

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, MRKH type I or isolated MRKH or Rokitansky sequence, MRKH type II or MURCS association (Müllerian duct aplasia, renal dysplasia and cervical somite anomalies), congenital absence of the uterus and vagina (CAUV), genital renal ear syndrome and Müllerian aplasia (MA).
1.2 OMIM# of the disease
277000 (MRKH, CAUV) and 601076 (MURCS).
1.3 Name of the analyzed genes or DNA/chromosome segments
1q21.1, 4q34-qter, 8p23.1, 10p14–15, 16p11.2, 17q12, 22q11.21 and Xpter-p22.32.
1.4 OMIM# of the gene(s)
Putative candidate genes: 189907 (TCF2),1, 2 601999 (LHX1),2, 3, 4, 5 312865 (SHOX),6 602427 (TBX6)2 609783 (ITIH5).7
1.5 Mutational spectrum
Maximal deletion in 1q21.1: 398.5 kb;3, 5 in 4q34-qter: 8 Mb;8 in 8p23.1: 1.2 Mb;7 in 10p14–15: 0.2 Mb;7 in 16p11.2: 600 kb;2 in 17q12: 1.5 Mb2, 3, 5 and in 22q11.21: 3 Mb.2, 3, 5, 7, 9, 10
Maximal duplication in 1q21.1: 200 kb.3
Partial duplication of the Xpter pseudoautosomal region 1.6
1.6 Analytical methods
Search for microrearrangements by means of multiplex ligation-dependent probe amplification (MLPA) using the SALSA P023 DiGeorge MLPA kit (MRC-Holland, Amsterdam, The Netherlands), comparative genomic hybridization array (CGH array), duplex PCR/liquid chromatography (DP/LC) and/or FISH.
1.7 Analytical validation
Validation of MLPA results or CGH array results by DP/LC or FISH.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
1 in 4500 female births.11
1.9 If applicable, prevalence in the ethnic group of investigated person
Not applicable.
1.10 Diagnostic setting

Comment: WNT4 syndrome is close but different from MRKH. It differs from this latter by an hyperandrogenism and gonadal affection.12 It thus needs to be considered in regards to differential diagnosis.13
2. TEST CHARACTERISTICS
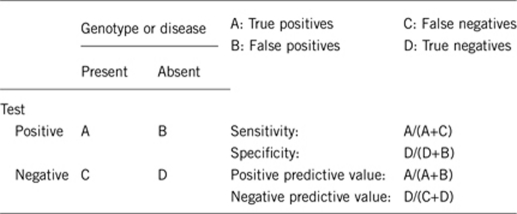
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Nearly 100% using analytical methods described above.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Nearly 100% using analytical methods described above.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
The synthesis of the literature2, 3, 4, 5, 9, 10 on the genome-wide analysis on MRKH patients was used to estimate that only ∼1% show a 1q21.1 rearrangement, ∼1% in 16p11.2, ∼6% in 17q12 and ∼4% in 22q11.21, from a total of 153 cases.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
100% of control subjects tested by MLPA (DiGeorge kit) showed negative results.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
Not applicable.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Not applicable.
Index case in that family had not been tested:
Not applicable.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: the tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient?
Initial clinical examination and transabdominal ultrasonography must be the first investigations in evaluating patients with suspected MA. If necessary, magnetic resonance imaging affords to clearly precise the malformation. A full check-up (transabdominal ultrasonography, spine radiography and/or heart echography, audiogram) should be undertaken to search for associated malformations in order to determine the MRKH type (I or II). Testosterone dosage should help orientating the diagnosis towards MRKH or WNT4 syndrome.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Alternative diagnosis methods described above are, at present, the only way to accurately diagnose MRKH syndrome, as the phenotype–genotype correlations still cannot be established. Clinical examination, imaging, and biological measurements, will in any case remain necessary to evaluate the level of care for patients.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: the tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
If the test result is negative (please describe):
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Can improve genetic counseling.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
No.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
In rare familial forms.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Acknowledgments
This work was supported by the EuroGentest, an EU-FP6 supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest.
References
- Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–2008. doi: 10.1093/hmg/8.11.2001. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Strick R, Storer M, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J Med Genet. 2011;48:197–204. doi: 10.1136/jmg.2010.082412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroki C, Krepischi-Santos AC, Szuhai K, et al. Genomic imbalances associated with Mullerian aplasia. J Med Genet. 2008;45:228–232. doi: 10.1136/jmg.2007.051839. [DOI] [PubMed] [Google Scholar]
- Bernardini L, Gimelli S, Gervasini C, et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis. 2009;4:25. doi: 10.1186/1750-1172-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. 2011;95:1589–1594. doi: 10.1016/j.fertnstert.2010.07.1062. [DOI] [PubMed] [Google Scholar]
- Gervasini C, Grati FR, Lalatta F, et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet Med. 2010;12:634–640. doi: 10.1097/GIM.0b013e3181ed6185. [DOI] [PubMed] [Google Scholar]
- Morcel K, Watrin T, Pasquier L, et al. Utero-vaginal aplasia (Mayer-Rokitansky-Kuster-Hauser syndrome) associated with deletions in known DiGeorge or DiGeorge-like loci. Orphanet J Rare Dis. 2011;6:9. doi: 10.1186/1750-1172-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendavid C, Pasquier L, Watrin T, et al. Phenotypic variability of a 4q34 → qter inherited deletion: MRKH syndrome in the daughter, cardiac defect and Fallopian tube cancer in the mother. Eur J Med Genet. 2007;50:66–72. doi: 10.1016/j.ejmg.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Cheroki C, Krepischi-Santos AC, Rosenberg C, et al. Report of a del22q11 in a patient with Mayer-Rokitansky-Kuster-Hauser (MRKH) anomaly and exclusion of WNT-4, RAR-gamma, and RXR-alpha as major genes determining MRKH anomaly in a study of 25 affected women. Am J Med Genet A. 2006;140:1339–1342. doi: 10.1002/ajmg.a.31254. [DOI] [PubMed] [Google Scholar]
- Uliana V, Giordano N, Caselli R, et al. Expanding the phenotype of 22q11 deletion syndrome: the MURCS association. Clin Dysmorphol. 2008;17:13–17. doi: 10.1097/MCD.0b013e3282ef97ee. [DOI] [PubMed] [Google Scholar]
- Folch M, Pigem I, Konje JC. Mullerian agenesis: etiology, diagnosis, and management. Obstet Gynecol Surv. 2000;55:644–649. doi: 10.1097/00006254-200010000-00023. [DOI] [PubMed] [Google Scholar]
- Biason-Lauber A, Konrad D. WNT4 and sex development. Sex Dev. 2008;2:210–218. doi: 10.1159/000152037. [DOI] [PubMed] [Google Scholar]
- Morcel K, Camborieux L, Guerrier D. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007;2:13. doi: 10.1186/1750-1172-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]