°C. Fresh floating sagittal sections (50 μm) were generated from the right hemispheres on a vibratome and collected in series in PBS. Sections were stored in 1% PFA/PBS at 4°C until immunofluorescence staining. For antigen retrieval, sections were incubated in 2N HCL for 1 h followed by 0.1 M borate buffer and PBS washes. Sections were incubated overnight at 4°C in primary antibodies while shaking on a tilt-rocker. BrdU antibody (rat anti-BrdU, 1 : 300, AbD Serotec, Raleigh, NC, USA), GFAP (rabbit anti-GFAP, 1 : 1000, Millipore, Billerica, MA, USA) and NeuN (1 : 500, Millipore) were diluted in PBS/10% goat serum/0.3% BSA/0.2% Triton X100. For BrdU single staining, anti-rat Rhodamine RedX was diluted 1 : 1000 in 0.3% BSA/0.2% TritonX100/PBS. For BrdU and GFAP double staining, anti-rat Rhodamine RedX and anti-rabbit Cy5 antibodies were diluted 1 : 1000 and 1 : 500, respectively. All stainings included primary antibody controls in which serial brain sections were treated identically except that the primary antibody was withheld. All antibody controls for BrdU and GFAP were negative. For apoptosis studies, TUNEL (in situ cell death detection kit, TMR Red, Roche) and cleaved casp 3 stainings were performed on the same type of free-floating sagittal brain sections.
Image acquisition and quantification
From each brain, serial sections were collected in sets of six, and the first-in-six section from each set was stained for BrdU, whereas the second section was double stained for BrdU and GFAP. Each of these single and double stained one-in-six series contained at least 11 sagittal brain sections, representing the full anatomical architecture of the hippocampus along its medio–lateral axis. A Zeiss LSM 510 confocal microscope system (Zeiss, Thornwood, NY, USA) with an Axiovert 200 M inverted microscope (Zeiss) was used to generate Z-stack images of the entire hippocampal thickness by scanning each section in intervals of 2 μm. To determine the number of total proliferating progenitors in WT and p73−/− brains, the total number of BrdU+/Nestin+ cells in the dentate SGZ of each hemisphere was quantified using the LSM Image Browser Software (Zeiss). To determine the total number of proliferating NSCs and neural progenitor cells, the total number of BrdU+/Nestin+/GFAP+ cells and BrdU+/Nestin+/GFAP− cells in the SGZ were quantified. Nestin+ cells that were located outside the SGZ, such as the hilus or in the granule cell layer were excluded in the quantification.
qRT-PCR
Semiquantitative and quantitative real-time RT-PCR assays were performed using total RNAs prepared from neurospheres as previously described.40 Primers are in Supplementary Data.
Behavioral tests
Animals were individually housed in laboratory cages with a 12-h light–dark cycle and free access to food and water throughout the study. Barnes maze test, tail suspension test, hind grip test, light–dark box test and open field test were performed as previously described using standardized conditions.41
Supplementary Material
Acknowledgments
This work was funded by grants from the National Cancer Institute (CA93853 to UMM), Deutsche Krebshilfe (108173 to UMM) and NINDS (NS42168 to SET). We thank Frank McKeon for the p73 knockout mice and G Enikolopov for the Nestin–GFP mice. We thank B Anderson for the support with the behavioral tests.
Abbreviations
- BrdU
bromodeoxyuridine
- CA
cornu ammonis
- CNS
central nervous system
- DG
dentate gyrus
- FACS
fluorescence-activated cell sorting
- GnRH
gonadotropin releasing hormone
- KO
knockout
- NCFC
neural colony-forming cell assay
- NSA
neurosphere assay
- NSC
neural stem cell
- SGZ
subgranular zone
- SVZ
subventricular zone
- WT
wild type
Footnotes
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Conflict of interest The authors declare no conflict of interest.
References
- 1.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–386. [PubMed] [Google Scholar]
- 2.Meyer G, Cabrera Socorro A, Perez GarciaC G, Martinez Millan L, Walker N, Caput D. Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J Neurosci. 2004;24:9878–9887. doi: 10.1523/JNEUROSCI.3060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 4.Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD. p73 is required for survival and maintenance of CNS neurons. J Neurosci. 2002;22:9800–9809. doi: 10.1523/JNEUROSCI.22-22-09800.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 6.Lee AF, Ho DK, Zanassi P, Walsh GS, Kaplan DR, Miller FD. Evidence that DeltaNp73 promotes neuronal survival by p53-dependent and p53-independent mechanisms. J Neurosci. 2004;24:9174–9184. doi: 10.1523/JNEUROSCI.1588-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tissir F, Ravni A, Achouri Y, Riethmacher D, Meyer G, Goffinet AM. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci USA. 2009;106:16871–16876. doi: 10.1073/pnas.0903191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, et al. Isoform-specific p73 knockout mice reveal a novel role for \{Delta\}Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549–560. doi: 10.1101/gad.1873910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 10.Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alonso M, Ortega-Perez I, Grubb MS, Bourgeois JP, Charneau P, Lledo PM. Turning astrocytes from the rostral migratory stream into neurons: a role for the olfactory sensory organ. J Neurosci. 2008;28:11089–11102. doi: 10.1523/JNEUROSCI.3713-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemajerova A, Palacios G, Nowak NJ, Matsui S, Petrenko O. Targeted deletion of p73 in mice reveals its role in T cell development and lymphomagenesis. PLoS One. 2009;4:e7784. doi: 10.1371/journal.pone.0007784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 14.Marshall GP, II, Reynolds BA, Laywell ED. Using the neurosphere assay to quantify neural stem cells in vivo. Curr Pharm Biotechnol. 2007;8:141–145. doi: 10.2174/138920107780906559. [DOI] [PubMed] [Google Scholar]
- 15.Louis SA, Rietze RL, Deleyrolle L, Wagey RE, Thomas TE, Eaves AC, et al. Enumeration of neural stem and progenitor cells in the neural colony-forming cell assay. Stem Cells. 2008;26:988–996. doi: 10.1634/stemcells.2007-0867. [DOI] [PubMed] [Google Scholar]
- 16.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 17.Wang TW, Stromberg GP, Whitney JT, Brower NW, Klymkowsky MW, Parent JM. Sox3 expression identifies neural progenitors in persistent neonatal and adult mouse forebrain germinative zones. J Comp Neurol. 2006;497:88–100. doi: 10.1002/cne.20984. [DOI] [PubMed] [Google Scholar]
- 18.Favaro R, Valotta M, Ferri AL, Latorre E, Mariani J, Giachino C, et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci. 2009;12:1248–1256. doi: 10.1038/nn.2397. [DOI] [PubMed] [Google Scholar]
- 19.Cavallaro M, Mariani J, Lancini C, Latorre E, Caccia R, Gullo F, et al. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development. 2008;135:541–557. doi: 10.1242/dev.010801. [DOI] [PubMed] [Google Scholar]
- 20.Agathocleous M, Iordanova I, Willardsen MI, Xue XY, Vetter ML, Harris WA, et al. A directional Wnt/beta-catenin-Sox2-proneural pathway regulates the transition from proliferation to differentiation in the Xenopus retina. Development. 2009;136:3289–3299. doi: 10.1242/dev.040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 22.Higuchi M, Kiyama H, Hayakawa T, Hamada Y, Tsujimoto Y. Differential expression of Notch1 and Notch2 in developing and adult mouse brain. Brain Res Mol Brain Res. 1995;29:263–272. doi: 10.1016/0169-328x(94)00257-f. [DOI] [PubMed] [Google Scholar]
- 23.Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell. 2007;27:647–659. doi: 10.1016/j.molcel.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 24.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 25.Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26:867–874. doi: 10.1093/carcin/bgh296. [DOI] [PubMed] [Google Scholar]
- 26.Ferron SR, Marques-Torrejon MA, Mira H, Flores I, Taylor K, Blasco MA, et al. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci. 2009;29:14394–14407. doi: 10.1523/JNEUROSCI.3836-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci USA. 2006;103:8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seri B, Garcia-Verdugo JM, Collado-Morente L, McEwen BS, Alvarez-Buylla A. Cell types, lineage, and architecture of the germinal zone in the adult dentate gyrus. J Comp Neurol. 2004;478:359–378. doi: 10.1002/cne.20288. [DOI] [PubMed] [Google Scholar]
- 29.Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- 31.Temple S, Alvarez-Buylla A. Stem cells in the adult mammalian central nervous system. Curr Opin Neurobiol. 1999;9:135–141. doi: 10.1016/s0959-4388(99)80017-8. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Mu Y, Gage FH. Development of neural circuits in the adult hippocampus. Curr Top Dev Biol. 2009;87:149–174. doi: 10.1016/S0070-2153(09)01205-8. [DOI] [PubMed] [Google Scholar]
- 33.Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, et al. Roles for p53 and p73 during oligodendrocyte development. Development. 2004;131:1211–1220. doi: 10.1242/dev.01035. [DOI] [PubMed] [Google Scholar]
- 34.Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 36.Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol. 2008;317:246–259. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- 38.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 39.Dimri GP, Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harb Symp Quant Biol. 1994;59:67–73. doi: 10.1101/sqb.1994.059.01.010. [DOI] [PubMed] [Google Scholar]
- 40.Wolff S, Talos F, Palacios G, Beyer U, Dobbelstein M, Moll UM. The alpha/beta carboxy-terminal domains of p63 are required for skin and limb development New insights from the Brdm2 mouse which is not a complete p63 knockout but expresses p63 gamma-like proteins. Cell Death Differ. 2009;16:1108–1117. doi: 10.1038/cdd.2009.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.