

Abstract
Tobacco smoke exposure stimulates the expression of genes that are likely to be involved in the metabolism of its combustion products such as polycyclic aromatic hydrocarbons (PAH). Four of the smoke induced genes are aldo-keto reductases (AKR), enzymes that metabolically activate PAH to PAH o-quinones. Alternatively, PAHs are metabolized to (±)-anti-diol epoxides, such as (±)-anti-benzo[a]pyrene diol epoxide ((±)-anti-BPDE)), by the combined action of P4501A1/1B1 and epoxide hydrolase. (±)-anti-BPDE forms DNA adducts directly, while PAH o-quinones cause DNA damage by oxidative stress through a futile redox cycle. To address the role of AKRs in PAH cytotoxicity, we compared the cytotoxicity of PAH metabolites and the effects of overexpressing AKR1A1 in lung cells. (±)-anti-BPDE and B[a]P-7,8-trans-dihydrodiol, an intermediate in (±)-anti-BPDE metabolism, are toxic to A549 cells at concentrations with an IC50 of ~2 μM. In contrast, the PAH o-quinone B[a]P-7,8-dione was about 10-fold less toxic to A549 cells with an IC50 > 20 μM. Similar differences in cytoxicity was observed with two other PAH o-quinones (benz[a]anthracene-3,4-dione and 7,12-dimethylbenz[a]anthracene-3,4-dione) compared with their respective diol-epoxide counterparts (BA-3,4-diol-1,2-epoxide and DMBA-3,4-diol-1,2-epoxide). In addition, both anti-BPDE and B[a]P-7,8-trans-dihydrodiol induced p53 expression ~6 hours post-treatment at concentrations as low as 1 μM consistent with extensive DNA damage. B[a]P-7,8-dione treatment did not induce p53 but generated reactive oxygen species (ROS) in A549 cells and induced the expression of oxidative response genes in H358 cells. We also observed that overexpression of AKR1A1 in H358 cells, which otherwise have low levels of AKR expression, protected cells 2–10 fold from the toxic effects of B[a]P-7,8-trans-dihydrodiol. These data suggest that overexpression of AKRs may protect lung cancer cells from the acute toxic effects of PAH.
Keywords: PAH o-quinones, Aldo-Keto Reductases, anti-BPDE
Introduction
Lung cancer is caused primarily by exposure to tobacco smoke 1. Tobacco smoke, and other combustion products, such as charbroiled food and automobile exhaust, contains polycyclic aromatic hydrocarbons (PAH) 2. However, PAH must be metabolically activated into ultimate carcinogens that can damage DNA 3–5. If left unrepaired, the DNA damage can cause mutations to initiate cancer.
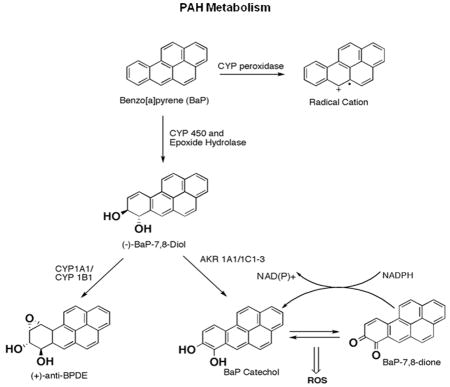
One of the most common PAH in tobacco smoke is Benzo[a]Pyrene (B[a]P). B[a]P can be converted into DNA reactive metabolites via three pathways. The first pathway utilizes enzymes from the cytochrome P450 family (CYP1A1 and CYP1B1) and epoxide hydrolases to form (±)anti-BPDE 6. (±)-anti-BPDE is highly mutagenic and forms bulky adducts with DNA 7, 8. The second pathway utilizes P450 peroxidase to form radical cations which react with DNA to yield depurinating adducts 9, 10. The third pathway employs aldo-keto reductases (AKRs) to oxidize B[a]P-7,8-trans-dihydrodiols to catechols which can undergo two spontaneous oxidation events to form PAH o-quinones 6. PAH o-quinones can form both stable and depurinating DNA adducts 11, 12. In addition, during the formation of PAH o-quinones, a futile redox cycle occurs in the presence of NADPH, which generates reactive oxygen species (ROS). Based on mutagenesis studies, measurement of ROS levels, and measurements of oxidized macromolecules in cells, it is likely that the redox cycling is the predominant cause of DNA damage by PAH o-quinones 13–17.
Efforts to trace the carcinogenic pathways leading to lung cancer have found evidence for all of these mechanisms of PAH activation. The diol epoxide pathway is supported by studies showing that B[a]P-7,8-trans-dihydrodiol epoxide adducts are found in smokers lungs and the locations of DNA adducts can be mapped to known hotspot codons on the tumor suppressor p5318. The PAH o-quinone pathway is supported by studies showing that smoking causes oxidative stress, which is characterized by low levels of antioxidants 19 and elevated levels of the oxidative lesion, 8-oxo-2’-deoxyguanosine (8-oxo-dGuo) 20–22. Expression of AKR1A1 reduced the levels of B[a]P-7,8-trans-dihydrodiol epoxide adducts in lung epithelial cells, suggesting that AKRs affect the metabolism of PAH 23. The products of radical cation damage and depurinating adducts have also been reported in PAH treated mice and cells, suggesting radical cations are also generated from tobacco metabolites 10, 23. Both B[a]P-7,8-trans-dihydrodiol epoxide adducts 24, 25 and 8-oxo-dGuo can cause G to T transversions 15, the predominant mutation in the tumor suppressor gene p53 in lung cancer 26, 27.
Microarray studies of the gene expression patterns associated with tobacco smoke exposure found that four AKR isoforms are among the genes that are smoking response genes 28–31. In one study, three AKR genes were among only nine genes consistently upregulated in smokers when compared with non-smokers and former smokers 30. Many of the genes induced by tobacco smoke have anti-oxidant response elements in their promoters and are thus likely to be responding to oxidative stress 32. These expression studies suggest that, by overexpressing AKRs, smokers divert some of the PAH into o-quinones, reducing the levels of other metabolites.
Another source of PAH is smoky coal, which is widely used for cooking and heating in Xuan Wei County, China. Non-smoking women from this area have elevated rates of lung cancer. Their lung cancers are characterized by G to T transversions in p53 and Ras, but the spectrum of mutations is different from the spectrum of mutations in smokers in that their is one hotspot, codon 249, which is not a preferred PAH adduct site. In addition, within this population there are single nucleotide polymorphisms (SNPs) in AKR1C3 and OGG1 (which repairs 8-oxo-dGuo) which are associated with increased lung cancer risk. These studies suggest a role of AKRs and oxidative damage in smoky coal-induced lung cancer 33, 34.
We hypothesized that AKRs protected cells from exposure to PAH. To address this hypothesis we first tested the toxicity of PAH o-quinones to other PAH metabolites in lung carcinoma cells (A549). Next we tested if an intermediate product of PAH metabolism, diols, would be more or less toxic to lung cancer cells that overexpress AKR1A1 (H358/AKR1A1) compared to cells with no AKRs (H358). To determine if cells were undergoing oxidative stress, we stained A549 cells treated with PAH o-quinones with a specific dye (H2DCFDA) used to detect ROS generation. Finally, we examined the expression levels of oxidative response genes in both A549 cells and H358 cells exposed to o-quinones to determine if ROS generated by PAH o-quinone treatment would induce the expression of oxidative response genes. These results suggest that AKR1A protects lung epithelial cells from the toxic effects of PAH metabolites.
Materials and Methods
Chemicals and Reagents
Reagents and media for cell culture were purchased from Invitrogen (Carlsdbad, CA). B[a]P and B[a]P metabolites (B[a]P-7,8-trans-dihydrodiol, (±)-anti-BPDE and B[a]P-7,8-dione) were obtained from NCI Chemical Carcinogen Standard Reference Repository (Midwest Research Institute, Kansas City, MO). The purity of all PAH-metabolites was assessed by LC/MS. Other reagents were of the highest grade available. Caution: All PAHs are potentially hazardous and should be handled in accordance with “NIH Guidelines for the Laboratory Use of Chemical Carcinogens”. Ponalrestat (Statil) was purchased from TOCRIS biosciences (Ellisville, MO). B[a]P and B[a]P metabolites were dissolved in DMSO in accordance with their solubility profiles.
Cell Culture and Toxicity Studies
Human lung adenocarcinoma cells (A549) and human bronchial epithelial cells (H358) were obtained from ATCC (Manassas, VA). A549 cells were cultured in DMEM (Invitrogen Carlsbad, CA) supplemented with 10% FBS and 100 μg/ml Penicillin/Streptomycin. H358 cells were cultured in RPMI 1640 media (Invitrogen Carlsbad, CA) supplemented with 10% FBS and 100 μg/ml Penicillin/Streptomycin. H358/AKR1A1 cells, which overexpress AKR1A1, were obtained from Dr. Trevor Penning (University of Pennsylvania, Philadelphia PA) and grown in RPMI 1640 media supplemented with 10% FBS, 100 μg/mL Penicillin/Streptomycin and Geniticin (G418–0.4 mg/mL). All cells were grown at 37°C in a humidified 6% CO2 incubator. For toxicity studies, cells were plated at an initial density of 1.0 × 105 cells on 24-well plates, grown for 24 hrs, treated with various P[a]H metabolites and incubated at 37°C for the specified time. Cells were then washed with PBS, trypsinized, stained with trypan blue and counted on a Countess cell counting machine (Invitrogen). All data points represent 3 independent experiments repeated in triplicates.
Western Blot Analysis
Cells were washed with phosphate buffered saline (PBS), trypsinized and lysed in a buffer containing 50 mM Tris-HCl, pH 7.4, 10% glycerol, 1 mM EDTA pH 8.0, 100 mM NaCl, .1% SDS, 1% Triton X-100, 1mM sodium orthovanadate, 5 mM sodium fluoride, 1mM phenylmethanesulfonylfluoride (PMSF) and protease inhibitor tablet (Roche, Indianapolis, IN). Protein concentration was determined by the Bradford method (Bio-Rad Hercules, CA) and a Nano-drop spectrophotometer (Thermo Scientific West Palm Beach, FL). Samples of extracts containing 25–50 μg of protein were subjected to SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Millipore, Billerica, MA). Membranes were next blocked in 5% milk in 1× Tris-Buffered Saline Tween (TBST) for 1 hr and incubated with a specific primary antibody overnight in 5% milk/TBST at a concentration suggested by the manufacturer. Blots were washed in 1 × TBST three times before addition of a secondary antibody [anti-mouse, anti-goat, or anti-rabbit (Sigma)] for 1 hr. Finally blots were washed and bands visualized by enhanced chemiluminescence Western Blot detection kit (GE healthcare, Piscataway, NJ). The following primary antibodies were used (1/1000 dilution): anti-NQO1 sc-16464, anti-HO-1 sc136960, anti-p53 sc-126, anti-Actin sc-1615 (all from Santa Cruz, Santa Cruz, CA), anti-p-p38 4511, anti-p-JNK/SAPK 4668, anti-JNK/SAPK 9258, anti-p38 9212 (all from Cell Signaling, Danvers, MA).
Detection of ROS
Formation of ROS was measured after treating with the fluorescent dye 2',7'-dichlorodihydrofluorescin diacetate (H2DCFDA) (Invitrogen, Carlsbad, CA). Briefly, 80–90% confluent A549 cells were washed with HBSS twice at 37°C for 5 min. Cells were then pre-incubated with 1 μM of H2DCFDA dye for 1hr, washed with HBSS and treated as indicated with various concentrations of B[a]P-7,8-dione or (±)-anti-BPDE. The cells were then incubated for 6 hours at 37°C, washed twice with HBSS 37°C for 5 min each time and observed under a fluorescent microscope to determine ROS generation (Nikon Eclipse T5100).
Realtime PCR and PCR arrays
Real-time quantitative reverse transcription and polymerase chain reaction was performed using the taq-man assay (AB Step One Plus, Applied Biosystems, Germany). The probes and primers were acquired from Applied Biosystems and are as follows: human NQO1 (Hs00168547_m1), HO-1 (Hs01110251_m1) and GAPDH (Hs99999905_m1). Each experiment was standardized with GAPDH primers as an internal control and consisted of three samples in triplicates. RT-PCR array was performed using the human oxidative stress and antioxidant defense kit from SA biosciences (Fredkerick, MD).
Statistical Analysis
Results are presented as mean ± standard error of the mean. The student t-test was used to determine p-values between samples.
Results
PAH o-quinones are less cytotoxic to lung epithelial cells than (±)-anti-BPDE and B[a]P-7,8-trans-dihydrodiol
To address the role of AKRs in lung cell viability, we first compared the toxicity of the AKR metabolites, PAH o-quinones, with other PAH metabolites in lung cells. We treated A549 cells with the metabolites and measured cell viability using the trypan blue exclusion method. (±)-anti-BPDE and B[a]P-7,8-trans-dihydrodiol were cytotoxic as shown in previous studies 35. The IC50 for both (±)-anti-BPDE and B[a]P-7,8- trans-dihydrodiol was ~ 2.0 μM at 24 hr (Fig. 1A). In contrast, all concentrations of the PAH o-quinone B[a]P-7,8-dione (BPQ), including concentrations near the limits of solubility (20 μM), reduced cell viability by ≤ 20% at 24 hours (Fig 1A) and this reduction was not statistically significant. To further evaluate the level of toxicity of the metabolites, we increased the period of treatment. There was virtually no cell death in A549 cells treated with B[a]P-7,8-dione up to 72 hrs of exposure compared to DMSO alone (Fig. 1D). At 5μM of (±)anti-BPDE, however, ≥80% of the cells died by 24 hr (Fig. 1D). Thus, B[a]P-7,8-dione is significantly less toxic to A549 cells than other PAH metabolites (±)-anti-BPDE and B[a]P-7,8-trans-dihydrodiol. To address the relative toxicity of other quinone/diol-epoxide pairs, we tested benz[a]anthracene-3,4-dione (BAQ) and BA-3,4-diol-1,2-epoxide (BADE) as well as 7,12-dimethylbenz[a]anthracene-3,4-dione (DMBAQ) and DMBA-3,4-diol-1,2-epoxide (DMBADE). In each pair we found that the quinone was significantly less cytotoxic than its diol epoxide (Fig 1B–C). We conclude that that PAH o-quinone metabolic activation pathways are less cytotoxic than diol-epoxide pathways.
Figure 1.

PAH o-quinones are less cytotoxic than PAH diols and PAH diol epoxides to A549 cells. A) A549 cells were seeded at density of 1.0 × 105 cells per well and allowed to attach for 24 hrs. Cells were then treated with varying concentrations of anti-BPDE, B[a]P-7,8-trans-dihydrodiol (Diol) and B[a]P-7,8-dione (BPQ) for 24 hrs, trypsinized and counted using the trypan blue exclusion method. B–C) Cell viability of A549 cells treated with (B) DMBAQ and DMBADE (C) BAQ and BADE for 24 hrs. D) A549 cells were treated with different concentrations of BPQ and anti-BPDE and allowed to grow for 72 hours. At each time point cells were trypsinized and counted using trypan blue exclusion method. Results are the mean of three independent experiments.
Overexpression of AKRs protect bronchial alveolar cells (H358) from the toxic effects of Bp 7,8 diol
To test the hypothesis that AKRs are metabolizing PAH to less toxic metabolites, we tested cells that overexpress AKRs. Because A549 cells express multiple AKR isoforms at high levels, it was not possible to manipulate the levels of AKRs in them. However, H358 cells express low levels of AKRS and can be engineered to express high levels of AKR1A1 and convert B[a]P-7,8-trans-dihydrodiols into PAH o-quinones36. Again, as in the A549 cells, we found that the PAH o-quinones were significantly less toxic than B[a]P-7,8-trans-dihydrodiol or (±)-anti-BPDE in H358 cells (Figure 2A). However, the cells that overexpress the AKR1A1 (H358/AKR1A1) were resistant to B[a]P-7,8- trans-dihydrodiol (Fig. 2B) suggesting that AKR1A1 was converting the B[a]P-7,8- trans-dihydrodiol to the less cytotoxic PAH o-quinones. To confirm that resistance was the result of AKR overexpression, we performed the toxicity studies in the presence of an AKR1A1 inhibitor, Statil. Treating cells with 50 and 100 μM of Statil reduced cell viability by 35 and 55 percent, respectively, enhancing the toxicity of B[a]P-7,8-trans-dihydrodiol (Fig. 2C). The viability of the treated cells was about the same as those that do not overexpress AKR1A1. These concentrations of statil did not affect viability of cells in the absence of B[a]P-7,8-trans-dihydrodiol. Collectively, these results support the hypothesis that overexpression of AKRs protect lung cancer cell lines from the toxic effects of B[a]P-7,8-trans-dihydrodiol.
Figure 2.

AKR1A1 overexpression protects lung cells from the toxic effects of PAH metabolites at the expense of increased oxidative stress. A) H358 cells were treated with various concentrations BPQ and anti-BPDE, grown for 24 hrs and cell viability determined using the trypan blue exclusion method. B) Cell viability of H358 and H358/AKR1A1 cells treated with various concentrations of B[a]P-7,8-trans-dihydrodiol for 24 hrs. C) H358/AKR1A1 cells were treated with B[a]P-7,8-trans-dihydrodiol and an AKR1A1 inhibitor (statil) for 24 hrs and cell viability determined. D) Western blot analysis showing overexpression of AKR1A1 in H358/AKR1A1 cells compared to H358 cells.
Response of Stress genes to PAH metabolites
We next determined if B[a]P-7,8-dione induced stress responses in cells. We first tested p53 induction, which is induced by DNA damage. p53 expression was stimulated in A549 cells exposed to 1 μM of (±)anti-BPDE for 6 hrs as well as 24 hrs (Fig. 3A). B[a]P induced p53 expression but with delayed kinetics compared to (±)anti-BPDE. This delay probably reflects the time required for B[a]P to be converted into DNA reactive metabolites. However, B[a]P-7,8-dione did not stimulate p53 expression even at concentrations up to 20 μM (Fig. 3A). To confirm that B[a]P-7,8-dione generated ROS, we pre-incubated A549 cells with 2',7'-dichlorodihydrofluorescin diacetate (H2DCFDA), a fluorescent marker that detects ROS, and treated A549 cells with 20uM B[a]P-7,8-dione for 6 hrs. B[a]P-7,8-dione generated high levels of ROS, as visualized by the increased fluorescent intensity, while (±)-anti-BPDE treated cells did not generate ROS (Fig. 3B).
Figure 3.

Response of Stress genes to PAH metabolites. A) A549 cells were treated with B[a]P, anti-BPDE or BPQ for 6 hours or 24 hours. Western analysis was performed on A549 total cell lysates with anti-p53 antibodies. B) A549 cells were incubated with H2DCFDA for 1 hr, washed with HBSS and treated for 6 hrs with DMSO, anti-BPDE or BPQ. Cells were then visualized under a fluorescence microscope and images documented. C) Western analysis using stress kinase antibodies was performed on A549 cells treated with DMSO, B[a]P-7,8-trans-dihydrodiol, anti-BPDE, or BPQ for 24 hrs.
We also examined other markers of stress, including phosphorylation of p38 and JNK/SAPK. The phosphorylation levels of both were elevated with (±)anti-BPDE and B[a]P-7,8-trans-dihydrodiol (Fig. 3C). However both p-p38 and p-JNK/SAPK showed little change with B[a]P-7,8-dione treatment. Similar results were observed in p-p38 and p-JNK/SAPK in H358 cells (data not shown). Together these experiments confirm that PAH o-quinones stimulate ROS, but interestingly do not induce double stranded breaks, which are required to induce p53 37, 38.
Induction of oxidative response genes by B[a]P-7,8-dione
Oxidative stress can be measured by examining the relative expression of a set of genes that have been determined to respond to various types of oxidative stress. To determine which oxidative response genes were elevated due to the exposure of o-quinones, we performed a PCR array on H358 cells treated with PAH o-quinones. A total of 84 genes were analyzed on an array. The highest level of induction was 3.4 fold for Sulfiredoxin 1 homolog (SRNX1-Data not shown).
We also analyzed two widely studied genes, NAD(P)H quinone oxidoreductase 1 (NQO1) and Heme Oxygenase 1 (HO-1) as markers for oxidative stress, individually, because they were not represented on the array. We analyzed the genes in both A549 and H358 cell lines. Using Real-Time-PCR on mRNA isolated from A549 cells treated with B[a]P-7,8-dione, we found that at 6 hrs NQO1 expression levels do not significantly change between control group (DMSO only) and treatment group (B[a]P-7,8-dione) (Fig. 4A). However, at 24 hrs, the expression of NQO1 and HO-1 mRNA levels were elevated by ~2 fold (p<.05 for NQO1; p<.005 for HO-1) (Fig. 4A,C). This suggests that oxidative response genes are upregulated in A549 cells after B[a]P-7,8-dione exposure. We also observed that NQO1 protein expression was also elevated relative to DMSO control in A549 cells after B[a]P-7,8-dione exposure (Fig 4B). These data suggest that treating A549 cells with B[a]P-7,8-dione induces the expression of oxidative response genes, which may be involved in cell protection.
Figure 4.

Increase of NQO1 and HO-1 mRNA levels in A549 cells treated with BPQ. A) Realtime PCR showing expression levels of NQO1 mRNA in A549 cells treated with BPQ for 6 hrs and 24hrs. C) Western blot analysis for NQO1 in A549 cells treated with PAH metabolites. D) Realtime PCR showing increase in HO-1 mRNA levels isolated from A549 cells treated with B[a]P-7,8-dione for 24 hrs. All realtime PCR experiments were performed with 3 biological replicates in triplicates for each sample and standardized to GAPDH expression levels.
Induction of HO-1 in H358 cells exposed to B[a]P-7,8-dione
A549 cells do not respond well to oxidative stress because they harbor a mutation in Kelch-like ECH-associated protein 1 (KEAP1), a negative regulator of oxidative response genes39, 40. As a result, these cells have elevated levels of oxidative response genes, which are only weakly stimulated by oxidative stress. Therefore we conducted additional stress studies in H358 cells. Note that H358 cells are p53 negative so we focused on oxidative stress response genes. Quantitative real-time PCR showed that HO-1 mRNA expression was elevated 20 fold in the H358 cells (Fig. 5A; p<.001) and 85 fold in H358/AKR1A1 cells (Fig. 5C; p<.001) in response to 20 μM of B[a]P-7,8-dione. In addition we also observed a concentration dependent increase in HO-1 protein levels exposed to 5, 10 and 20 μM of B[a]P-7,8-dione in both H358 (Fig. 5B) cells and H358/AKR1A1 cells (Fig. 5D). Interestingly, B[a]P-7,8-trans-dihydrodiol did not induce an increase of HO-1 induction up to 20 μM.
Figure 5.

Increased expression of HO-1 mRNA in H358 cells and H358/AKR1A1 cells exposed to BPQ. A) Realtime PCR reveals highly elevated levels of HO-1 mRNA in H358 cells exposed to BPQ for 24 hrs. B) Dose dependent increase of HO-1 protein expression in H358 cells exposed to increasing concentrations of BPQ C) Increase of HO-1 mRNA levels in H358/AKR1A1 cells exposed to BPQ for 24 hrs. D) Increase in HO-1 protein levels in H358/AKR1A1 cells exposed to increasing concentration of BPQ for 24 hrs.
Induction HO-1 by PAH o-quinones is ROS dependent
To address if quinone induction of HO-1 is dependent on ROS, we treated H358/AKR1A1 cells with the anti-oxidants α-tocopherol (Vitamin E) or ascorbic acid (Vitamin C) (Fig. 6) prior to quinone induction. Both compounds prevented HO-1 induction, suggesting that B[a]P-7,8-dione induces HO-1 through ROS.
Figure 6.

Antioxidants abate HO-1 induction in H358/AKR1A1 cells treated with BPQ. Western blot analysis for HO-1 on H358/AKR1A1 cells exposed to BPQ and 250 μM of α-tocopherol and ascorbic acid.
Discussion
Smokers, and mice exposed to smoke, are under oxidative stress resulting in reduced levels of plasma antioxidants, increased expression of oxidative response genes and elevated levels of 8-oxo-dGuo in their DNA and in their urine 20–22, 41, 42. One source of oxidants are quinones, which can be generated by combustion or alternatively when PAH are metabolized by AKRs. The o-quinones are not mutagenic by themselves, but become highly mutagenic in the presence of NADPH because they undergo redox cycling to generate ROS 14, 43–45. ROS oxidizes DNA to cause 8-oxo-dGuo, a mutagenic lesion that is misread to cause G to T transversions, which are characteristic of tobacco induced lung cancers 14–16, 46, 47.
Microarray analysis identified genes that are induced in the epithelia of smokers, the oral mucosa of smokers and in cells that are exposed to cigarette smoke condensate 28–31. While multiple studies found AKRs induced, the study from Zhang et. al., also examined former smokers and non-smokers and found that nine genes were induced in smokers but not in former smokers or non-smokers 30. These genes were: ALDH3A1, CYP1B1, MUC5AC, NQO1 and SCGB1A1. Most of the induced genes are likely to have protective roles. ALDH3A1 (aldehyde dehydrogenase), CYP1B1, NQO1 (NAD(P)H quinone oxidoreductase 1), AKR1C2, AKR1B10, AKR1C1 and AKR1C3 all metabolize tobacco combustion products 31 while MUC5AC, encodes mucin, a major component of mucus. There are some direct examples of these genes having protective roles. For example, NQO1 knockout mice are more susceptible to the toxic effects of quinones 48. Although knockouts of CYP1B1 and CYP1A1 found some unexpected toxicities, the outcomes suggest that CYP1B1 and CYP1A1 both metabolize benzopyrene, depending on expression levels of each in different tissues 49. Since PAH are metabolized by AKRs into PAH o-quinones, we addressed the role of AKRs by overexpression, as well as by comparing the toxicity of PAH o-quinones relative to other PAH metabolites.
We hypothesized that AKRs are part of the protective response to PAH induced stress. We measured genotoxic stress by p53 induction, oxidative stress by ROS generation, and induction of oxidative response genes. As documented previously, we found that PAH diol-epoxides ((±)-anti-BPDE) efficiently induced genotoxic stress while PAH o-quinones showed signs of oxidative stress32, 50. Perhaps most striking was the observation that PAH o-quinones were not cytotoxic despite evidence of oxidative stress and induction of HO-1. Overexpression of AKRs in a cell that expresses low levels of AKRs also reduced the toxicity of the B[a]P-7,8-trans-dihydrodiols. Together these data suggest a protective role for AKRs in response to PAH exposure.
We propose that cells expressing higher levels of AKRs direct a portion of the PAH from being metabolized into the more toxic (±)-anti-BPDE. In adduct studies, when H358 cells were treated with (±)-B[a]P-7,8-dihydrodiol, (±)-anti- trans-B[a]P DE-N2-dGuo was the major DNA adduct observed. However, overexpression of AKR1A1 reduced the levels of this DNA adduct from 46 ± 4 adducts/106 bases to 26 ± 2 adducts/106 bases. Thus AKR1A1 prevents the accumulation of diol epoxide DNA adducts, most likely by converting diols into other metabolites such as B[a]P-7,8-dione 51. In the CYP1B1 and CYP1A1 knockout mice studies, adduct levels also correlated with toxicity 49. These adduct studies support our cytotoxicity observations, since we find that (±)-anti-BPDE, which forms (±)-anti- trans- B[a]PDE-N2-dGuo adducts is more cytotoxic than B[a]P-7,8-dione.
Although B[a]P-7,8-dione stimulated ROS in A549 cells, it did not stimulate p53 expression. This may be because the ROS generated by B[a]P-7,8-dione exposure does not cause enough double strand breaks, which are required to induce p53 37. ROS damages DNA through formation of oxidation products such as 8-oxo-deoxyguanosine, while (±)-anti-BPDE forms bulky adducts. The (±)-anti-BPDE adducts are probably causing double-stranded breaks, perhaps through the extensive activation of repair pathways.
While we observed a strong induction of HO-1 in H358 cells, we found high constitutive levels of HO-1 expression and only a small induction in A549 cells. HO-1 transcription is activated by the transcription factor NRF-2, which upon oxidation is released by the inhibitory factor KEAP1. A549 cells have a mutation in KEAP-1, which leads to the high constitutive levels of NRF-2 leading to high levels of HO-1 and other oxidative response genes 39, 40.
We note that while the short term effects of AKR expression may be protective to cells, AKR derived PAH o-quinones can undergo futile redox cycling to generate ROS, which is likely to cause oxidative stress. Although we did not detect induction of HO-1 from B[a]P-7,8-trans-dihydrodiol treatment, oxidatively damaged DNA has been reported in A549 cells 50. The differences may be due to the expression of different isoforms and differences in the oxidative response genes between the two cell lines.
Acknowledgments
Funding Support
This work was supported by grant R01 GM48241 and R01 NIEHS to J.F and P30 ES013508. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
We thank the NCI Chemical Carcinogen Standard Reference Repository for (±)-anti-BPDE. We thank Dr. Trevor Penning for helpful discussions and Sarah Ahmed for technical assistance.
Abbreviations
- (±)-anti-BPDE
(±)anti-benzo[a]pyrene 7,8-diol 9,10-epoxide
- BP-7,8-diol or Diol
B[a]P-7,8-trans-dihydrodiol
- BPQ
benzo[a]pyrene-7,8-dione
- BAQ
benz[a]anthracene-3,4-dione
- DMBAQ
7,12-dimethylbenz[a]anthracene-3,4-dione
- BADE
benz[a]anthracene-3,4-diol-1,2-epoxide
- DMBADE
7,12-dimethylbenz[a]anthracene-3,4-diol-1,2-epoxide
- PAH
polycyclic aromatic hydrocarbons
- H2DCFDA
2',7'-dichlorodihydrofluorescin diacetate
References
- 1.Jemal A, Thun MJ, Ries LAG, Howe HL, Weir HK, Center MM, Ward E, Wu XC, Eheman C, Anderson R, Ajani UA, Kohler B, Edwards BK. Annual Report to the Nation on the Status of Cancer, 1975–2005, Featuring Trends in Lung Cancer, Tobacco Use, and Tobacco Control. Journal of the National Cancer Institute. 2008;100(23):1672–1694. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 3.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. G.H.A Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 4.Gelboin HV. Benzo[a]pyrene metabolism, activation and carcinogenesis: Role and regulation of mixed function oxidases and related enzymes. Physiol Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 5.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicity. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 6.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 7.Jennette KW, Jeffery AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Nucleoside adducts from the in vitro reaction of benzo[a]pyrene- 7,8-dihydrodiol-9,10-oxide or benzo[a]pyrene-4,5-oxide with nucleic acids. Biochemistry. 1977;16:932–938. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]
- 8.Koreeda M, Moore PD, Wislocki PG, Levin W, Conney AH, Yagi H, Jerina DM. Binding of benzo[a]pyrene-7,8-diol-9,10-epoxides to DNA, RNA and protein of mouse skin occurs with high stereoselectivity. Science. 1978;199:778–781. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- 9.Cavalieri EL, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 10.Chakravarti D, Pelling JC, Cavalieri EL, Rogan EG. Relating aromatic hydrocarbon-induced DNA adducts and c-H-ras mutations in mouse skin papillomas: the role of apurinic sites. Proc Natl Acad Sci USA. 1995:10422–10426. doi: 10.1073/pnas.92.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCoull KD, Rindgen D, Blair IA, Penning TM. Synthesis and characterization of polycyclic aromatic hydrocarbon o-quinone depurinating N7-guanine adducts. Chem Res Toxicol. 1999;12:237–246. doi: 10.1021/tx980182z. [DOI] [PubMed] [Google Scholar]
- 12.Shou M, Harvey RG, Penning TM. Reactivity of benzo[a]pyrene-7,8-dione with DNA. Evidence for the formation of deoxyguanosine adducts. Carcinogenesis. 1993;14:475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- 13.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox-active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 14.Yu D, Berlin JA, Penning TM, Field J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem Res Toxicol. 2002;15(6):832–42. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- 15.Shen YM, Troxel AB, Vedantam S, Penning TM, Field J. Comparison of p53 Mutations Induced by PAH o-Quinones with Those Caused by anti-Benzo[a]pyrene Diol Epoxide in Vitro: Role of Reactive Oxygen and Biological Selection. Chem Res Toxicol. 2006;19(11):1441–1450. doi: 10.1021/tx0601206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JH, Gelhaus S, Vedantam S, Oliva AL, Batra A, Blair IA, Troxel AB, Field J, Penning TM. The pattern of p53 mutations caused by PAH o-quinones is driven by 8-oxo-dGuo formation while the spectrum of mutations is determined by biological selection for dominance. Chem Res Toxicol. 2008;21(5):1039–49. doi: 10.1021/tx700404a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JH, Troxel AB, Harvey RG, Penning TM. Polycyclic Aromatic Hydrocarbon (PAH) o-Quinones Produced by the Aldo-Keto-Reductases (AKRs) Generate Abasic Sites, Oxidized Pyrimidines, and 8-Oxo-dGuo via Reactive Oxygen Species. Chem Res Toxicol. 2006;19(5):719–28. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denissenko MF, Pao A, Tang MS, Pfeifer GP. Preferential formation of Benzo[a]pyrene adducts at lung cancer mutation hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 19.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(17):7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loft S, Vistisen K, Ewertz M, Tjonneland A, Overvad K, Poulsen HE. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis. 1992;13(12):2241–7. doi: 10.1093/carcin/13.12.2241. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki J, Inoue Y, Suzuki S. Changes in the urinary excretion level of 8-hydroxyguanine by exposure to reactive oxygen-generating substances. Free Radic Biol Med. 1995;18(3):431–6. doi: 10.1016/0891-5849(94)00152-a. [DOI] [PubMed] [Google Scholar]
- 22.Prieme H, Loft S, Klarlund M, Gronbaek K, Tonnesen P, Poulsen HE. Effect of smoking cessation on oxidative DNA modification estimated by 8-oxo-7,8-dihydro-2'-deoxyguanosine excretion. Carcinogenesis. 1998;19(2):347–51. doi: 10.1093/carcin/19.2.347. [DOI] [PubMed] [Google Scholar]
- 23.Jiang H, Gelhaus SL, Mangal D, Harvey RG, Blair IA, Penning TM. Metabolism of benzo[a]pyrene in human bronchoalveolar H358 cells using liquid chromatography-mass spectrometry. Chem Res Toxicol. 2007;20(9):1331–41. doi: 10.1021/tx700107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei SJ, Chang RL, Bhachech N, Cui XX, Merkler KA, Wong CQ, Hennig E, Yagi H, Jerina DM, Conney AH. Dose-dependent differences in the profile of mutations induced by (+)-7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo(a)pyrene in the coding region of the hypoxanthine (guanine) phosphoribosyltransferase gene in Chinese hamster V-79 cells. Cancer Res. 1993;53(14):3294–301. [PubMed] [Google Scholar]
- 25.Wei SJ, Chang RL, Hennig E, Cui XX, Merkler KA, Wong CQ, Yagi H, Jerina DM, Conney AH. Mutagenic selectivity at the HPRT locus in V-79 cells: comparison of mutations caused by bay-region benzo[a]pyrene 7,8-diol-9,-10-epoxide enantiomers with high and low carcinogenic activity. Carcinogenesis. 1994;15(8):1729–35. doi: 10.1093/carcin/15.8.1729. [DOI] [PubMed] [Google Scholar]
- 26.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 27.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22(3):367–74. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 28.Boyle JO, Gumus ZH, Kacker A, Choksi VL, Bocker JM, Zhou XK, Yantiss RK, Hughes DB, Du B, Judson BL, Subbaramaiah K, Dannenberg AJ. Effects of Cigarette Smoke on the Human Oral Mucosal Transcriptome. Cancer Prevention Research. 2010;3(3):266–278. doi: 10.1158/1940-6207.CAPR-09-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gumus ZH, Du B, Kacker A, Boyle JO, Bocker JM, Mukherjee P, Subbaramaiah K, Dannenberg AJ, Weinstein H. Effects of Tobacco Smoke on Gene Expression and Cellular Pathways in a Cellular Model of Oral Leukoplakia. Cancer Prev Res. 2008;1(2):100–111. doi: 10.1158/1940-6207.CAPR-08-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Lee JJ, Tang H, Fan YH, Xiao L, Ren H, Kurie J, Morice RC, Hong WK, Mao L. Impact of Smoking Cessation on Global Gene Expression in the Bronchial Epithelium of Chronic Smokers. Cancer Prev Res. 2008;1(2):112–118. doi: 10.1158/1940-6207.CAPR-07-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penning TM, Lerman C. Genomics of Smoking Exposure and Cessation: Lessons for Cancer Prevention and Treatment. Cancer Prev Res. 2008;1(2):80–83. doi: 10.1158/1940-6207.CAPR-08-0047. [DOI] [PubMed] [Google Scholar]
- 32.Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenases. Cancer Res. 1999;59:607–614. [PubMed] [Google Scholar]
- 33.Lan Q, Mumford JL, Shen M, Demarini DM, Bonner MR, He X, Yeager M, Welch R, Chanock S, Tian L, Chapman RS, Zheng T, Keohavong P, Caporaso N, Rothman N. Oxidative damage-related genes AKR1C3 and OGG1 modulate risks for lung cancer due to exposure to PAH-rich coal combustion emissions. Carcinogenesis. 2004;25(11):2177–81. doi: 10.1093/carcin/bgh240. [DOI] [PubMed] [Google Scholar]
- 34.DeMarini DM, Landi S, Tian D, Hanley NM, Li X, Hu F, Roop BC, Mass MJ, Keohavong P, Gao W, Olivier M, Hainaut P, Mumford JL. Lung tumor KRAS and TP53 mutations in nonsmokers reflect exposure to PAH-rich coal combustion emissions. Cancer Res. 2001;61(18):6679–81. [PubMed] [Google Scholar]
- 35.Caino MC, Oliva JL, Jiang H, Penning TM, Kazanietz MG. Benzo[a]pyrene-7,8-dihydrodiol Promotes Checkpoint Activation and G2/M Arrest in Human Bronchoalveolar Carcinoma H358 Cells. Mol Pharmacol. 2007;71(3):744–750. doi: 10.1124/mol.106.032078. [DOI] [PubMed] [Google Scholar]
- 36.Jiang H, Vudathala DK, Blair IA, Penning TM. Competing roles of aldo-keto reductase 1A1 and cytochrome P4501B1 in benzo[a]pyrene-7,8-diol activation in human bronchoalveolar H358 cells: role of AKRs in P4501B1 induction. Chem Res Toxicol. 2006;19(1):68–78. doi: 10.1021/tx0502488. [DOI] [PubMed] [Google Scholar]
- 37.Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol. 1994;14(3):1815–23. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venkatachalam S, Denissenko M, Wani AA. Modulation of (+/−)-anti-BPDE mediated p53 accumulation by inhibitors of protein kinase C and poly(ADP-ribose) polymerase. Oncogene. 1997;14(7):801–9. doi: 10.1038/sj.onc.1200890. [DOI] [PubMed] [Google Scholar]
- 39.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10):e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S. Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Research. 2008;68(5):1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 41.Ames B, Gold L, Willett W. The Causes and Prevention of Cancer. Proc Natl Acad Sci USA. 1995;92(12):5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blake DJ, Singh A, Kombairaju P, Malhotra D, Mariani TJ, Tuder RM, Gabrielson E, Biswal S. Deletion of Keap1 in the Lung Attenuates Acute Cigarette Smoke-Induced Oxidative Stress and Inflammation. Am J Respir Cell Mol Biol. 2010;42(5):524–536. doi: 10.1165/rcmb.2009-0054OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chesis PL, Levin DE, Smith MT, Ernster L, Ames BN. Mutagenicity of quinones: pathways of metabolic activation and detoxification. Proceedings of the National Academy of Sciences. 1984;81(6):1696–1700. doi: 10.1073/pnas.81.6.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flowers L, Bleczinski WF, Burczynski ME, harvey RG, Penning TM. Disposition and biological activity of Benzo[a]pyrene-7,8-dione. A genotoxic metabolite generated by dihydrodiol dehydrogenase. Biochemistry. 1996;35:13664–13672. doi: 10.1021/bi961077w. [DOI] [PubMed] [Google Scholar]
- 45.Flowers-Geary L, Bleczinski W, Harvey RG, Penning TM. Cytotoxicity and mutagenicity of polycyclic aromatic hydrocarbon o-quinones produced by dihydrodiol dehydrogenase. Chemico-Biological interactions. 1996;99:55–72. doi: 10.1016/0009-2797(95)03660-1. [DOI] [PubMed] [Google Scholar]
- 46.Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol. 2006;19(4):491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 47.Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res. 1998;400:99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 48.Joseph P, Long DJ, 2nd, Klein-Szanto AJ, Jaiswal AK. Role of NAD(P)H:quinone oxidoreductase 1 (DT diaphorase) in protection against quinone toxicity. Biochem Pharmacol. 2000;60(2):207–14. doi: 10.1016/s0006-2952(00)00321-x. [DOI] [PubMed] [Google Scholar]
- 49.Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, Gonzalez FJ, Nebert DW. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol. 2006;69(4):1103–14. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- 50.Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, Penning TM. Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc Natl Acad Sci USA. 2008;105(19):6846–51. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruan Q, Gelhaus SL, Penning TM, Harvey RG, Blair IA. Aldo-keto reductase- and cytochrome P450-dependent formation of benzo[a]pyrene-derived DNA adducts in human bronchoalveolar cells. Chem Res Toxicol. 2007;20(3):424–31. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]