

Abstract
In the CNS, interleukin-1β (IL-1β) is synthesized and released during injury, infection, and disease, mediating inflammatory responses. However, IL-1β is also present in the brain under physiological conditions, and can influence hippocampal neuronal function. Several cell-specific IL-1-mediated signaling pathways and functions have been identified in neurons and astrocytes, but their mechanisms have not been fully defined. In astrocytes, IL-1β induced both the p38 MAPK and NF-κB (nuclear factor κB) pathways regulating inflammatory responses, however in hippocampal neurons IL-1β activated p38 but not NF-κB. Additionally, IL-1β induced Src phosphorylation at 0.01 ng/ml in hippocampal neurons, a dose 1000-fold lower than that used to stimulate inflammatory responses. IL-1 signaling requires the type 1 IL-1 receptor and the IL-1 receptor accessory protein (IL-1RAcP) as a receptor partner. We previously reported a novel isoform of the IL-1RAcP, IL-1RAcPb, found exclusively in CNS neurons. In this study, we demonstrate that AcPb specifically mediates IL-1β activation of p-Src and potentiation of NMDA-induced calcium influx in mouse hippocampal neurons in a dose-dependent manner. Mice lacking the AcPb, but retaining the AcP, isoform were deficient in IL-1β regulation of p-Src in neurons. AcPb also played a modulatory role in the activation of p38 MAPK, but had no effect on NF-κB signaling. The restricted expression of AcPb in CNS neurons, therefore, governs specific neuronal signaling and functional responses to IL-1β.
Introduction
Interleukin-1 (IL-1) is a proinflammatory cytokine that is expressed in multiple cell types in the brain (Breder et al., 1988; Hunter et al., 1992; Pitossi et al., 1997; Sims and Smith, 2010). Its expression is particularly high during injury and neurodegenerative disease (Loddick et al., 1998), as it plays a major role in coordinating the inflammatory response to these conditions. IL-1β is rapidly synthesized and released, primarily by microglia (Giulian et al., 1986; Pearson et al., 1999) and astrocytes (Davies et al., 1999; Pearson et al., 1999). Under pathophysiological conditions, IL-1 mediates inflammatory activity by stimulating release of additional proinflammatory cytokines and growth factors from astrocytes and microglia (Merrill and Benveniste, 1996). In the hippocampus, high concentrations of IL-1 act upon neurons to inhibit synaptic strength and long-term potentiation (LTP) (Katsuki et al., 1990; Bellinger et al., 1993; Murray and Lynch, 1998). In contrast, physiological levels of IL-1 promote LTP and memory formation (Yirmiya et al., 2002). The distinct functions of IL-1 are mediated through the same type 1 IL-1 receptor (IL-1RI), which is expressed and regulated by IL-1 in both hippocampal neurons and astrocytes (Friedman, 2001). However, the signaling pathways for IL-1 in these two cell types differ (Srinivasan et al., 2004). The precise mechanism governing the cell type-specific IL-1 signaling is unknown and remains to be investigated. Two classical downstream signaling pathways are activated following IL-1 binding to its receptors (Ninomiya-Tsuji et al., 1999). One pathway involves phosphorylation and degradation of the IκB subunit of NF-κB (nuclear factor κB), leading to the release and nuclear translocation of the active NF-κB transcription factor and subsequent gene expression. Another pathway involves activation of the three MAPKs, ERK, JNK, and P38, and additional transcription factors such as ATF (activating transcription factor) and AP1, leading to nuclear gene transcription (O'Neill et al., 1990; Zhang et al., 1996; Ninomiya-Tsuji et al., 1999). Among these, NF-κB and p38 MAPK are known to be activated by IL-1β in hippocampal astrocytes, but only p38 MAPK is activated in neurons (Srinivasan et al., 2004). In addition, an alternative signaling mechanism involving Src kinase has been reported to be activated by IL-1β in hippocampal neurons (Viviani et al., 2003).
IL-1R accessory protein (IL-1RAcP) is a required receptor partner in IL-1 signaling (Cullinan et al., 1998) and is expressed ubiquitously. AcPb, a novel isoform of IL-1RAcP, was recently identified and is mainly expressed in CNS neurons (Smith et al., 2009), but its precise function in mediating neuronal responses to IL-1 remains unclear. Here, we investigated IL-1β-mediated cell type-specific signaling pathways in hippocampal neurons and astrocytes, and the role of AcPb in each pathway. We demonstrate that AcPb governs neuron-specific functional responses to IL-1β involving Src phosphorylation and enhanced Ca2+ influx in response to NMDA receptor activation.
Materials and Methods
Animals.
Mice lacking either all AcP isoforms (total AcP−/−) or only the AcPb isoform (AcPb−/−) were generated as previously described (Smith et al., 2009) and maintained on a fully backcrossed C57BL/6 background. Briefly, the original total AcP knock-out mice were generated by deletion of exons 4 and 5, encoding crucial extracellular domains, and insertion of a neomycin sequence; therefore, both AcP and AcPb were functionally knocked out. AcPb-specific knock-out mice were generated by replacing the variant AcPb-associated exon 12b with a neomycin resistance gene, while the upstream 12 exons were left intact to maintain expression of normal full-length AcP (Smith et al., 2009). All mice were genotyped using primers covering either the inserted neomycin or the knocked out region before being mated. All animal studies were conducted using the NIH guidelines for the ethical treatment of animals with approval of the Rutgers Animal Care and Facilities Committee.
Astrocyte culture.
Pregnant mice were killed by exposure to CO2 and soaked in 80% ethanol for 10 min. Embryonic day 19 (E19) fetuses of either sex were removed under sterile conditions and kept in PBS on ice. Hippocampi were dissected, dissociated by trituration, and plated on poly-d-lysine (0.1 mg/ml)-precoated flasks in NM15 medium (Eagle's MEM with Earle's salts and 2 mm l-glutamine, 15% heat-inactivated fetal bovine serum, 6 mg/ml glucose, 0.5 μg/ml penicillin and 0.5 U/ml streptomycin). Astrocytes were isolated by shaking flasks on an orbital shaker at 450 rpm for 10 min. Medium was replaced with fresh NM15 and the flasks were returned to the incubator for 2 h before being shaken overnight at 225 rpm to remove neurons, microglia and oligodendrocytes. The confluent astrocytes were exposed to cytosine arabinoside (0.1 mm) for 3 d to eliminate any remaining non-astrocyte cell populations. Finally, the astrocytes were trypsinized and replated at subconfluent density. The purity of astrocyte cultures was >99%, confirmed by immunostaining.
Neuronal culture.
E16 hippocampi were dissected from mice of either sex, dissociated, and plated in serum-free medium on poly-d-lysine (0.01 ng/ml)-precoated plates. The medium consists of 1:1 mixture of Eagle's MEM (Invitrogen) and Ham's F12 supplemented with glucose (6 mg/ml), insulin (25 μg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5U/ml) and streptomycin (0.5 μg/ml). Neurons were cultured for 5 d in serum-free medium and subjected to cytokine treatments. The purity of neuronal cultures was >95%, confirmed by immunostaining.
Total RNA isolation and PCR: RNA was isolated with TRIzol reagent (Invitrogen) as recommended by the manufacturer. Briefly, 1 × 106 cells were lysed in 1 ml of TRIzol reagent, followed by chloroform extraction and isopropanol precipitation. The RNA was resuspended in nuclease-free water and quantified spectrophotometrically at 260/280 nm. One microgram of RNA was reverse transcribed at 42°C for 2 h in a 20 μl reaction mixture using SuperScript II reverse transcriptase (Invitrogen). cDNA was amplified and quantified by regular PCR and real-time PCR. The following primer sequences were used: IL1R (forward: 5′-CTACTTGGGTTCATTTGTCTCATTGTGCC-3′, reverse: 5′-TTCCACTTCCAGTAGACAAGGTCGGTGAAC-3′); mouse AcP-specific primers (forward 5′-TGTTTCCTATGCAAGAAATGTGGAAGAAGAGG-3′, rev: 5′-TGCTTGTCATTGCTAGACCACCTGG-3′) and mouse AcPb-specific primers (forward: 5′-TGTTTCCTATGCAAGAAATGTGGAAGAAGAGG-3′, rev: 5′-ATGGGGTTGCTCAAGCGACGGTACTCCAC-3′); mouse IL-6 (forward 5′-GGAGAGGAGACTTCACAGAGG-3′, reverse: 5′-GGAAATTGGGGTAGGAAGGA-3′); actin (forward: 5′-TCATGAAGTGTGACGTTGACATCCGT-3′, reverse: 5′-CTTAGAAGCATTTGCGGTGCACGATG-3′); GAPDH (forward: 5′-TTCTTGTGCAGTGCCAGCC-3′, reverse: 5′-CACCGACCTTCACCATCTTGT-3′). PCRs were performed in a 20 μl volume with goTaq Green Master Mix (Promega) and 1 μm primers according to manufacturer's recommendation. PCR conditions for IL1R, AcP and AcPb were: 95°C 5 min; 95°C 30 s, 50°C 30 s, 72°C 30 s for 35 cycles; and 72°C 7 min. Reaction conditions for IL-6, actin, and GAPDH were identical except that the annealing temp was 60°. PCR products were resolved on 1% agarose gel. For real-time PCR, Roche 480 SYBR Green Mastermix was used according to the manufacturer's recommendation.
Western blots.
Cells were harvested in lysis buffer containing 120 mm Tris, pH 6.8, 2% SDS, and 10% glycerol supplemented with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail (Roche). Proteins were quantified using the Bradford assay (Bio-Rad). Equal amounts of protein were run on SDS polyacrylamide gels and transferred to nitrocellulose membrane. The blots were blocked in 5% nonfat milk in TBST for 1 h, and incubated with primary antibody (1: 1000 in 5% milk in TBST) overnight at 4°C. The primary antibodies are anti-p-IκB α (Cell Signaling Technology, catalog #9246), anti-IκB α (Cell Signaling Technology, catalog #9242), anti-p-p38 (Thr 180/Tyr 182) (Cell Signaling Technology, catalog #9215), anti-p38 (Cell Signaling Technology, catalog #9212), anti-p-Src (Tyr 416) (Cell Signaling Technology, catalog #2101), anti-Src (Cell Signaling Technology, catalog #2110), anti-p-NR2B (Cell Signaling Technology catalog #4208), anti-NR2B (Cell Signaling Technology catalog #4212) anti-β actin (Sigma catalog #A5441). The blots were then washed 3 times with TBST, and incubated with secondary antibody (goat anti-mouse, or goat anti-rabbit labeled with horseradish peroxidase, Thermo Fisher Scientific, 1:5000) for 1 h at room temperature. The membranes were developed using the Pierce ECL Western blot substrate kit (Thermo Fisher Scientific). Blots were stripped in 62.5 mm Tris-HCl ph6.8, 2%SDS and 100 mm β-mercaptoethanol at 65°C for 5 min. Quantification of immunoblots was performed on scanned images of the films using Adobe Photoshop software.
Immunoprecipitation/coimmunoprecipitation-Western blot analysis.
Cultured neurons or astrocytes were lysed in TNE buffer (50 mm Tris, pH 7.3, 137 mm NaCl, 2 mm EDTA) containing 0.2% Triton, 60 mm octylglucoside, and Protease Inhibitor Cocktail. Proteins were quantified using the Bradford assay (Bio-Rad). Protein (500 μg) was incubated with primary antibody anti-AcP/b M215 (10 ng/ml, Amgen) overnight on a rocking platform at 4°C. Protein G-Sepharose was added to the lysates and kept for an additional 3 h at 4°C. The lysates were then centrifuged at 14,000 rpm for 15 min and washed with TNE buffer three times and distilled water once. The pellets were resuspended in 1× loading buffer with β-mercaptoethanol. The samples were then run by SDS-PAGE, transferred to nitrocellulose membrane, and probed using anti-AcP P2 anti-rabbit antibody (1:1000, Amgen) which detects both AcP and AcPb. Astrocytes transfected with AcP or AcPb plasmids were used as positive controls.
Cell transfection.
Primary mouse astrocytes were transfected using the Mouse Astrocyte Nucleofector kit (Amaxa Biosystems), program T-20 by Amaxa according to the manufacturer's instructions. In brief, 2–4 million astrocytes were suspended in 100 μl of supplemented Nucleofector solution in each transfection. The cell suspension was then mixed with 3–5 μg of AcP, AcPb, empty vector DNA plasmids, 3–5 μg of κB-driven-luciferase plasmid and/or 1 μg of GFP plasmid (Amaxa Biosystems). Within 15 min the cell suspension was transfected with program T-20. Cells were then resuspended with warm NM-15 medium and replated on poly-d-lysine precoated dishes. Two hours after transfection, fresh NM-15 medium was applied to the cultures. One to 3 d after transfection, cells were exposed to IL-1β treatment (generously provided by Dr. Ronald Hart, Department of Cell Biology and Neuroscience, Rutgers University, Newark, NJ).
Luciferase activity assay.
Twenty-four hours after transfection, astrocytes were treated with IL-1β (10 ng/ml) for 4 h. Protein lysates were then collected in reporter lysis buffer (Promega, E 4030). Protein lysates were centrifuged at 14,000 rpm for 2 min at 4°C and supernatants were stored at −80°C. Lysates were warmed at room temperature for 30 min and equal amount of protein was mixed with luciferase substrate and subjected to luminescence reading immediately.
Immunocytochemistry.
Primary neurons and astrocytes were cultured in poly-d-lysine-precoated-Lab-tek chamber slide (Nunc). Cells were treated with IL-1β (10 ng/ml) for 2 h, followed by fixation with 4% paraformaldehyde for 30 min. Fixed cells were blocked in PBS/10% goat serum and permeabilized with PBS/0.3%Triton X-100, then exposed to anti-p65 (1:500, Santa Cruz Biotechnology) overnight at 4°C in PBS. Slide wells were then washed 3 in PBS, exposed for 1 h at room temp to goat anti-rabbit secondary antibodies coupled to Alexa 555 (1:500). After secondary antibody, cells were washed 3 times in PBS, followed by DAPI (10 μg/ml, Invitrogen) application. Cells were then coverslipped with anti-fading medium (ProLong Gold, Invitrogen) and imaged on a Nikon TE200 Inverted Epifluorescent microscope.
Calcium imaging.
Hippocampal neurons were cultured on poly-d-lysine (0.1 mg/ml)-coated glass coverslips in SFM for 12 d before calcium imaging. Neurons were maintained in an extracellular medium (ECM) containing 33 mm glucose in Hanks' balanced salt solution buffer (in mmol/L: 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4) at 37°C during imaging. Fluorescent imaging measurement of intracellular Ca2+ ([Ca2+]i) was performed using fura-2 acetoxymethyl ester (Teflabs). Hippocampal neurons were loaded with 2.5 μm fura-2 acetoxymethyl ester in ECM for 20 min (37°C). Cells were washed twice with ECM to remove free fura-2 and transferred to a thermostatically regulated microscope chamber (37°C). Fura-2 fluorescent images were acquired every 5 s by alternating excitation at 340 and 380 nm, and emissions (420–600 nm) were collected using a cooled, charge-coupled device camera. For each experiment, baseline was established by recording for 4 min, followed with or without IL-1 treatment for 6 min. NMDA (10 μm) was then applied for 7 min before washing for 1–4 min. At the end of each recording, KCl (30 mm) was applied to ensure calcium influx and cell viability. Only cells that responded to KCl were picked for quantification. Changes in [Ca2+]I fluctuation in response to NMDA were assessed for 7 min after NMDA application by calculating the integrated area under curve (AUC) using Origin 7.0 software (OriginLab).
Statistical analysis.
All data are expressed as mean ± SEM of three or more independent experiments conducted on different culture preparations. Student's t test was used to analyze two part comparisons. One-way ANOVA followed by Tukey–Kramer post hoc tests were used for analysis of groups of three or more. For all statistical analyses, a value of p < 0.05 was considered significant.
Results
AcPb is expressed in hippocampal neurons but not astrocytes
Previous reports from our laboratory indicated that IL-1RI was present in both neurons and astrocytes from the rat hippocampus (Friedman, 2001), and that IL-1β stimulated different signaling pathways in these two distinct cell types (Srinivasan et al., 2004). IL-1RAcP is a known, required receptor partner in IL-1 signaling (Cullinan et al., 1998). A novel isoform of AcP, termed AcPb, that is expressed exclusively in CNS neurons was recently identified. AcPb was able to form a complex with IL-1 and the IL-1 receptor and was found to abrogate some gene expression induced by IL-1 in cortical neuron cultures (Smith et al., 2009). Thus, we investigated whether AcPb was responsible for the distinct IL-1β signaling seen in neurons and astrocytes.
To characterize the role of AcPb in IL-1β signaling, we first examined the expression of IL-1RI and AcP/b in mouse neurons and astrocytes. In accordance with previous results in the rat, IL-1RI mRNA was detected by reverse transcriptase-PCR in mouse hippocampal neurons and astrocytes (Fig. 1A). The mRNA levels of AcP and AcPb were determined by PCR analysis using primers designed specifically against the last two exons. AcP was detected in both neurons and astrocytes, but AcPb was found only in neurons (Fig. 1A). Real-time PCR quantification showed that hippocampal neurons express both AcP and AcPb at approximately equal, but low, levels (Fig. 1B). In contrast, AcPb mRNA was not detected in astrocytes, and AcP levels were 7 times above that in neurons. To examine protein expression of AcP isoforms, immunoprecipitation (IP)-Western blot analysis was performed with anti-pan-AcP antibodies that recognize both AcP and AcPb proteins at different sizes. Only AcP was detected in astrocytes, however both AcP and AcPb proteins were present in neurons, but very low levels (Fig. 1C), consistent with the relatively low levels of AcP and AcPb mRNA in neurons. It is not clear why the AcP/b proteins migrate slightly differently in neurons and astrocytes, but differential glycosylation may underlie some of the differences as reported by Smith et al. (2009).
Figure 1.

Expression of IL-1R1, AcP and AcPb in primary mouse hippocampal neurons and astrocytes. A, Reverse transcriptase-PCR amplification of AcP, AcPb and IL-1RI mRNAs from hippocampal neurons and astrocytes, β actin was used as control. NTC was control without cDNA for PCR. B, Relative expression of AcP and AcPb mRNA levels in hippocampal neurons and astrocytes quantified by real-time PCR. All mRNAs are expressed relative to the neuronal expression level of AcP, which was normalized to 1, using actin as an internal control (data from three different experiments with triplicate loading each). C, IP-Western blots for AcP and AcPb in hippocampal neurons and astrocytes. Protein (50 μg) from astrocytes transfected with AcP or AcPb was used as positive control in the left two lanes. A total of 500 μg of protein from primary neurons or astrocytes was immunoprecipitated with a pan-AcP monoclonal antibody and immunoblots were analyzed with a pan-AcP antiserum.
AcPb does not influence NF-κB activation by IL-1 in hippocampal neurons and astrocytes
The NF-κB signaling pathway is known to be activated by IL-1β in hippocampal astrocytes but not in neurons (Srinivasan et al., 2004). Our previous study showed that expression of AcPb in addition to AcP inhibited IL-1-regulated gene induction compared with expression of AcP alone (Smith et al., 2009), suggesting that the presence of AcPb might inhibit IL-1β-mediated activation of NF-κB. Therefore we examined whether IL-1β could activate NF-κB in the absence of AcPb.
Cultured astrocytes were prepared from wild-type, total AcP−/−, and AcPb−/− mice. Cultures were treated with IL-1β and probed for p-IκB and total IκB by Western blot analysis. In WT astrocytes, IκB was phosphorylated within 5–10 min, accompanied by degradation of total IκB (Fig. 2A). Total AcP deletion in astrocytes abolished IκB phosphorylation and degradation induced by IL-1β. However, astrocytes from AcPb−/− mice showed a similar IκB response compared with WT, consistent with the lack of AcPb expression in astrocytes. Since astrocytes only express AcP, this suggests that only classic AcP but not AcPb was required for NF-κB activation in astrocytes.
Figure 2.
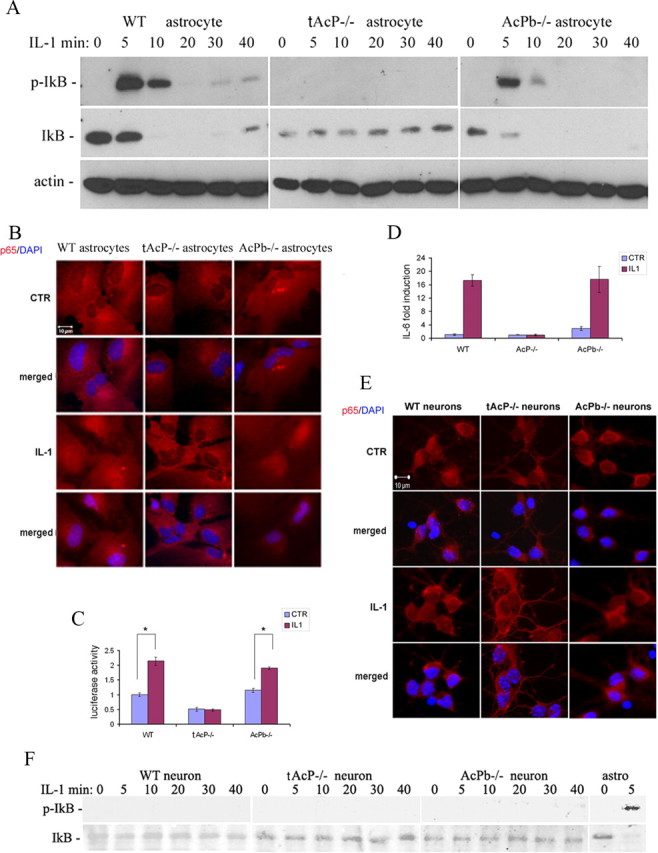
AcPb does not regulate NF-κB signaling. A, IL-1β induced phosphorylation of IκB in WT and AcPb−/− astrocytes but not total AcP−/− astrocytes. Cultured hippocampal astrocytes were treated with IL-1β (10 ng/ml) for the indicated times and lysates were analyzed by Western blot for phospho-IκB, stripped and reprobed for total IκB and β actin. The blot shown is representative of three independent experiments. B, p65 NF-κB subunit translocated to the nucleus in WT and AcPb−/− astrocytes but not AcP−/− astrocytes. Astrocytes were treated with IL-1β (10 ng/ml) for 2 h, fixed with 4%PFA, and immunostained for p65 (red). Cell nuclei were labeled with DAPI (blue). C, κB-luciferase activity is induced by IL1β in WT and AcPb−/−astrocytes, but not AcP−/− astrocytes. Astrocytes were transfected with κB-driven luciferase reporter plasmids. Twenty-four hours after transfection, cells were treated with IL-1β (10 ng/ml) for 4 h. Protein lysates were analyzed for luciferase activity. Data are from four different experiments with triplicate loading each. Student's t test was used to evaluate data from cells treated with or without IL-1β. *p < 0.01. D, IL-6 mRNA was induced by IL1β (10 ng/ml) in WT and AcPb−/− astrocytes but not total AcP−/− astrocytes by real-time PCR analysis. Graph depicts 6 h IL-1β treatment. mRNA levels were normalized to wild-type control. E, No nuclear translocation of p65 was induced by 2 h IL-1β (10 ng/ml) in hippocampal neurons. F, No phosphorylation of IκB was detected in WT, total AcP−/− or AcPb−/− neurons in response to IL-1β (10 ng/ml). p-IκB in astrocytes was used as positive control. The blot shown is representative of three independent experiments.
NF-κB subunits p65 and p50, have previously been shown to translocate to the nucleus following IL-1β stimulation in astrocytes (Friedman et al., 1996). To confirm that NF-κB was translocated to nuclei after IκB phosphorylation, cultured astrocytes were immunostained for the NF-κB p65 subunit and imaged using fluorescence microscopy. Nuclear translocation of the p65 subunit was observed in WT and AcPb−/− astrocytes, but not in total AcP−/− astrocytes (Fig. 2B), confirming the role of AcP but not AcPb in activating NF-κB in astrocytes.
To further confirm the role of AcP in mediating IL-1-induced NF-κB activity in astrocytes, WT, total AcP−/− and AcPb−/− astrocytes were transfected with a κB-driven-luciferase plasmid. Twenty-four hours after plasmid transfection, astrocytes were treated with IL-1β (10 ng/ml) for 4 h, and equal amounts of protein were used for luciferase assay. Luciferase activity was induced by IL-1β in WT astrocytes; this induction was abolished in total AcP−/− astrocytes but remained unchanged in AcPb−/− astrocytes (Fig. 2C), confirming the essential role of classical AcP, but not AcPb, in IL-1β-induced NF-κB activity in astrocytes.
IL-1β induces expression of a number of genes via activation of NF-κB, including IL-6 (Libermann and Baltimore, 1990) and NGF (Friedman et al., 1996). To determine the roles of AcP and AcPb in IL-1β-mediated signaling in hippocampal astrocytes, real-time PCR was used to quantify IL-6 mRNA expression from WT, total AcP−/− or AcPb−/− astrocytes treated with or without IL-1β (10 ng/ml) for 6 h. IL-1β elicited a 17-fold induction of IL-6 in WT astrocytes, and the absence of total AcP completely abolished this induction (Fig. 2D). Consistent with our previous findings, IL-6 induction by IL-1β in AcPb−/− astrocytes was similar to WT astrocytes.
In contrast to astrocytes, no p65 nuclear translocation (Fig. 2E) or IκB phosphorylation (Fig. 2F) was induced by IL-1β in hippocampal neurons of any genotype, including the AcPb−/−, indicating that removing AcPb had no effect on this pathway.
Neurons lacking AcPb did not show P-IκB and activation of NF-κB in response to IL-1β, suggesting that the presence of AcPb was not the determining factor for the absence of NF-κB induction by IL-1β in hippocampal neurons. To confirm that the presence of this protein did not suppress activation of NF-κB, we examined whether overexpression of AcPb in astrocytes would inhibit IκB phosphorylation. AcPb was transfected into WT astrocytes and no dramatic effect was observed on IL-1β-induced IκB phosphorylation (Fig. 3), indicating that AcPb does not play a crucial role in the regulation of NF-κB by IL-1β.
Figure 3.

Transfection of AcPb into WT astrocytes does not inhibit IκB phosphorylation induced by IL-1β. WT astrocytes were transfected with AcPb or an empty vector. Forty-eight hours after transfection, cells were treated with IL-1β (10 ng/ml) for 5 min. Protein lysates were immunoblotted for p-IκB and AcPb, then stripped and reprobed for IκB and actin as control. The blot shown is representative of four independent experiments.
Role of AcPb in IL-1β-induced p38 MAPK signaling in hippocampal neurons and astrocytes
In WT astrocytes, p38 phosphorylation was strongly induced by IL-1β (Fig. 4A). No phosphorylation of p38 was induced by IL-1β in total AcP−/− astrocytes, but astrocytes lacking only AcPb were similar to WT, indicating an essential role for AcP but not AcPb in IL-1β-induced-p38 signaling in astrocytes, as with NF-κB.
Figure 4.
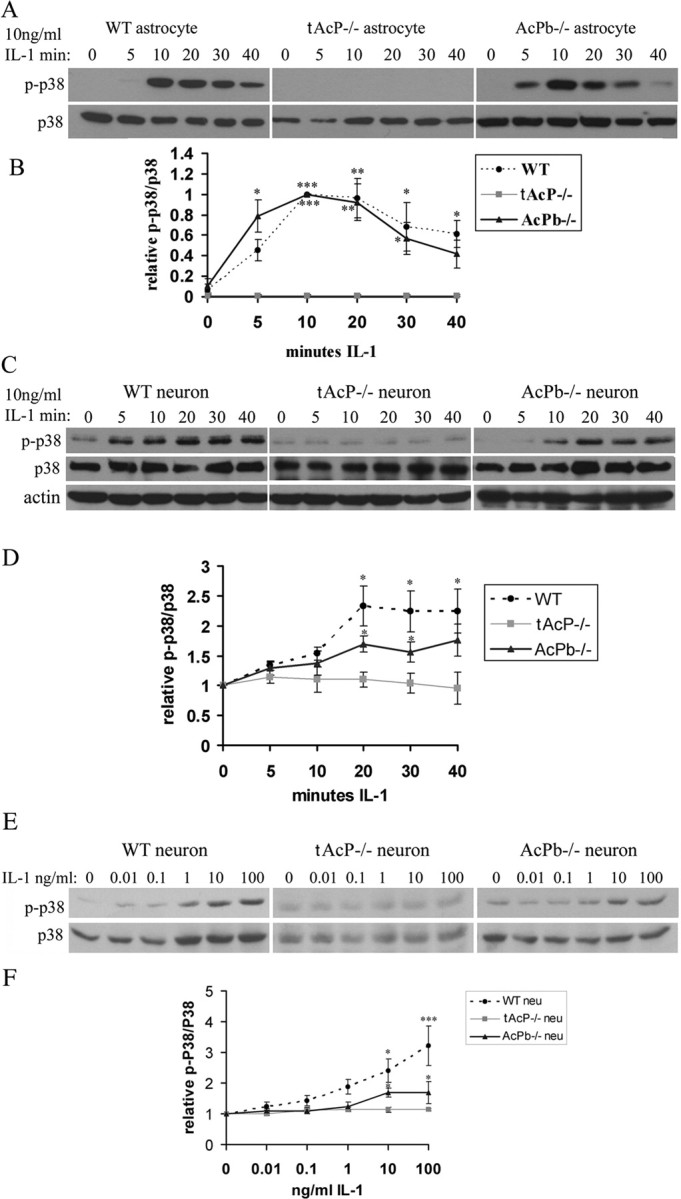
IL-1β induced p38 phosphorylation in hippocampal astrocytes and neurons. A, Time course of IL-1β (10 ng/ml)-induced p38 phosphorylation in WT, total AcP−/−, and AcPb−/− astrocytes. B, Quantification of blots from four different experiments as in A. Densitometric values were expressed as p-p38/total p38, and then normalized to the treatment at 10 min (peak value). P-p38 was not detected in control samples, therefore the values were set at 0. C, Time course of IL-1β (10 ng/ml)-induced p38 phosphorylation in WT, total AcP−/−, and AcPb−/− neurons. D, Quantification of blots from four different experiments as in C. Densitometric values were expressed as p-p38/total p38, and then normalized to untreated cells (0 min) for each genotype. E, Dose–response of IL-1β-induced p38 phosphorylation after 20 min in WT, total AcP−/−, and AcPb−/− neurons. F, Quantification of blots from three different experiments as in E. Densitometric values were expressed as p-p38/total p38, and then normalized to untreated cells (0 ng/ml IL-1) for each genotype. The significance was determined by one-way ANOVA with Tukey's post hoc analysis. The p value equals to 0.0023 between WT and AcPb−/− neurons treated with 100 ng/ml IL-1. Asterisks indicate values significantly different from control at *p < 0.05, **p < 0.001, and ***p < 0.0001.
Consistent with previous studies from our lab using rat neurons (Srinivasan et al., 2004), p38 phosphorylation was induced by IL-1β at 5 min and was maximal by 20 min in WT mouse neurons (Fig. 4C). Eliminating total AcP abolished this induction, while eliminating AcPb weakened but did not eliminate this regulation (Fig. 4C,D). A dose-dependent induction of p-p38 by IL-1β was also observed in WT and AcPb−/− neurons, but not total AcP−/− neurons (Fig. 4E,F). The weakening of IL-1β-induced p38 activation in the AcPb−/− neurons suggests that AcPb may play a modulatory role in regulating p38 activation by IL-1β in brain neurons.
Src phosphorylation by IL-1 in neurons requires AcPb
An atypical IL-1β signaling pathway that regulates Src phosphorylation has been reported in hippocampal neurons induced by low doses of IL-1β (0.05 ng/ml) (Viviani et al., 2003). To investigate a possible role for AcPb in regulating this pathway, cultured E16 mouse hippocampal WT neurons were exposed to IL-1β at different doses for 20 min and examined for p-Src (Tyr416). IL-1β induced Src phosphorylation at a low dose (0.01 ng/ml), but not at higher doses (Fig. 5A). To test whether AcPb plays a role in IL-1-induced Src activation, WT, total AcP−/− and AcPb−/− neurons were treated with IL-1β (0.01 ng/ml/) for different time periods. This low dose of IL-1β induced Src phosphorylation in WT neurons within 5 min, peaked at 20 min and stayed stable for at least 40 min. Similar to the other pathways examined, removal of total AcP eliminated IL-1β activation of Src (Fig. 5B). However, in contrast to the other pathways investigated, the absence of AcPb alone was sufficient to abrogate the induction of p-Src by IL-1β (Fig. 5B), despite the continued presence of AcP, suggesting that AcPb was necessary for IL-1β to activate p-Src.
Figure 5.

Src phosphorylation by IL-1β (0.01 ng/ml) in hippocampal neurons is dependent on AcPb. A, Dose–response treatment of IL-1β applied to cultured WT mouse hippocampal neurons for 20 min. Protein lysates were immunoblotted for p-Src and reprobed for total Src. Quantification of blots from three different experiments is shown below the blot. Densitometric values were expressed as p-Src/total Src, and then normalized to untreated cells (0 ng/ml). B, Time course of Src phosphorylation induced by IL-1β in mouse hippocampal neurons. At low concentration (0.01 ng/ml), Src is phosphorylated by IL-1β in WT neurons, but not in total AcP−/− or AcPb−/− neurons. Quantification of blots from three different experiments is shown below the blot. The significance was determined by one-way ANOVA with Tukey's post hoc analysis. Asterisks indicate values significantly different from control at *p < 0.05, ***p < 0.0001.
Expression of exogenous AcPb in astrocytes induces Src phosphorylation by IL-1
To determine whether p-Src is activated by IL-1β in astrocytes that do not express AcPb, WT astrocytes were treated with IL-1β at different doses and different time points. None of these treatments with IL-1β induced Src phosphorylation in WT astrocytes (Fig. 6A). However, when WT astrocytes were transfected with AcPb, IL-1β was able to activate p-Src (Fig. 6A), demonstrating that expression of AcPb in WT astrocytes is sufficient to induce p-Src.
Figure 6.

Effects of overexpressing AcPb in astrocytes on IL-1β-induced p-Src and p-p38 signaling. A, Time course of IL-1β (0.01 ng/ml) treatment of WT astrocytes with and without AcPb transfection. Expression of AcPb in WT astrocytes enabled IL-1β to induce Src phosphorylation. B, IL-1 (0.01 ng/ml)-induced p-Src in total AcP−/− astrocytes restored with AcPb but not AcP. In contrast, restoration of AcP, but not AcPb was sufficient to restore IL-1β induction of p-p38 MAPK, even at the low dose. C and D show quantification of p-Src and p-p38 MAPK, respectively, from blots from three different experiments. Densitometric values were expressed as p-Src/total Src (C) or p-p38/total p38 (D), and then normalized to untreated cells (0 min). The significance was determined by one-way ANOVA with Tukey's post hoc analysis. Asterisk indicates values significantly different from control at p < 0.05. E, High dose of IL-1 (10 ng/ml)-induced p-p38 in total AcP−/− astrocytes restored with either AcP or AcPb, but only induced p-IκB in total AcP−/− astrocytes restored with AcP. F, Quantification of p-p38 induction in the tAcP−/− astrocytes transfected either with AcP or AcPb and treated with the high dose (10 ng/ml) of IL-1 for 5 min. Note in B and E that although equal amounts of plasmid DNA for AcP or AcPb were used for transfections, expression of AcPb is always lower than AcP. Thus, when normalized to the amount of AcP or AcPb expressed, the level of p-p38 activation was similar.
Since WT astrocytes express AcP, transfection of AcPb yields expression of both isoforms, as in neurons. To assess whether AcPb alone would be sufficient for IL-1β to induce p-Src, we examined total AcP−/− astrocytes transfected with AcP alone or AcPb alone. Restoration of AcPb alone, but not AcP alone, in AcP−/− astrocytes was sufficient for IL-1β (0.01 ng/ml) to induce p-Src (Fig. 6B,C). In addition, the same experiments showed that this low dose of IL-1β (0.01 ng/ml) induced p-p38 in astrocytes expressing AcP alone but not AcPb alone (Fig. 6B,D), confirming the role of these distinct accessory protein isoforms in activating distinct signaling events. We further examined whether the high dose (10 ng/ml) of IL-1β was able to induce p-p38 or p-IκB in astrocytes expressing AcPb alone. When total AcP−/− astrocytes were reconstituted with AcPb alone, a high dose of IL-1β (10 ng/ml) induced p-p38, although to a lesser degree when compared with astrocytes expressing AcP alone (Fig. 6E). However, expression of exogenously transfected AcPb was always lower than AcP, despite use of the same amount of plasmid, therefore we normalized the relative p-P38/p38 to the level of AcP or AcPb protein, and found no significant difference for IL-1β (10 ng/ml) activation of p-p38 after normalization (Fig. 6F). In contrast, AcPb alone was completely unable to restore activation of p-IκB by IL-1β (Fig. 6E), while AcP restored robust activation of P-IκB. Thus, replacement of AcP with AcPb reconstituted the ability to activate p38 MAPK but did not restore NF-κB signaling in astrocytes.
AcPb is required for IL-1-potentiated calcium influx induced by NMDA in hippocampal neurons
A functional consequence of IL-1β-induced Src activation is to phosphorylate the NMDA receptor NR2B subunit, which colocalizes with IL-1RI in hippocampal neurons, resulting in potentiation of calcium influx induced by NMDA (Viviani et al., 2003). The same low dose of IL-1 that evoked Src phosphorylation elicited a sharp peak in phosphorylation of NR2B in WT hippocampal neurons after 5 min of treatment (Fig. 7). To assess whether the effects of IL-1β on Src and NR2B phosphorylation might regulate calcium influx, preincubation of hippocampal neurons with IL-1β (0.01 ng/ml) potentiated NMDA (10 μm)-induced calcium influx (Fig. 8A,D), consistent with previous reports. In addition, preexposing neurons to the Src kinase inhibitor, PP2 (2.5 μm), blocked this effect. Consistent with our signaling results on Src phosphorylation, higher doses of IL-1β (10 ng/ml) had no effect on NMDA-induced calcium influx (Fig. 8A,D). To determine whether AcPb was necessary for the IL-1β effect on NMDA-induced calcium influx, WT, total AcP−/− and AcPb−/− neurons were compared. Removal of total AcP (tAcP−/−) eliminated the IL-1β potentiation of calcium influx induced by NMDA, as expected, however removal of AcPb alone (AcPb−/−) was sufficient to abrogate the IL-1 effect (Fig. 8B,C), suggesting that AcPb is required for IL-1β-induced Src phosphorylation and potentiation of NMDA-mediated calcium influx.
Figure 7.

IL-1β (0.01 ng/ml) induced transient phosphorylation of NR2B within 5 min in WT neurons. Graph shows quantification of two independent experiments.
Figure 8.

AcPb is required for IL-1β (0.01 ng/ml)-potentiated calcium influx induced by NMDA in hippocampal neurons. A–C, Representative changes in [Ca2+]i measured by Fura-2 in response to IL-1β and NMDA in WT, total AcP−/− and AcPb−/− neurons. Data are shown as the ratio of 340/380 fluorescence emission. A, WT neurons were pretreated with or without 0.01 ng/ml IL-1β, PP2 + 0.01 ng/ml IL-1β, or 10 ng/ml IL-1β before the addition of NMDA. B, C, AcPb−/− neurons (B) or total AcP−/− neurons (C) were pretreated with or without IL-1β (0.01 ng/ml) before NMDA addition. Baseline recordings were established for 4 min followed by treatment with or without IL-1β for 6 min. NMDA (10 μm) was applied for 7 min and then washed away. At the end of each recording, KCl (30 mm) was applied to ensure calcium response and cell viability. D, Quantification of NMDA-induced calcium influx from 3 to 5 individual cultures. Area under the curve (AUC) from each group was normalized to WT neuron control, which was set at 100%. Total number of neurons analyzed for a given group is as follows: WT neuron, control (CTR), 300; WT neuron, 0.01 ng/ml IL-1β, 377; WT neuron, PP2 + 0.01 ng/ml IL-1β, 125; WT neuron, 10 ng/ml IL-1β, 73; total AcP−/− neuron, CTR, 155; total AcP−/− neuron, 0.01 ng/ml IL-1β, 158; AcPb−/− neuron, CTR, 315; AcPb−/− neuron, 0.01 ng/ml IL-1β, 271.
Discussion
AcPb in neuronal IL-1β signaling and function
IL-1 is a proinflammatory cytokine that influences many mammalian functions. IL-1 binds the IL-1RI/IL-1RAcP receptor complex, however distinct signaling cascades have been described in different cell types with no mechanistic explanation. Within the CNS IL-1β activates different signaling pathways in hippocampal neurons and astrocytes (Viviani et al., 2003; Srinivasan et al., 2004). The discovery of AcPb, a new CNS-restricted isoform of AcP (Smith et al., 2009), provides a novel mechanism governing distinct responses to IL-1β in CNS neurons. Mice lacking either total AcP or only AcPb were used to define neuron-specific effects on IL-1 signaling and function. Although the germ-line deletions may have developmental effects that influence IL-1 signaling, these studies revealed a specific role for AcPb in the regulation of Src activation and NMDA function.
IL-1RAcPb and Src
Src family kinases (SFKs) can phosphorylate ion channels (Salter and Kalia, 2004) and directly modulate neuronal responses, bypassing the need for de novo gene expression. Src activity has long been implicated in regulating neuronal long-term potentiation (LTP) (Lu et al., 1998; Salter, 1998; Huang et al., 2001). SFKs can phosphorylate NMDA receptors and regulate their cell surface trafficking. Phosphorylation of the NR2B subunit has been specifically associated with trafficking to synaptic sites (Goebel-Goody et al., 2009). We found that both Src and NR2B were phosphorylated in hippocampal neurons by a low dose of IL-1β (0.01 ng/ml), suggesting that IL-1 may facilitate trafficking of NMDA receptors to synaptic sites resulting in the enhanced NMDA-induced Ca2+ influx that we observed. Consistent with these data, IL-1 was shown to phosphorylate SFKs and NR2B in vivo, eliciting hyperexcitability and potentiation of kainic acid-induced seizures (Balosso et al., 2008). Our current study demonstrates that AcPb specifically mediates Src activation by IL-1.
AcPb differs from AcP with a 140 aa extension in the C-terminal region. Interestingly, a related protein, IL-1-receptor accessory protein-like 1 (IL1RAPL1), also has an extended C-terminal region, which interacts with the neuronal calcium sensor-1 protein (NCS-1) to regulate N-type calcium channels (Gambino et al., 2007). IL1RAPL1 also associates with the major excitatory postsynaptic density protein, PSD-95 that plays a role in the development and organization of dendritic spines. Mutations in IL1RAPL1 lead to a decrease in the number of spines and subtle alterations in hippocampal LTP (Pavlowsky et al., 2010), and have been associated with mental retardation and autism (Piton et al., 2008). Although the C-terminal extension in AcPb has no apparent similarity to the analogous region of IL1RAPL1, the presence of this extended domain, and the enhancement of NMDA-induced calcium influx, suggests a similar role for AcPb in modulating excitatory neurotransmission.
Dose-dependent actions of IL-1 signaling in neurons
IL-1 has previously been shown to have a biphasic effect on hippocampal LTP (Goshen et al., 2007). Physiologically low levels of IL-1 (1–10 pm; Desson and Ferguson, 2003) facilitate hippocampus-dependent memory formation (Yirmiya et al., 2002), possibly by inducing Src activity followed by rapid phosphorylation of the NMDA receptor. Moreover, blocking IL-1 signaling impairs normal maintenance of LTP and memory consolidation (Avital et al., 2003; Spulber et al., 2009). These effective doses of IL-1 actions are below the reported Kd for the IL-1R1; however, the presence of AcP and AcPb in the receptor complex yields a lower Kd than IL-1R1 alone. Moreover, previous studies have demonstrated functional effects of IL-1 at doses below the measured Kd (Bomsztyk et al., 1989). In contrast, at pathophysiological levels, IL-1 inhibits LTP and impairs novel memory formation (Murray and Lynch, 1998) in part via activation of the p38 MAPK pathway (Kelly et al., 2003). Consistent with this, the high dose of IL-1β (10 ng/ml) failed to elicit Src activation and NR2B phosphorylation, but did induce p38 MAPK phosphorylation in neurons, which required both AcP and AcPb. Deletion of AcPb weakened, but did not suppress, induction of p-p38 in neurons, suggesting that AcPb partially contributes to activation of this pathway. Moreover, in astrocytes lacking total AcP, overexpression of either AcP alone or AcPb alone restored IL-1β-induced p38 phosphorylation.
The mechanisms and functional consequences for the distinct neuronal responses to IL-1β at physiological and pathophysiological doses remain unclear. One possible consequence of Src activation by IL-1β at low concentrations may be to constitutively maintain a certain level of neuronal responsiveness. Several studies have suggested a physiological role for IL-1β (1 pm or 0.017 ng/ml) in enhancing neuronal excitability (Viviani et al., 2003; Desson and Ferguson, 2003). However, the increased excitability may also explain why preinjection of low doses of IL-1β prolonged bicuculline-induced seizures in mice (Vezzani et al., 2000). Thus, it is likely that at low concentrations IL-1β induces p-Src to maintain a certain level of neuronal excitability, but may also increase vulnerability to insults.
In vivo, IL-1β does not directly induce neuronal death, but can exacerbate injury-induced neuronal damage (Stroemer and Rothwell, 1998). In our system, treatment of hippocampal neurons with high concentrations of IL-1β (10–100 ng/ml) induced phosphorylation of p38, but not JNK or ERK, followed by activation of the transcription factor CREB (Srinivasan et al., 2004). CREB regulates expression of numerous genes, including calcium channels, AMPA receptors, and neurotransmitters. Interestingly, we found that IL-1β also induced phosphorylation of p38 at the low dose, but the role of this pathway remains unclear. It does not necessarily require AcPb, and it is not neuron specific. p38 MAPK has been implicated in mediating neuronal death induced by glutamate, and inhibiting p38 can attenuate neuronal death (Cao et al., 2004). Therefore, the activation of this pathway even with the low dose of IL-1β may contribute to increased neuronal vulnerability to subsequent insults.
AcPb does not regulate NF-κB
Since IL-1β activates NF-κB in many cell types, but not hippocampal neurons, one possibility was that AcPb suppressed activation of this pathway. However, neurons lacking AcPb did not show NF-κB activation, and transfection of AcPb into astrocytes did not suppress NF-κB activation, suggesting that the expression of AcPb does not explain the inability of IL-1 to induce NF-κB signaling in neurons. Previous results from our lab showed that TRAF6, a key adaptor protein in IL-1-mediated NF-κB activation (Cao et al., 1996b), was abundant in hippocampal astrocytes but not detected in neurons (Srinivasan et al., 2004). TRAF6 interacts with IRAK (Cao et al., 1996a), and activates IκB kinases through TAK1, NIK, and MEKK1 in response to IL-1 (Lee et al., 1997; Ninomiya-Tsuji et al., 1999), suggesting an essential role for TRAF6 in the NF-κB pathway. Further, dominant-negative TRAF6 blocked NF-κB activation induced by IL-1β but not by TNFα (Cao et al., 1996b), consistent with our studies showing that TNFα but not IL-1β activated NF-κB in hippocampal neurons (Choi and Friedman, 2009). Thus, it is likely the absence of TRAF6, rather than the presence of AcPb, that explains why IL-1β does not induce NF-κB in hippocampal neurons.
The role of AcPb versus AcP
The complexities of IL-1 signaling to elicit the broad spectrum of functional effects are not fully understood. The association of the receptor with a specific accessory protein is one mechanism for the activation of distinct pathways, particularly in brain neurons expressing both AcP and AcPb. We show here that low concentrations of IL-1β elicit Src phosphorylation via AcPb, while higher concentrations activate p38, but not Src, requiring AcP. It is possible that the higher IL-1 concentrations elicit a more stable IL1R/AcP complex that may allow an association-dependent conformational change to occur which interferes with formation of the AcPb-dependent signaling complex. Alternatively, it is possible that the higher concentration of IL-1β leads to the formation of other signaling complexes on the receptor heterodimer over time that sterically prevent association of the Src phosphorylating complex.
AcP can associate with other receptors of the IL-1R family, such as the IL-33 receptor ST2, and the orphan receptor IL-1 receptor-related protein 2 (IL-1Rrp2). It was possible that AcPb might associate with one of these other receptors to modulate signaling in neurons. However, PCR analysis revealed ST2 expression in astrocytes but not neurons (data not shown), consistent with a previous report (Andre et al., 2005). Similarly, IL-1Rrp2 is expressed in brain glia but not neurons (Berglöf et al., 2003). Thus, it is unlikely that the neuron-specific AcPb interacts with either of these other potential coreceptors in these cells.
The link between IL-1R1 and Src in neurons has not previously been identified, but our results indicate that it requires AcPb. Since the extended C-terminal domain of the AcPb protein is unique, it may recruit distinct adapter proteins that activate Src instead of the classical MAPK or NF-κB pathways, which may explain why overexpression of AcPb, even in astrocytes from total AcP−/− mice, was able to induce p-Src. Indeed, biochemical studies have shown that AcPb associates with IL-1RI in a ligand-dependent manner (Smith et al., 2009), but fails to recruit MyD88 and IRAK4, resulting in failure to activate the classical IL-1 signaling pathways including the JNK and ERK MAPK pathways (Smith et al., 2009), as well as NF-κB, as shown here.
IL-1 is a pleiotropic cytokine that can activate multiple signaling pathways in different cell types with distinct functional consequences. The discovery of AcPb, a novel, neuron-specific IL-1 signaling protein, provides mechanistic insight into cell-specific IL-1 signaling and function in the brain.
Footnotes
This work was supported by the New Jersey Commission for Brain Injury Research and NIH/NINDS Grant 1R21NS076867. D.E.S. and J.E.S. are employees and shareholders of Amgen Corporation. We thank Barry Levin and Ambrose Dunn-Meynell for assistance with the calcium imaging, and Matthew Wilkins for excellent technical assistance.
References
- Andre R, Lerouet D, Kimber I, Pinteaux E, Rothwell NJ. Regulation of expression of the novel IL-1 receptor family members in the mouse brain. J Neurochem. 2005;95:324–330. doi: 10.1111/j.1471-4159.2005.03364.x. [DOI] [PubMed] [Google Scholar]
- Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, Yirmiya R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13:826–834. doi: 10.1002/hipo.10135. [DOI] [PubMed] [Google Scholar]
- Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, Vezzani A. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. 2008;131:3256–3265. doi: 10.1093/brain/awn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR. Interleukin 1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- Berglöf E, Andre R, Renshaw BR, Allan SM, Lawrence CB, Rothwell NJ, Pinteaux E. IL-1Rrp2 expression and IL-1F9 (IL-1H1) actions in brain cells. J Neuroimmunol. 2003;139:36–43. doi: 10.1016/s0165-5728(03)00130-9. [DOI] [PubMed] [Google Scholar]
- Bomsztyk K, Stanton TH, Smith LL, Rachie NA, Dower SK. Properties of interleukin-1 and interferon-γ receptors in B lymphoid cell line. J Biol Chem. 1989;264:6052–6057. [PubMed] [Google Scholar]
- Breder CD, Dinarello CA, Saper CB. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science. 1988;240:321–324. doi: 10.1126/science.3258444. [DOI] [PubMed] [Google Scholar]
- Cao J, Semenova MM, Solovyan VT, Han J, Coffey ET, Courtney MJ. Distinct requirements for p38alpha and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem. 2004;279:35903–35913. doi: 10.1074/jbc.M402353200. [DOI] [PubMed] [Google Scholar]
- Cao Z, Henzel WJ, Gao X. IRAK: A kinase associated with the interleukin-1 receptor. Science. 1996a;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996b;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Choi S, Friedman W. Inflammatory cytokines IL-1β and TNF-α regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN Neuro. 2009;1:pii, e00010. doi: 10.1042/AN20090009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan EB, Kwee L, Nunes P, Shuster DJ, Ju G, McIntyre KW, Chizzonite RA, Labow MA. IL-1 receptor accessory protein is an essential component of the IL-1 receptor. J Immunol. 1998;161:5614–5620. [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1999;19:87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- Desson SE, Ferguson AV. Interleukin 1beta modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J Physiol. 2003;550:113–122. doi: 10.1113/jphysiol.2003.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Thakur S, Seidman L, Rabson AB. Regulation of nerve growth factor mRNA by interleukin-1 in rat hippocampal astrocytes is mediated by NFkappaB. J Biol Chem. 1996;271:31115–31120. doi: 10.1074/jbc.271.49.31115. [DOI] [PubMed] [Google Scholar]
- Gambino F, Pavlowsky A, Béglé A, Dupont JL, Bahi N, Courjaret R, Gardette R, Hadjkacem H, Skala H, Poulain B, Chelly J, Vitale N, Humeau Y. IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca2+-channel and neurite elongation. Proc Natl Acad Sci U S A. 2007;104:9063–9068. doi: 10.1073/pnas.0701133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ, Shih LC, Lachman LB. Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med. 1986;164:594–604. doi: 10.1084/jem.164.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32:1106–1115. doi: 10.1016/j.psyneuen.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- Hunter CA, Roberts CW, Murray M, Alexander J. Detection of cytokine mRNA in the brains of mice with toxoplasmic encephalitis. Parasite Immunol. 1992;14:405–413. doi: 10.1111/j.1365-3024.1992.tb00015.x. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1B inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1β on long-term potentiation in rat dentate gyrus. J Biol Chem. 2003;278:19453–19462. doi: 10.1074/jbc.M301938200. [DOI] [PubMed] [Google Scholar]
- Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loddick SA, Liu C, Takao T, Hashimoto K, De Souza EB. Interleukin-1 receptors: cloning studies and role in central nervous system disorders. Brain Res Brain Res Rev. 1998;26:306–319. doi: 10.1016/s0165-0173(97)00037-4. [DOI] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1B is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase Tak1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Bird TA, Saklatvala J. Interleukin 1 signal transduction. Immunol Today. 1990;11:392–394. doi: 10.1016/0167-5699(90)90155-3. [DOI] [PubMed] [Google Scholar]
- Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M, Valnegri P, Rezai X, Bassani S, Brambilla D, Kumpost J, Blahos J, Roux MJ, Humeau Y, Chelly J, Passafaro M, Giustetto M, Billuart P, Sala C. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol. 2010;20:103–115. doi: 10.1016/j.cub.2009.12.030. [DOI] [PubMed] [Google Scholar]
- Pearson VL, Rothwell NJ, Toulmond S. Excitotoxic brain damage in the rat induces interleukin-1beta protein in microglia and astrocytes: correlation with the progression of cell death. Glia. 1999;25:311–323. [PubMed] [Google Scholar]
- Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, Champagne N, Lafrenière RG, Hamdan FF, Joober R, Fombonne E, Marineau C, Cossette P, Dubé MP, Haghighi P, Drapeau P, Barker PA, Carbonetto S, Rouleau GA. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet. 2008;17:3965–3974. doi: 10.1093/hmg/ddn300. [DOI] [PubMed] [Google Scholar]
- Pitossi F, del Rey A, Kabiersch A, Besedovsky H. Induction of cytokine transcripts in the central nervous system and pituitary following peripheral administration of endotoxin to mice. J Neurosci Res. 1997;48:287–298. doi: 10.1002/(sici)1097-4547(19970515)48:4<287::aid-jnr1>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Salter MW. Src, N-methyl-d-aspartate (NMDA) receptors, and synaptic plasticity. Biochem Pharmacol. 1998;56:789–798. doi: 10.1016/s0006-2952(98)00124-5. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, Huang Y, Friedman WJ, Boissonneault V, Plante MM, Rivest S, Sims JE. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity. 2009;30:817–831. doi: 10.1016/j.immuni.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spulber S, Mateos L, Oprica M, Cedazo-Minguez A, Bartfai T, Winblad B, Schultzberg M. Impaired long term memory consolidation in transgenic mice overexpressing the human soluble form of IL-1ra in the brain. J Neuroimmunol. 2009;208:46–53. doi: 10.1016/j.jneuroim.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type specific interleukin-1β signaling in the central nervous system. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J Cereb Blood Flow Metab. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, De Simoni MG, Sperk G, Andell-Jonsson S, Lundkvist J, Iverfeldt K, Bartfai T. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A. 2000;97:11534–11539. doi: 10.1073/pnas.190206797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78:379–389. doi: 10.1006/nlme.2002.4072. [DOI] [PubMed] [Google Scholar]
- Zhang P, Miller BS, Rosenzweig SA, Bhat NR. Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J Neurosci Res. 1996;46:114–121. doi: 10.1002/(SICI)1097-4547(19961001)46:1<114::AID-JNR14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]