

Abstract
Until recently, ammonia had rarely succumbed to catalytic transformations with homogeneous catalysts, and the development of such reactions that are selective for the formation of single products under mild conditions has encountered numerous challenges. However, recently developed catalysts have allowed several classes of reactions to create products with nitrogen-containing functional groups from ammonia. These reactions include hydroaminomethylation, reductive amination, alkylation, allylic substitution, hydroamination, and cross-coupling. This Minireview describes examples of these processes and the factors that control catalyst activity and selectivity.
Keywords: alkylation, ammonia, cross-coupling reaction, hydroamination, reductive amination
1. Introduction
The introduction of functional groups through reactions that occur under mild conditions with unactivated reagents while generating few byproducts has been a theme of modern synthesis. In this vein, the direct synthesis of nitrogen-containing molecules from the inexpensive commodity chemical ammonia is an important goal. Most ammonia (83%)[1] is used as fertilizer, but many basic chemical reactions of ammonia are conducted on large scale, including those to form urea, ethanolamines, and even hydrazine rocket fuel.[1,2]
Although conducted on large scales, these reactions typically require high temperatures or pressures, and most are not selective for formation of a single product. For instance, methylamine is synthesized from methanol and ammonia over silica–alumina catalysts at 300–430°C, but dimethylamine and triethylamine are also produced.[3] To improve the reactivity and selectivity of ammonia toward many desired transformations, catalysts based on transition-metal complexes have been studied.
Unfortunately, many common reactions catalyzed by transition-metal complexes do not occur with ammonia. This lack of reactivity can be attributed to several factors. First, the catalyst is often deactivated by formation of stable Werner ammine complexes. Second, the strength of the N–H bond in ammonia (107 kcalmol−1) makes “N–H activation” by the metal center challenging. Third, the moderate basicity and low acidity of ammonia disfavor proton exchanges, either to or from ammonia. Finally, handling high pressures of ammonia requires special equipment.
Nevertheless, reactions of ammonia catalyzed by transition-metal complexes have recently been developed. These reactions can be divided into three classes. The first comprises tandem processes in which ammonia reacts during an uncatalyzed step of the sequence and is tolerated by the metal during the catalytic steps. The second involves a catalytic transformation of ammonia in which the ammonia reacts with a coordinated ligand, rather than the metal center. The third involves a catalytic process in which ammonia reacts with the metal center to form a transition-metal–amido complex. This Minireview presents examples of these three clases of reactions, the challenges confronted during their development, and the limitations they currently possess.
2. Reactions in which the Catalysts Tolerate the Presence of Ammonia
During some reactions catalyzed by transition-metal complexes, the catalyst tolerates ammonia, but does not react directly with it. Many of these reactions occur with soft, low-valent, late transition-metal complexes that bind ammonia weakly and that possess chelating ligands to discourage the binding of ammonia. Three examples of such reactions are described.
2.1. Hydroaminomethylation
Hydroaminomethylation is a tandem reaction in which hydroformylation of an olefin produces an aldehyde, and this aldehyde undergoes reductive amination. The hydroformylation and hydrogenation steps are catalyzed by transition-metal complexes. Many hydroaminomethylation reactions have been reported with primary and secondary amines. Hydroaminomethylations with ammonia to form primary amines would be an important development, but few reports of such a process have been published.
Hydroaminomethylation in its current form with any amine has limitations. The reaction often occurs with modest selectivity for linear or branched products (n vs. iso), and isomerization of the starting olefin has been shown to occur in competition with hydroaminomethylation. In addition, selectivity for formation of the primary amine over a series of alcohol and condensation byproducts is often poor.
The hydroaminomethylation of olefins with ammonia was first disclosed in a 1950 patent by workers at DuPont.[4] A variety of amine products were reported to be obtained in ca. 40% total yield by the reaction of olefins with 100–2000 bar of syngas and ammonia in the presence of a metallic cobalt catalyst in diethyl ether. For this process, optimum temperatures exceeded 250°C. A patent from 1988 by Lin and Knifton disclosed a phosphine-ligated cobalt octacarbonyl catalyst that reacted with olefins in dioxane to produce primary and secondary amines in a ratio just over 1:1.[5] The reaction required temperatures in excess of 150°C and gas pressures over 138 bar.
In 1999, Beller et al. reported hydroaminomethylation reactions that are more selective for primary amines and occur under more moderate conditions.[6] The reaction was conducted with two catalysts: a rhodium catalyst for hydroformylation and an iridium catalyst for hydrogenation of the intermediate imine. The selectivity for primary amines was improved by conducting the reactions with a water-soluble catalyst in a biphasic system in which the product amine and the catalyst would reside in different phases. The reaction of an aqueous solution of ammonia containing the combination of 10 mol% of tppts (trisodium 3,3′,3″-phosphandiyltris(benzenesulfonate)) or binas (sulfonated 2,2′-bis(diphenylphosphinomethyl)-1,1′-binapthyl), 0.03 mol% [{Rh(cod)Cl}2] (cod = 1,5-cyclooctadiene), 0.2 mol% [{Ir(cod)Cl}2], and an organic phase of MTBE (methyl tert-butyl ether) formed the hydroaminomethylation products from terminal olefins at 130°C under a syngas pressure of 78 bar (Scheme 1).
Scheme 1.
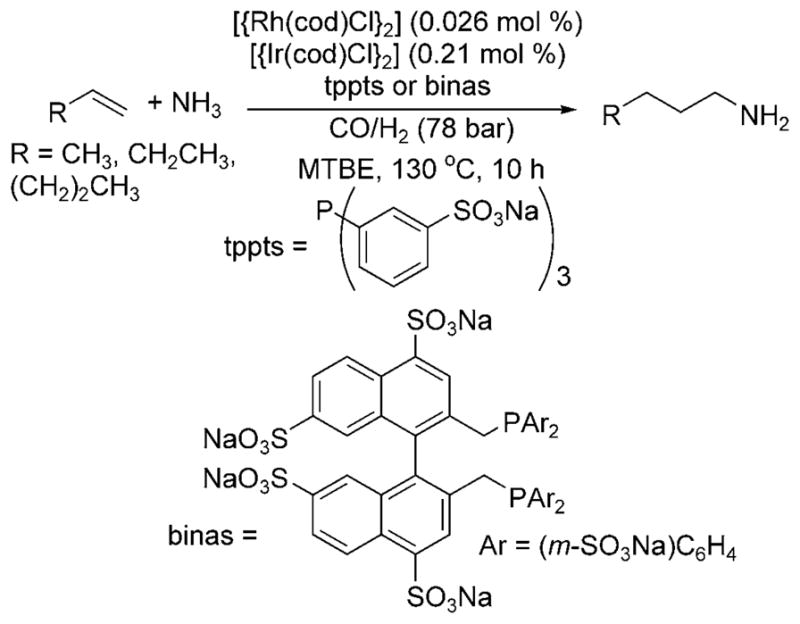
Rh-catalyzed hydroaminomethylation with ammonia.
Although constituting progress toward selective hydroaminomethylation with ammonia, these reactions were limited to a narrow range of terminal olefins, and the best selectivity for formation of primary amines over secondary amines was 10:1. In addition, this process required temperatures of 110–130°C, more than 60 total bar of pressure and a total quantity of ligand exceeding 20 mol%. Thus, this process suggests that the appropriate choice of catalyst and reaction conditions can lead to the formation of primary amines from alkenes, ammonia and carbon monoxide, but much progress must be made before this reaction becomes a viable process.
2.2. Reductive Amination of Ketones
Like hydroaminomethylation, reductive amination with ammonia could be an efficient, tandem process for the synthesis of primary amines. In this case, aldehydes or ketones react with ammonia to form an imine, and the imine undergoes metal-catalyzed hydrogenation to form an amine. Until recently, this process with ammonia had not formed primary amines in high yield.
In 2000, Börner et al. reported the first general homogeneous catalytic reductive amination of aldehydes with primarily secondary amines as nucleophiles and hydrogen gas as the reductant.[7] The rhodium complexes [Rh(PPh3)3Cl] and [{Rh(dppb)Cl}2] (dppb = 1,4-bis(diphenylphosphino)butane) catalyzed the reaction of alkyl- and aryl-substituted ketones and aldehydes at room temperature under 50 bar of hydrogen to form the expected amine product from reductive amination (Scheme 2).
Scheme 2.
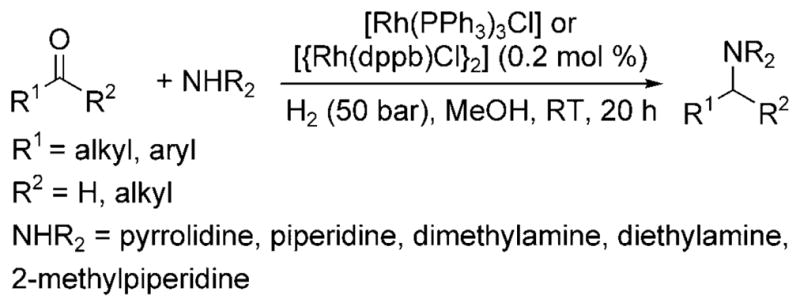
Rh-catalyzed reductive amination with amines.
Beller et al. then reported a rhodium-catalyzed reductive amination of aldehydes with ammonia.[8] As was used to improve the selectivity for hydroaminomethylations with ammonia, a biphasic system was used to segregate the amine product from the water-soluble rhodium catalyst and ammonia nucleophile. The reaction of benzaldehyde with aqueous ammonia and hydrogen in the presence of 0.05 mol% of [{Rh(cod)Cl}2] and 1.3 mol% of the water-soluble phosphine tppts (see Scheme 1) at 135°C formed benzylamine, as well as a small amount of benzyl alcohol byproduct (Scheme 3). Most of the reported reactions were conducted with aromatic aldehydes. The reductive amination of aliphatic aldehydes with ammonia was also reported, but higher temperatures and higher catalyst loadings were required, and reactions of these aldehydes formed a significant amount of secondary amines and condensation products. Therefore, the design of a complex that catalyzes the reductive amination of aliphatic aldehydes at lower temperatures and with higher turnover numbers remains a current goal.
Scheme 3.
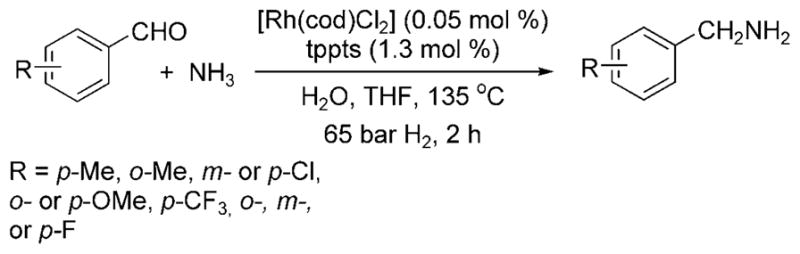
Rh-catalyzed reductive amination of aldehydes with ammonia.
Reductive amination with ammonia was first shown by Fukuzumi and co-workers to form α-amino acids from α-keto acids in aqueous solvent.[9] The iridium(III) complex [{Cp*Ir-(bpy)H}2]SO4 (bpy = bipyridine, Cp* = pentamethylcyclo-pentadienyl) catalyzed the reduction of α-imino acids produced in situ from the acid-catalyzed reaction of ammonia with α-keto acids (Scheme 4). At low pH, protonation of ammonia prevented reaction with the α-keto acid and led to the formation of α-hydroxy carboxylic acid byproducts; at higher pH, the inactive iridium hydroxo species [Cp*Ir-(bpy)(OH)]+ formed. Thus, careful control of the reaction conditions was necessary, and the highest yields of α-amino acids were achieved with ammonium formate and a pH between 5 and 6.5.
Scheme 4.
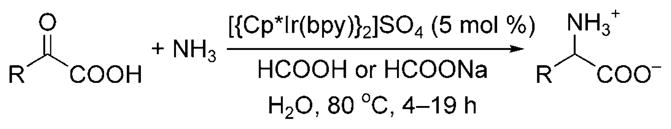
Synthesis of α-amino acids through reductive amination with ammonia.
The first asymmetric reductive amination involving an ammonium salt was discovered by workers at Merck and applied to the synthesis of the Type II Diabetes drug, Januvia. Reaction of a 1,3-diketone with ammonium acetate formed the unprotected enamine. Asymmetric hydrogenation of the enamine with [{Rh(cod)Cl}2] and the Josiphos ligand PPF-tBu formed Januvia with greater than 99% conversion and 95% ee (Scheme 5). Deuterium labeling studies indicated that hydrogenation occurred from the imine tautomer of the enamine.
Scheme 5.

Asymmetric reductive amination in the synthesis of Januvia.
2.3. Alkylation of Ammonia with Alcohols
Many alkylamines are prepared on large scale by the reaction of an alcohol with ammonia.[10,11] These processes are conducted with heterogeneous catalysts and require high pressures and temperatures. Significant amounts of alkanes and alkenes, as well as secondary and tertiary amine products form. Thus, a more selective coupling of alcohols with amines under milder conditions would be valuable.
Reactions in which soluble transition-metal complexes catalyze the alkylation of amines with alcohols are now known.[12] In these processes, the alcohol undergoes dehydrogenation to form a ketone, which reacts with an amine to generate an imine in situ. This imine then undergoes catalytic hydrogenation to form the product amine. Again, the formation of secondary and tertiary amines, as well as the low reactivity of ammonia toward formation of imines, complicates the development of the alkylation of ammonia with alcohols.
For example, the N-alkylation of ammonium salts with alcohols to form a mixture of secondary and tertiary amines was reported for the first time by Fujita and co-workers in 2007.[13] The group showed that [{Cp*IrCl2}2] catalyzes the multiple alkylation of ammonium acetate with benzylic and aliphatic alcohols and a catalytic amount of sodium carbonate at 130–140°C to form tertiary amines (Scheme 6). However, the reaction of ammonium tetrafluoroborate with aliphatic alcohols under the same conditions formed secondary amines. The monoalkylated product was not obtained under either of these conditions.
Scheme 6.
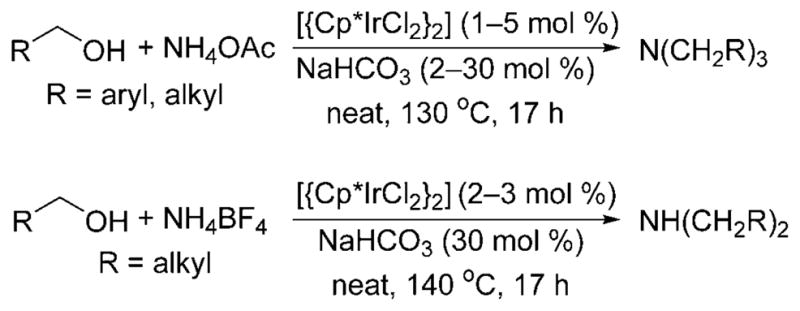
Multiple alkylation of ammonium salts catalyzed by [{Cp*IrCl2}2].
The alkylation of ammonia with alcohols was recently achieved by Milstein and Gunanathan.[14] The ruthenium PNP pincer complex (Ru cat., Scheme 7) catalyzed the reaction of alcohols with 7.6 bar of ammonia. Benzylic alcohols, hetero-arylmethyl alcohols, and aliphatic alcohols reacted in good yields with high selectivity for formation of the primary amine. The corresponding imine was the major side product of the reaction when conducted in toluene, and the carboxylic acid was the major side product when conducted in water.
Scheme 7.
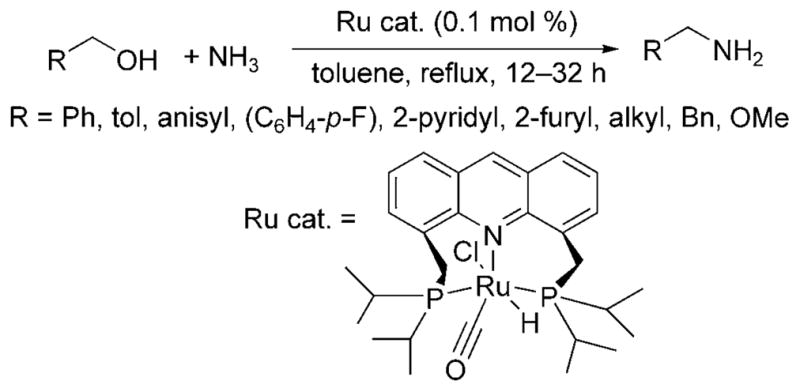
Ru-catalyzed alkylation of ammonia with alcohols.
Shortly thereafter, Mizuno et al. disclosed a ruthenium-catalyzed formation of nitriles from alcohols and ammonia.[15] Benzylic or allylic alcohols, as well as aliphatic, aromatic and α,β-unsaturated aldehydes, reacted with a THF solution of ammonia in the presence of the heterogeneous ruthenium hydroxide catalyst Ru(OH)x/Al2O3 at 120 °C to generate a nitrile product (Scheme 8). Presumably the reaction occurred by a similar “hydrogen-borrowing”[12] strategy employed for the N-alkylation of ammonia. The imine was converted further to a nitrile through metal-catalyzed aerobic oxidation.
Scheme 8.
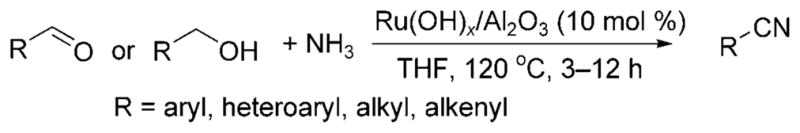
Alkylation of ammonia and oxidation to a nitrile.
3. Catalytic Reactions of Ammonia Occurring without Direct Coordination to the Metal
Coordination of unsaturated groups to transition metals often causes them to be electrophilic and to be susceptible to nucleophilic attack. The coordination sphere of the metal can then affect the regioselectivity and steroselectivity of the addition step. Catalytic processes involving such nucleophilic additions to coordinated ligands are well known and include olefin oxidation, olefin hydroamination, and allylic substitution.[16] Although these reactions are known to occur with water, alcohols and many nitrogen-containing reagents as nucleophiles, such reactions were reported only recently with ammonia. The following examples illustrate catalytic processes in which iridium and palladium π-allyl intermediates and gold–alkyne complexes react with ammonia to form primary amines, imines and enamines.
3.1. Allylic Substitution and Telomerization
Allylic aminations with nitrogen nucleophiles form linear or branched amines, sulfonamides, and imides, depending on the identity of the catalyst.[17] In contrast, allylic aminations with ammonia to form primary allylamines were reported only recently. In 2007 Hartwig and co-workers demonstrated that a cyclometallated iridium complex catalyzes the allylation of ammonia.[18] In this initial system, however, secondary amines were the exclusive products of the catalytic process.
In 2009, Nagano and Kobayashi reported the reactions of allylic acetates and allylic carbonates with ammonia to form primary amines.[19] In the presence of aqueous ammonia in dioxane, [Pd(PPh3)4] catalyzed the amination of allylic acetates and carbonates at room temperature (Scheme 9). The feasibility of developing an asymmetric version of the reaction was demonstrated by the formation of 1,3-dipheny-lallylamine in 71% yield and 87% ee from the reaction of 1,3-diphenylallyl acetate with ammonia catalyzed by the combination of [{PdCl(Allyl)}2] and (R)-binap (Scheme 10). Although this reaction was the first allylic substitution with ammonia to form a primary allylic amine product with substantial ee, it required high catalyst loadings (20 mol%), was conducted under dilute conditions, required a relatively long reaction time (18 h), and resulted in moderate enantio-selectivity.
Scheme 9.
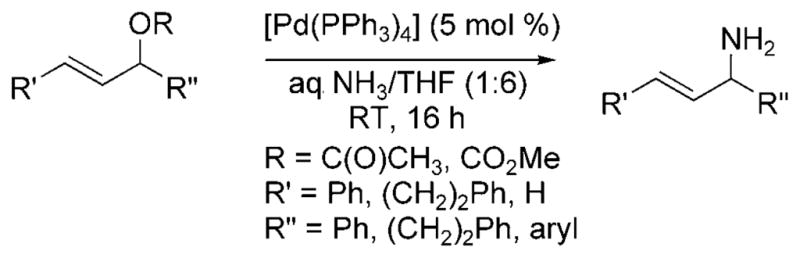
Allylic amination of allylic acetates and carbonates with ammonia.
Scheme 10.
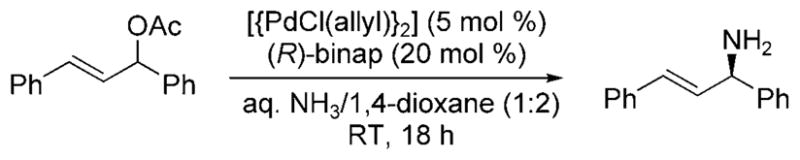
Enantioselective amination of allylic acetate.
The iridium(I) cyclometalated complex (Ir cat., L = (R,R,R)-L1, Scheme 11) developed by Hartwig and co-workers catalyzed a broad range of asymmetric allylic substitutions with ammonia.[20] The ethylene-bound precursor of the active catalyst (Ir cat., L = C2H4, Scheme 11) was stable to 2500 equivalents of ammonia (relative to the iridium catalyst). The monoallylamine products were obtained with an ee exceeding 96% and reaction times of 4–12 h at 30°C. In addition, acylation of the primary amine product forms the corresponding N-allyl amide, a product inaccessible by allylic substitution with amide nucleophiles.
Scheme 11.
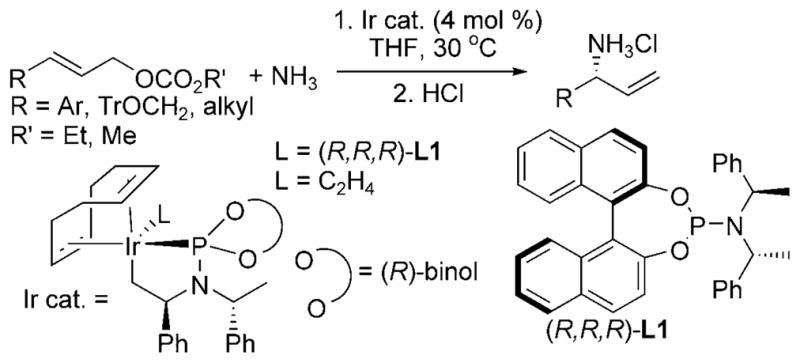
Ir-catalyzed asymmetric allylation of ammonia. Tr = triphe-nylmethyl.
The telomerization of dienes with amines occurs through allyl intermediates that are related to those in allylic substitution reactions. Prinz and Driessen-Hölscher reported the biphasic telomerization of butadiene with ammonia catalyzed by Pd(OAc)2 and tppts or the isolated palladium–ammine complex 1 with added monophosphine (Scheme 12).[21] The reaction likely occurs by substitution of a monophosphine for an ammonia ligand, followed by oxidative coupling of two dienes to form an allyl intermediate. Outer-sphere attack of ammonia on the allylpalladium intermediate then forms two isomeric primary amine products, 2 and 3 (Scheme 12). The ratio of the combination of the primary amines to the combination of secondary and tertiary amines was high (>90–95% in most cases).
Scheme 12.
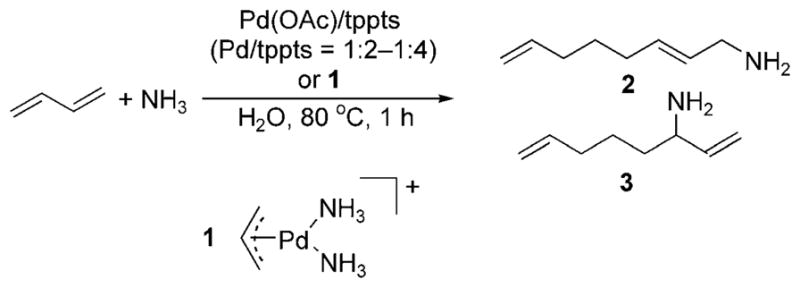
Telomerization of ammonia and butadiene.
3.2. Hydroamination with Ammonia
Hydroamination is the addition of the N–H bond of an amine across an unsaturated C–C bond. The transformation is thermodynamically favorable for the reaction of ammonia with ethylene to form ethylamine but is less favorable for the reaction of ammonia or alkylamines with substituted alkenes.[10] Hydroaminations of alkenes with ammonia occurs with heterogeneous catalysts, but the reaction conditions require high temperatures and high pressures.[10]
The hydroamination of alkenes with ammonia has not been achieved, but Lavallo et al. reported the hydroamination of alkynes and allenes with ammonia in 2008.[22] Several alkynes and allenes reacted over 3.5 h at 160–175°C with excess ammonia in the presence of 5–10 mol% of a gold(I)–CAAC complex (CAAC = cyclic alkyl aminocarbene, Au cat., Scheme 13) to form imines, nitrogen heterocycles and allylamines. The type of product depended on the reagent; terminal alkynes formed ketimines, internal alkynes formed N-vinyl ketimines, and diynes reacted with ammonia to form pyrroles. Reactions of allenes formed mixtures of primary, secondary and tertiary allylic amines, the ratio of which depended on the ratio of ammonia to allene.
Scheme 13.
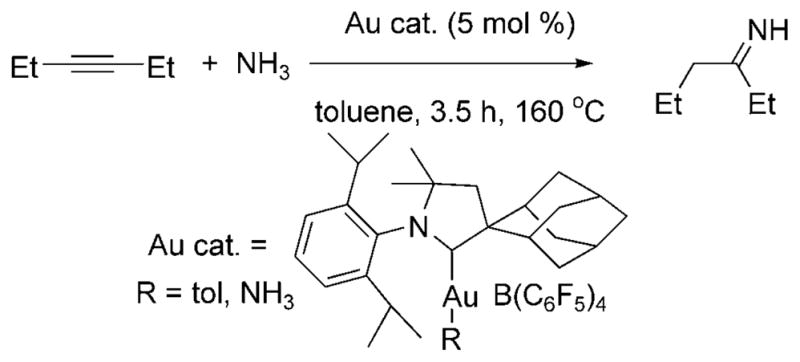
Au-catalyzed hydroamination of 3-hexyne with ammonia.
To probe the mechanism of the hydroamination of ammonia, the authors characterized a series of gold complexes formed in the presence of ammonia and 3-hexyne (Scheme 14). Reaction of the gold arene precursor 4 with 3-hexyne formed the cationic gold–alkyne complex 5. This complex was not stable in the presence of excess ammonia; the alkyne was replaced by ammonia to form the Werner-type gold–ammine complex 6. Consistent with this observation, the reagent alkyne did not displace the ammine. Rather, the reaction of the ammine complex with excess of 3-hexyne formed the gold–imine complex 7. This complex was proposed to form by intramolecular addition of the N–H bond across the alkyne, in part because this reaction occurred in an open system. Although the gold-catalyzed hydroamination of alkynes with ammonia is a novel transformation, practical applications of such a reaction requires that it occur with less reactive substrates, including alkenes, at lower temperatures, and with greater control of selectivity for primary amine products.
Scheme 14.

Au–NH3 and Au–alkyne complexes in hydroamination.
4. Catalytic Reactions of Ammonia Occurring through Metal–Amido Complexes
Parent amido complexes (M-NH2) of late transition metals are rare, and few catalytic reactions have been discovered that proceed through this type of intermediate. In the one known example of a catalytic reaction occurring through a parent amido complex, an arylpalladium halide ligated by an electron-rich bisphosphine reacts with ammonia and base to form such a species.[23] This amido complex then undergoes reductive elimination to form the arylamine product. A number of examples of palladium-catalyzed arylations of ammonia have been reported,[23–27] and an amido complex of palladium is presumed to be an intermediate in all of these reactions. Several copper-catalyzed cross-couplings of aryl halides with ammonia have also been reported.[28–31] Because monomeric amido complexes of copper have been documented,[32] a Cu–NH2 intermediate could be formed during these catalytic reactions. However, the formation of such a species during these processes and the reactivity of such intermediates have not yet been reported.
4.1. Palladium-Catalyzed Coupling of Ammonia with Aryl Halides
The coupling of haloarene electrophiles with ammonia to produce monoarylamines in the presence of copper catalysts has been conducted on an industrial scale at high temperatures and pressures.[33] However, the formation of hydrodehalogenated arenes, biaryl compounds, and constitutional isomers limit the utility of these protocols.[33] To avoid these undesired side products, more recent cross-coupling reactions of aryl halides with ammonia surrogates have been developed.[34–36] These reactions proceed in high yield under mild conditions, but the ammonia surrogates are more expensive than ammonia, and this approach requires deprotection of the initial coupling product.
In 2006, Shen and Hartwig published the first palladium-catalyzed coupling of aryl halides with ammonia.[23] Aryl chlorides, bromides, iodides, and triflates coupled with ammonia in high yield and with high selectivity for the monoarylamine product (Scheme 15) under 5.5 bar of ammonia pressure with NaOtBu base and 1 mol% of the preformed Josiphos-ligated palladium(II) complex [(CyPF-tBu)PdCl2] as catalyst. LiNH2 also coupled with aryl halides under similar reaction conditions, albeit with slightly lower selectivities for the monoarylamine. Shortly thereafter, Vo and Hartwig reported the formation of primary arylamines from reactions with the highly active, air-stable combination of [Pd(P-o-tol3)2] and the CyPF-tBu Josiphos ligand as catalyst. These reactions occurred with catalyst loadings as low as 0.1 mol% and just 5 equivalents of ammonia (Scheme 16).[26] Under optimized conditions, a wider variety of substituents on the aryl halide were tolerated than in the initial report, including base-sensitive functional groups. Reactions of aryl chlorides occurred with this catalyst, and reactions of relatively stable aryl tosylates occurred for the first time.
Scheme 15.
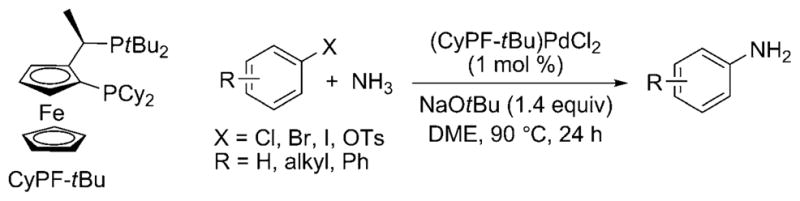
Pd-catalyzed coupling of ammonia and aryl halides. Cy = cyclohexyl; Ts = p-toluenesulfonyl; DME = dimethoxyethane.
Scheme 16.
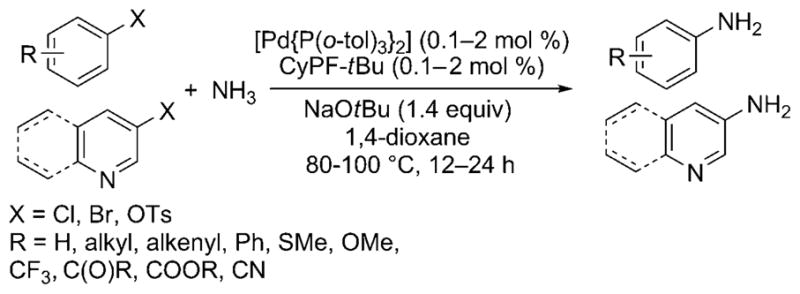
Improved conditions for Pd-catalyzed ammonia arylation.
In 2007, Buchwald and co-workers reported a similar transformation catalyzed by the combination of [Pd2(dba)3] (dba = dibenzylideneacetone) and the biarylphosphane ligand in Scheme 17.[24] Five substrates—chlorobenzene, 3,5-di-tert-butyl bromobenzene, 2-phenylbromobenzene, and two protected bromophenols—were converted to the corresponding arylamines. The selectivity for the monoarylamine remained high when the reactions were conducted with low pressures of ammonia. The authors also demonstrated that slight changes in the reaction conditions, namely, the concentration of substrate and equivalents of ammonia, could bias the reaction toward formation of di- and triarylamines. Evaporation of excess ammonia from the reaction vessel allowed for a one-pot synthesis of unsymmetrical di- and triarylamines.
Scheme 17.
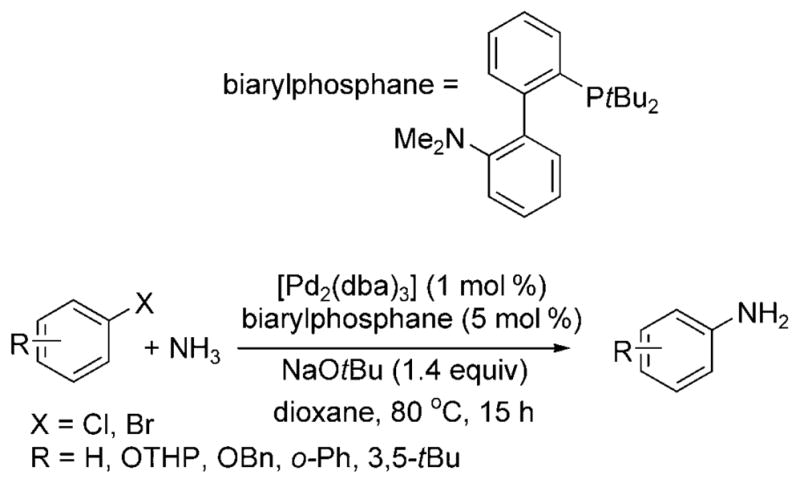
Pd-catalyzed arylation of ammonia with biarylphosphane ligand. OTHP = tetrahydropyranyl ether.
In 2009, Beller et al. also disclosed a palladium-catalyzed protocol for the arylation of ammonia with aryl chlorides.[25] The modular imidazole-based monophosphine ligands formed robust, air-stable catalysts with palladium that coupled ammonia with numerous aryl bromides and chlorides in high yields with high selectivity for the primary arylamine (Scheme 18). The method was conducted with the same 0.5M solution in dioxane reported by Buchwald. However, these reactions were conducted at the high reaction temperature of 140°C, and a 4:1 ratio of ligand to palladium (2–4 mol% palladium) was needed for high yields with less reactive substrates. In addition, a high pressure of an inert gas was used with the ammonia.
Scheme 18.
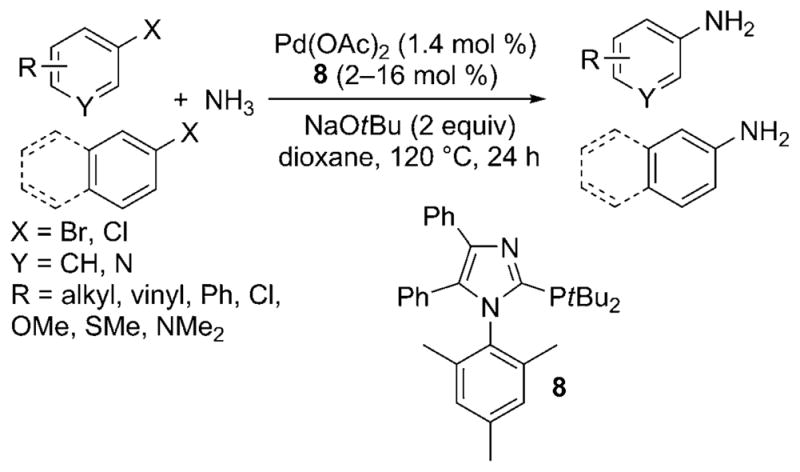
Imidazole-based ligands for Pd-catalyzed cross-coupling of ammonia with aryl bromides and chlorides.
More recently, Stradiotto and co-workers have developed a catalyst for the cross-coupling of amines and ammonia with aryl chlorides and tosylates.[27] This catalyst contains a structurally simple P,N phenylene ligand, Mor-DalPhos. With a modest catalyst loading, the monoarylation of ammonia proceeded at low pressures and temperatures as low as room temperature with high selectivity for the monoarylamine (Scheme 19). The substrate scope encompasses a variety of aryl chlorides, including those with both electron-rich and electron-poor ortho, meta and para substituents. However, the reaction is limited to only a few electron-neutral or sterically bulky aryl tosylates, and reactions of substrates with base-sensitive functional groups were not reported.
Scheme 19.
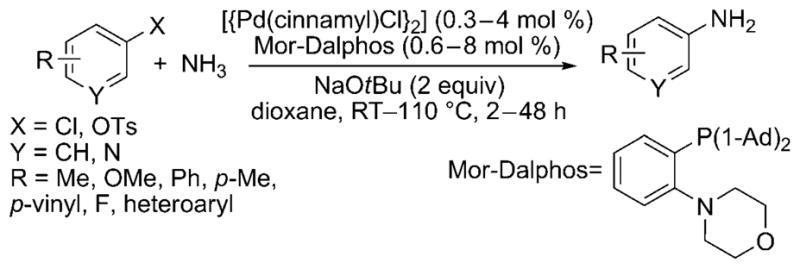
Mor-Dalphos-ligated Pd catalyst for amination of aryl chlorides and tosylates with ammonia. 1-Ad = 1-adamantyl, Ts = toluene-4-sulfonyl.
4.2. Copper-Catalyzed Coupling of Ammonia with Aryl Halides and Boronic Acids
Copper salts have been used for many years as reagents or catlysts for Ullmann and Goldberg reactions of aryl halides with amines and other nitrogen nucleophiles.[33] These systems are attractive because of their low cost. Traditional strategies for arylamine syntheses with copper require stoichiometric amounts of the metal and high temperatures,[33] and, until recently, ammonia was rarely used as the nucleophile in these processes.
In 2001, researchers at Merck reported the first copper-catalyzed arylation of ammonia at low pressures and temperatures.[37] The reaction proceeded with excellent selectivity for the monoarylamine with a ligandless copper oxide catalyst in an 8M solution of ammonia in ethylene glycol at 80°C (Scheme 20). The scope of these reactions was largely limited to electron-poor heteroaromatic halides, such as halopyridines, halothiazoles, and haloquinolines, and electron-poor arenes, such as 4-bromobenzophenone and 1-bromo-4-tri-fluoromethylbenzene. Aryl- and heteroaryl chlorides did not react. The factors controlling selectivity for formation of primary vs secondary amines are not clear. 2-Bromopyridine gave a 4:1 ratio of primary to secondary amine, while 3-bromo and 4-bromopyridine gave 20:1 to 30:1 ratios of the two products.
Scheme 20.
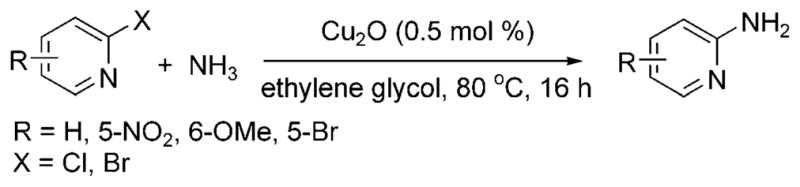
Coupling of heteroaryl halides with ammonia catalyzed by ligandless Cu.
Subsequent reports have focused on expanding the scope of the reaction and developing systems that tolerate milder reaction conditions. Kim and Chang showed that a combination of CuI and L-proline with solid NH4Cl or aqueous ammonia and K2CO3 in a solvent consisting of 5% water in DMSO led to the amination of aryl iodides and activated aryl bromides at room temperature (Scheme 21).[38] Electron-poor aryl iodides reacted to give the highest arylamine yields, especially those with nitro, alkoxycarbonyl, and trifluoro-methyl substituents. However, 2-methyliodobenzene and 4-iodoanisole were less reactive under these conditions, yielding only 7% and 32% of the expected arylamine products, respectively. Electron-poor aryl bromides formed the primary amine product, but the electron-rich 4-bromoanisole formed the monoarylamine product in only ca. 40% yield after extended reaction times.
Scheme 21.
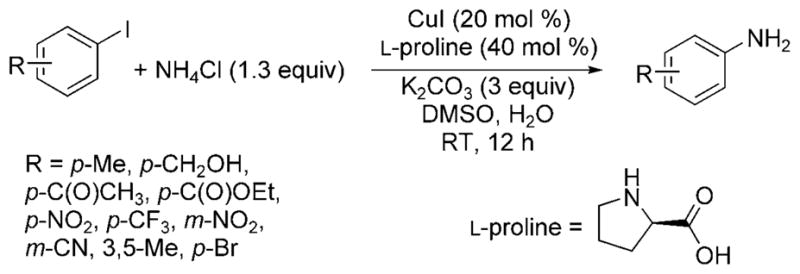
Amination of aryl iodides at room temperature catalyzed by Cu and L-proline.
Taillefer and Xia showed that these less reactive substrates couple with ammonia in the presence of [Cu(acac)2], the readily available 2,4-pentadione as ligand, and Cs2CO3 as a base (Scheme 22).[29] In the presence of this catalyst, a variety of aryl iodides, as well as unactivated aryl bromides, coupled with aqueous ammonia in DMF at elevated temperatures to form monoarylamines.
Scheme 22.
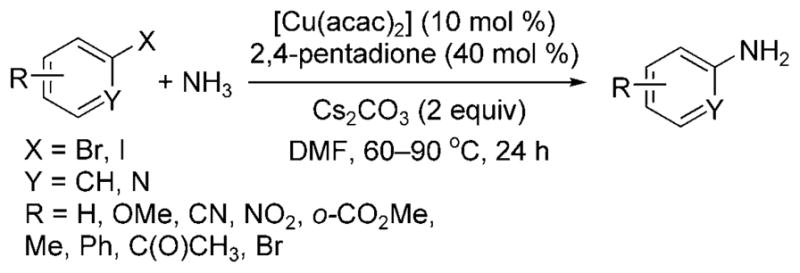
Arylation of ammonia with aryl bromides and iodides catalyzed by Cu and 2,4-pentadione. acac = acetyl acetonate.
Subsequently, Wang et al. reported copper-catalyzed coupling of aryl iodides with an aqueous solution of ammonia at room temperature.[30] The combination of 5 mol% CuBr and a 2-pyridinyl-β-ketone as ligand precursor (9, Scheme 23) with K3PO4 in DMSO led to the isolation of monoarylamines from aryl iodide substrates containing electron-rich, electron-poor, ortho, and heteroaryl substitutents. In a few cases, a temperature of 80°C was required to achieve optimum yields.
Scheme 23.

Room-temperature reaction of aryl iodides with ammonia catalyzed by CuI and 2-pyridinyl-β-ketone.
Wolf and Xu further simplified the copper-catalyzed cross-coupling with aqueous ammonia by introducing a ligandless process that operated under air and required no additional base additives.[28] With 5 mol% of Cu2O in aqueous ammonia and NMP (N-methylpyrrolidine), aryl iodides, and bromides containing a large range of functional groups, as well as bulky ortho substituents, were reported to be converted into the corresponding monoarylamines with high selectivity (Scheme 24). Even aryl chlorides were shown to be viable substrates for the cross-coupling reaction but required microwave heating for acceptable conversion and yield.
Scheme 24.
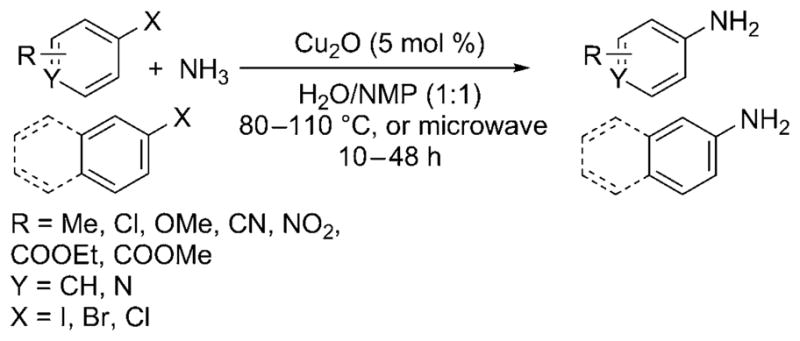
Amination of aryl iodides, bromides, and chlorides with ammonia catalyzed by ligandless Cu.
Wu and Darcel have published a set of related data and introduced the coupling of aryl iodides with aqueous ammonia catalyzed by a combination of iron and copper (Scheme 25).[31] In contrast to the report by Wolf and Xu[28] on the amination of aryl halides with ligandless copper oxide, Wu and Darcel stated that a maximum yield of 30% of the monoarylamine product was observed for reaction of iodo-benzene with CuI as catalyst in the absence of an iron cocatalyst. The reaction proceeded with 10 mol% of both FeIII and CuI salts in air in ethanol solvent with added sodium hydroxide. Both electron-poor and electron-rich aryl iodides formed the arylamine in high yields, but aryl bromides did not form arylamines under the reaction conditions.
Scheme 25.
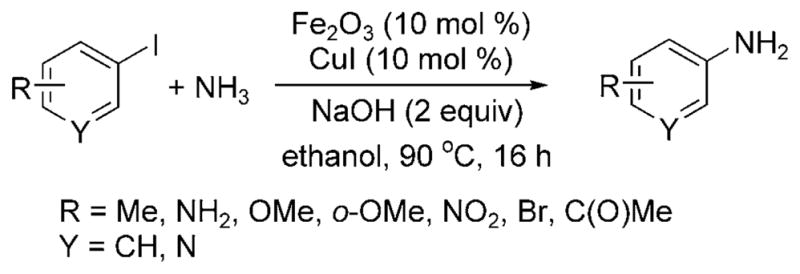
Arylation of ammonia with aryl iodides co-catalyzed by Cu and Fe.
The copper-catalyzed amination with ammonia has also been used in the synthesis of an aniline-containing variant of ifenprodil, a molecule that binds to the NMDA receptor in the nervous system.[39] A number of methods were explored for the coupling of aryl bromide 10 with aqueous ammonia to produce 11, but the best results were achieved with 10 mol% of CuI and 60 mol% of ligands 12 or 13. With added Cs2CO3 in DMF, the reaction completed in 18 h with a 65% yield of 11 (Scheme 26).
Scheme 26.

Cu-catalyzed amination in the synthesis of an ifenprodil analogue.
To expand the scope of copper-catalyzed cross-coupling with ammonia further, Rao et al. reported the conversion of boronic acids to monoarylamines by reaction of aqueous ammonia with 10 mol% of a ligandless Cu2O catalyst at room temperature under air (Scheme 27a).[40] Electron-rich, electron-poor, and even more sterically demanding boronic acids were all claimed to be reactive. Lower yields were obtained with the substrates containing electron-withdrawing substitutents, a trend opposite of that observed for copper-catalyzed coupling of ammonia with aryl halides.
Scheme 27.
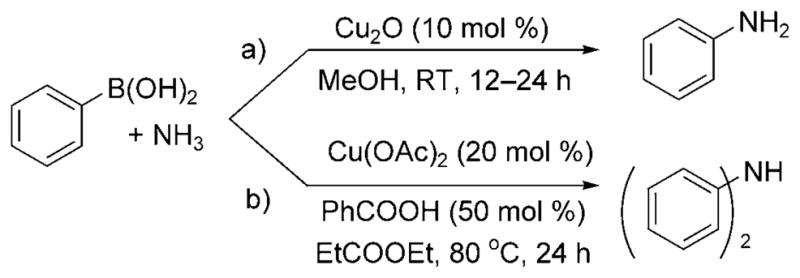
Cu-catalyzed coupling of ammonia with boronic acids.
Subsequent work indicates that this reaction is more complex than published. Zhou et al. demonstrated that reactions of boronic acids with aqueous solutions of ammonia and the copper(II) catalyst, Cu(OAc)2, generated exclusively diarylamine products (Scheme 27b).[41] Moreover, the reaction required an elevated temperature of 80°C and a benzoic acid additive to achieve acceptable yields of the diarylamines.
Thus, the copper-catalyzed coupling of ammonia with aryl halides and aryl boronic acids has been studied extensively, but a number of challenges remain. These challenges include identifying systems that react with high turnover numbers, identifying catalysts that react at lower temperatures, identifying systems that react in less toxic and more easily-separable solvents, and developing a system that reacts in general with ortho-substituted aryl halides and with chloroarenes.
5. Conclusions
Until recently, homogeneous reactions of ammonia catalyzed by transition-metal complexes were rare. Prior to the emergence of many of the reactions described in this Minireview, reactions of ammonia surrogates were used to prepare primary amines. Ammonia equivalents such as silylazides[35] or silazanes,[42] benzophenone imine,[34,36] sulfonamides,[43] and allyl-[36] or benzylamines[44] were used, but additional steps are then needed to generate the free primary amine. By developing catalytic reactions of ammonia itself, synthetic chemists can now access primary amines from alkenes, alcohols and aryl halides or psuedohalides. In some cases, relatively inexpensive combinations of metal complexes and ligand catalyze reactions of ammonia under mild conditions with little or no additional manipulations of functional groups. Much information remains to be gleaned about the reactivity of the amido complexes that are likely intermediates in some of these processes, but it is clear that the goal of demonstrating that reactions with ammonia can be catalyzed by late transition metals has been rapidly achieved.
Acknowledgments
We thank the NIH (NIGMS-55382) and the Department of Energy for support of our studies on the catalytic and fundamental chemistry of ammonia involving transition-metal complexes.
Biographies

John Hartwig received his AB from Princeton University conducting research with Maitland Jones, obtained his PhD from U.C. Berkeley with Bob Bergman and Richard Andersen, and conducted an American Cancer Society postdoctoral fellowship at MIT with Stephen Lippard. In 1992 he began his independent career at Yale University and became the Irenée P. DuPont Professor in 2004. In 2006, he moved to the University of Illinois and is the Kenneth L. Rinehart Jr. Professor of Chemistry. His group has contributed to the discovery and mechanistic analysis of several classes of cross-coupling reactions, C–H bond functionalization, olefin hydroaminations, and asymmetric allylic substitutions.

Jessica Klinkenberg received her BS from the University of Virginia in 2007 under the research direction of Cassandra Fraser. She is currently an NSF predoctoral fellow in the Hartwig group studying the mechanisms and methodologies of palladium-catalyzed reactions with ammonia.
References
- 1.M. Appl in Ullmann’s Encyclopedia of Industrial Chemistry. 7. Wiley; New York: Ammonia. online. [Google Scholar]
- 2.Schirmann J-P, Bourdauducq P. Ullmann’s Encyclopedia of Industrial Chemistry. 7. Wiley; New York: Hydrazine. online. [Google Scholar]
- 3.van Gysel AB, Musin W, editors. Ullmann’s Encyclopedia of Industrial Chemistry. 7. Wiley; New York: Methylamines. online. [Google Scholar]
- 4.E. I. du Pont de Nemours & Co. 2497310. United States: US Patent. 1950
- 5.Lin J-J, Knifton JF. 4794199. N. Texaco Inc. (White Plains); United States: US Patent. 1988
- 6.Zimmermann B, Herwig J, Beller M. Angew Chem. 1999;111:2515–2518. doi: 10.1002/(sici)1521-3773(19990816)38:16<2372::aid-anie2372>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1999;38:2372–2375. doi: 10.1002/(sici)1521-3773(19990816)38:16<2372::aid-anie2372>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 7.Tararov VI, Kadyrov R, Riermeier TH, Börner A. Chem Commun. 2000:1867–1868. [Google Scholar]
- 8.Gross T, Seayad AM, Ahmad M, Beller M. Org Lett. 2002;4:2055–2058. doi: 10.1021/ol0200605. [DOI] [PubMed] [Google Scholar]
- 9.Ogo S, Uehara K, Abura T, Fukuzumi S. J Am Chem Soc. 2004;126:3020–3021. doi: 10.1021/ja031633r. [DOI] [PubMed] [Google Scholar]
- 10.Roundhill DM. Chem Rev. 1992;92:1–27. [Google Scholar]
- 11.Lawrence SA. Amines: Synthesis Properties, and Application. Cambridge University Press; Cambridge: 2004. [Google Scholar]
- 12.Haniti M, Hamid SA, Slatford PA, Williams JMJ. Adv Synth Catal. 2007;349:1555–1575. [Google Scholar]
- 13.Yamaguchi R, Kawagoe S, Asai C, Fujita K-i. Org Lett. 2008;10:181–184. doi: 10.1021/ol702522k. [DOI] [PubMed] [Google Scholar]
- 14.Gunanathan C, Milstein D. Angew Chem. 2008;120:8789–8792. doi: 10.1002/anie.200803229. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:8661–8664. doi: 10.1002/anie.200803229. [DOI] [PubMed] [Google Scholar]
- 15.Oishi T, Yamaguchi K, Mizuno N. Angew Chem. 2009;121:6404–6406. doi: 10.1002/anie.200900418. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:6286–6288. doi: 10.1002/anie.200900418. [DOI] [PubMed] [Google Scholar]
- 16.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito: 2010. pp. 417–451. [Google Scholar]
- 17.a) Johannsen M, Jørgensen KA. Chem Rev. 1998;98:1689–1708. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]; b) Miyabe H, Takemoto Y. Synlett. 2005:1641–1655. [Google Scholar]; c) Lu Z, Ma SM. Angew Chem. 2008;120:264–303. [Google Scholar]; Angew Chem Int Ed. 2008;47:258–297. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 18.Pouy MJ, Leitner A, Weix DJ, Ueno S, Hartwig JF. Org Lett. 2007;9:3949–3952. doi: 10.1021/ol701562p. [DOI] [PubMed] [Google Scholar]
- 19.Nagano T, Kobayashi S. J Am Chem Soc. 2009;131:4200–4201. doi: 10.1021/ja900328x. [DOI] [PubMed] [Google Scholar]
- 20.Pouy MJ, Stanley LM, Hartwig JF. J Am Chem Soc. 2009;131:11312–11313. doi: 10.1021/ja905059r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prinz T, Driessen-Hölscher B. Chem Eur J. 1999;5:2069–2076. [Google Scholar]
- 22.Lavallo V, Frey GD, Donnadieu B, Soleilhavoup M, Bertrand G. Angew Chem. 2008;120:5302–5306. doi: 10.1002/anie.200801136. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:5224–5228. doi: 10.1002/anie.200801136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:10028–10029. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]
- 24.Surry DS, Buchwald SL. J Am Chem Soc. 2007;129:10354–10355. doi: 10.1021/ja074681a. [DOI] [PubMed] [Google Scholar]
- 25.Schulz T, Torborg C, Enthaler S, Schäffner B, Dumrath A, Spannenberg A, Neumann H, Börner A, Beller M. Chem Eur J. 2009;15:4528–4533. doi: 10.1002/chem.200802678. [DOI] [PubMed] [Google Scholar]
- 26.Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049–11061. doi: 10.1021/ja903049z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. Angew Chem. 2010;122:4165–4168. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:4071–4074. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]
- 28.Xu H, Wolf C. Chem Commun. 2009:3035–3057. doi: 10.1039/b904188e. [DOI] [PubMed] [Google Scholar]
- 29.Xia N, Taillefer M. Angew Chem. 2009;121:343–345. doi: 10.1002/anie.200802569. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:337–339. doi: 10.1002/anie.200802569. [DOI] [PubMed] [Google Scholar]
- 30.Wang DP, Cai Q, Ding K. Adv Synth Catal. 2009;351:1722–1726. [Google Scholar]
- 31.Wu XF, Darcel C. Eur J Org Chem. 2009:4753–4756. [Google Scholar]
- 32.a) Blue ED, Davis A, Conner D, Gunnoe TB, Boyle PD, White PS. J Am Chem Soc. 2003;125:9435–9441. doi: 10.1021/ja0353659. [DOI] [PubMed] [Google Scholar]; b) Goj LA, Blue ED, Munro-Leighton C, Gunnoe TB, Petersen JL. Inorg Chem. 2005;44:8647–8649. doi: 10.1021/ic0517624. [DOI] [PubMed] [Google Scholar]
- 33.Lindley J. Tetrahedron. 1984;40:1433–1456. [Google Scholar]
- 34.a) Wolfe JP, Ahman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron Lett. 1997;38:6367–6370. [Google Scholar]; b) Grasa GA, Viciu MS, Huang JK, Nolan SP. J Org Chem. 2001;66:7729–7737. doi: 10.1021/jo010613+. [DOI] [PubMed] [Google Scholar]; c) Mann G, Hartwig JF, Driver MS, Fernandez-Rivas C. J Am Chem Soc. 1998;120:827–828. [Google Scholar]
- 35.a) Lee DY, Hartwig JF. Org Lett. 2005;7:1169–1172. doi: 10.1021/ol050141b. [DOI] [PubMed] [Google Scholar]; b) Huang XH, Buchwald SL. Org Lett. 2001;3:3417–3419. doi: 10.1021/ol0166808. [DOI] [PubMed] [Google Scholar]
- 36.Jaime-Figueroa S, Liu YZ, Muchowski JM, Putman DG. Tetrahedron Lett. 1998;39:1313–1316. [Google Scholar]
- 37.Lang FR, Zewge D, Houpis IN, Volante RP. Tetrahedron Lett. 2001;42:3251–3254. [Google Scholar]
- 38.Kim J, Chang S. Chem Commun. 2008:3052–3054. doi: 10.1039/b804637a. [DOI] [PubMed] [Google Scholar]
- 39.Bouteiller C, Becerril-Ortega J, Marchand P, Nicole O, Barre L, Buisson A, Perrio C. Org Biomol Chem. 2010;8:1111. doi: 10.1039/b923255a. [DOI] [PubMed] [Google Scholar]
- 40.Rao HH, Fu H, Jiang YY, Zhao YF. Angew Chem. 2009;121:1134–1136. [Google Scholar]; Angew Chem Int Ed. 2009;48:1114–1116. doi: 10.1002/anie.200805424. [DOI] [PubMed] [Google Scholar]
- 41.Zhou CF, Chen F, Yang DP, Jia XF, Zhang LX, Cheng J. Chem Lett. 2009;38:708–709. [Google Scholar]
- 42.Barluenga J, Aznar F, Valdes C. Angew Chem. 2004;116:347–349. doi: 10.1002/anie.200352808. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:343–345. [Google Scholar]
- 43.a) Weihofen R, Dahnz A, Tverskoy O, Helmchen GN. Chem Commun. 2005:3541–3545. doi: 10.1039/b505197e. [DOI] [PubMed] [Google Scholar]; b) Weihofen R, Tverskoy E, Helmchen G. Angew Chem. 2006;118:5673–5676. doi: 10.1002/anie.200601472. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:5546–5549. doi: 10.1002/anie.200601472. [DOI] [PubMed] [Google Scholar]
- 44.Lim CW, Lee SG. Tetrahedron. 2000;56:5131–5136. [Google Scholar]