

Abstract
Activity-dependent redistribution of ion channels mediates neuronal circuit plasticity and homeostasis, and could provide pro-epileptic or compensatory anti-epileptic responses to a seizure. Thalamocortical neurons transmit sensory information to the cerebral cortex and through reciprocal corticothalamic connections are intensely activated during a seizure. Therefore, we assessed whether a seizure alters ion channel surface expression and consequent neurophysiologic function of thalamocortical neurons. We report a seizure triggers a rapid (≤2 hrs) decrease of EPSC-like current-induced phasic firing associated with increased transient A-type K+ current. Seizures also rapidly redistributed the A-type K+ channel subunit Kv4.2 to the neuronal surface implicating a molecular substrate for the increased K+ current. Glutamate applied in vitro mimicked the effect, suggesting a direct effect of glutamatergic transmission. Importantly, LGI1, a secreted synaptic protein mutated to cause human partial epilepsy, regulated this seizure-induced circuit response. Human epilepsy-associated dominant negative truncated mutant LGI1 inhibited the seizure-induced suppression of phasic firing, increase of A-type K+ current, and recruitment of Kv4.2 surface expression (in vivo and in vitro). The results identify a response of thalamocortical neurons to seizures involving Kv4.2 surface recruitment associated with dampened phasic firing. The results also identify impaired seizure-induced increases of A-type K+ current as an additional defect produced by the autosomal dominant lateral temporal lobe epilepsy gene mutant might contribute to the seizure disorder.
Keywords: LGI1, Kv4.2, epilepsy, ADLTE, A-type K+ current
Introduction
Autosomal dominant lateral temporal lobe epilepsy (ADTLE) is an inherited epilepsy disorder characterized by frequent partial seizures (two to five per month) associated with auditory or other sensory auras during a relatively normal electroencephalogram (EEG). Most affected individuals also suffer generalized tonic-clonic seizures (one per year) sometimes elicited by an environmental trigger. Mutations in leucine-rich glioma-inactivated-1 (LGI1) explain approximately 30% of ADTLE (Gu et al. 2002, Kalachikov et al. 2002, Morante-Redolat et al. 2002). Interestingly, LGI1 is an example of a limited number of non-ion channel human epilepsy genes (Noebels 2003). Instead, LGI1 is a 64-kDa, secreted (Senechal et al. 2005, Sirerol-Piquer et al. 2006) protein recently shown critical to postnatal glutamate synapse maturation and developmental pruning (Zhou et al. 2009, Anderson 2010).
LGI1 and its receptors, a disintegrin and metalloproteinase domains 22, 23, and 11 (ADAM 22, 23, and 11), co-immunoprecipitate with both postsynaptic density protein 95 (PSD95) and Kv1.1 potassium channels (Fukata et al. 2006, Fukata et al. 2010, Schulte et al. 2006) suggesting pre- and post-synaptic associations. LGI1 also shows high affinity binding to NOGO receptor to enhance neuronal growth on myelin-based inhibitory substrates (Thomas et al. 2010). Homozygous LGI1 knockout mice develop seizures and ultimately die within 2-3 weeks of birth (Chabrol et al. 2010, Fukata et al. 2010, Yu et al. 2010) at the approximate time when LGI1 expression increases (Zhou et al. 2009). Heterozygous LGI1 knockout mice show enhanced audiogenic kindling of seizures consistent with the reports of auditory-triggered seizures in ADLTE patients (Chabrol et al. 2010). We recently established that a dominant negative, truncated, ALDTE-associated mutant form of LGI1 (mLGI1), expressed as a full-length gene in transgenic mice, inhibits the normal postnatal developmental down-regulation of glutamatergic synapses in hippocampus. mLGI1 inhibited the normal developmental decrease of presynaptic release probability and NMDA receptor NR2B/NR2A ratio, increased excitatory synaptic transmission, and caused seizure susceptibility (Zhou et al. 2009). No effects on postsynaptic excitability were reported.
Seizure-induced redistribution of ion channels plays an important role in both pro-epileptic and anti-epileptic responses to seizures (Noebels 2003). Seizures induce both structural and biochemical changes in neurons, in some cases leaving the brain more susceptible, while in other cases initiating an anti-epileptic homeostatic response to inhibit future seizures. We hypothesized that, in addition to its effect in preventing normal postnatal glutamatergic synapse maturation and pruning, ADLTE mLGI1 might also disrupt adaptive homeostatic responses of glutamatergic synapses to a seizure. Consequently, in mice or humans carrying pathogenic LGI1 mutations, the brain may fail to generate the normal adaptive homeostatic response needed to inhibit future seizures.
We focus on the thalamus because during a seizure it is intensely activated (Blumenfeld et al. 2009, Paz et al. 2007, Tyvaert et al. 2009), and can display severe damage, with reactive gliosis, chronic atrophy, and interictal hypometabolism (Borges et al. 2003, Juhasz et al. 1999, Hashiguchi et al. 2007). Therefore, we suspected the thalamus would display prominent adaptive homeostatic responses. We found that a sustained seizure event rapidly inhibited phasic firing in thalamocortical neurons. This inhibition resulted from increased transient A-type K+ current with voltage-gating properties typical of the Kv4 family. As anticipated, seizures in vivo and glutamate in vitro induced a rapid surface recruitment of Kv4.2 channels in neurons of wild-type mice, but failed to do so in mLGI1 transgenics. Overall, our results establish that mutant LGI1 inhibits the normal seizure-induced dampening of phasic firing generated by glutamatergic synaptic transmission associated with the recruitment of A-type K+ currents and surface Kv4.2. The finding that ADLTE-associated mutant LGI1 blocks this homeostatic neuronal response identifies an additional mechanism of seizure susceptibility in ADLTE patients.
Methods and Materials
Animals
Mice studied were male adult (2-6 months old) mutant LGI1 transgenics or their littermate controls previously created and bred in house in an FVB genetic background (Zhou et al. 2009). Seizures were induced by subcutaneously injecting 1 mg/kg Scopolamine (Sigma), followed 30 minutes later by an intraperitoneal injection of 275 mg/kg pilocarpine. Both drugs were dissolved in sterile saline. Seizures were scored as in (Racine 1972) and (Schauwecker & Steward 1997) with slight modifications: Stage 0, normal activity; Stage 1: immobility and rigid posture; Stage 2: stiffened or Straub tail; Stage 3: partial- or mild-whole-body clonic seizures while retaining posture; Stage 4: rearing, whole body continuous clonic seizures while retaining posture; Stage 5: tonic–clonic seizures with loss of posture or jumping. Only mice that attained stage 4 or 5 seizures were used for electrophysiology. All procedures were approved by the Beth Israel Hospital IACUC and every effort was made to minimize animal suffering.
Electrophysiology
Brain slices were prepared as previously described (Kasten et al. 2007). Briefly, mice were deeply anesthetized with isofluorane and decapitated. The brain was quickly removed and placed in ice cold, oxygenated (95% O2, 5% CO2) sucrose cutting solution containing (in mM): 234 sucrose, 5 KCl, 5 MgSO4, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. Coronal brain slices (230-280 μm) were cut using a Vibratome 3000 tissue slicer in zero Z-axis vibration mode. Slices were transferred to oxygenated (5% CO2) artificial cerebrospinal fluid (ACSF) for 40-60 min at 37°C and then stored at room temperature for 1-9 hours before recording or for half an hour before in vitro treatments. ACSF contained (in mM): 124 NaCl, 3.5 KCl, 1 MgCl2, 1.2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose. Ventromedial nucleus thalamocortical relay neurons just lateral to the mammillothalamic tract were visually identified for recording using a 40× objective and infrared DIC optics on an upright Olympus BX-51WI microscope (Olympus, Tokyo, Japan). Recording pipettes were pulled from 1.5 mm OD capillary tubing (A-M Systems, Carlsborg, Washington) using a Flaming/Brown P-97 pipette puller (Sutter Instruments, Novato, California) and had tip resistances of 2-5 MΩ when filled with internal solution. Internal solution contained (in mM): 135 potassium methanesulfonate, 10 HEPES, 4 KCl, 2 NaCl, 4 Mg-ATP, 0.3 Tris-GTP, 7 phosphocreatine, 0.1 EGTA, 0.01 CaCl2 (pH=7.25, osmolarity ∼ 290 mOsm). All recordings were made with a HEKA triple EPC-10 patch-clamp amplifier utilizing Patchmaster 2 software (HEKA Instruments, Southboro, MA) at room temperature (∼22°C) in oxygenated (5% CO2) ACSF. After achieving a gigaohm seal, a stable recording was obtained with the cell in whole-cell configuration at Vc = -70 mV. Recordings were accepted for cells with a series resistance < 12 MΩ, most cells demonstrated series resistance in the range of 7-10 MΩ. Series resistance was 80% compensated. The liquid junction potential was calculated as 10.32mV, which was rounded to 10mV and corrected during data analysis. Data were filtered at 2.9 kHz and sampled at 20 kHz.
Tonic and phasic firing
Excitatory postsynaptic potential-spike coupling (E-S coupling) measures spiking in response to brief, naturalistic EPSC-like stimuli. Short EPSC-like pulses of depolarizing current (0-1000 pA in 200pA steps) were injected into patched cells as previously reported (Kasten et al. 2007). In summary, we examined sample EPSCs in thalamocortical neurons and developed a current injection protocol that mimicked the time course of the EPSC. The EPSC-like current injection consisted of a 0.5 ms linear rise to maximum, a 1 ms linear decay to 40% of maximum and a further 5 ms linear decay to baseline. The current amplitude at which a cell fired an action potential (AP) was determined. To asses tonic firing, longer 1s current pulses (0-100pA in 20pA steps) were used. The current amplitude at which a cell fired an action potential (AP) was again determined. AP half maximum spike width and threshold were measured for the first three spikes in the first current injection which elicited at least three spikes.
Potassium current measurements
Since multiple potassium currents are present in thalamocortical neurons, voltage clamp protocols and multiple ion channel blockers were used to isolate the Kv4-like channel currents described here. Despite the presence of a large number of potassium channel antagonists (see pharmacology section below), two major potassium channel current subtypes persisted, a rapidly-activating, rapidly-inactivating Kv4-like current (sensitive to millimolar 4-aminopyridine) and a slowly activating, slowly and partially-inactivating Kv2-like current (insensitive to millimolar 4-aminopyridine, partially sensitive to millimolar TEA). Voltage protocols consisted of clamping the neuron at (junction-potential-corrected) voltages between Vc = -70mV and +30 mV for 200ms, followed by a 200 ms step to Vc = +30 mV. In order to remove the slow Kv2-like currents, we subtracted the Vc = -10 mV (a voltage at which Kv4.2 channels are inactivated) from the Vc = -70 mV trace for each neuron to determine total Kv4.2 current. The total slow (Kv2-like) K current was measured by sampling the current amplitude at 190-200ms after the onset of the first depolarizing pulse, a time at which Kv4.2 currents had inactivated. The decay kinetic time constant (tau) for the subtracted Kv4.2 traces was calculated using curve fitting (Igor pro software).
Pharmacology
Drugs were bath applied using a gravity perfusion system in oxygenated (95% O2, 5% CO2) ACSF. All recordings were performed in the presence of antagonists of the AMPA/kainate and GABA-A receptors, 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μM, Tocris, Elleville, MO) and picrotoxin (100 μM, Sigma, St. Louis, MO), respectively. In voltage-clamp studies, the following drugs were used to block other Na+, K+, and Ca2+ currents: TTX (500 nM), 4-aminopyridine (4-AP, 500 μM), XE 991 (10 μM), TEA (10 mM), NiCl2 (2 mM), and CsCl (4 mM). To confirm channel specificity, 4-AP (5 mM) was used in some experiments to block Kv4 A-type K+ channels.
In vitro slice stimulation
Slices were cut as for electrophysiology, and were treated similarly. After recovery at 37° for 30 minutes, ACSF was replaced with either fresh ACSF (control), high-KCl ACSF (ACSF with 60 mM KCl and only 67.5 NaCl) or Glutamate-ACSF with 10 μM Glutamate (Sigma). After 20 min at 37°, slices were fixed in ice-cold 4% PFA in PBS for 15 minutes, and then placed in 20% sucrose overnight. Slices were then mounted in OCT, sectioned on a cryostat at 20 μm, and consecutive sections were mounted side-by-side on glass slides (Superfrost plus, Fisher).
Staining of brain sections
For each experiment, sections were stained in a single batch. Sections were circled with a PAP pen (Fisher), rehydrated in PBS and blocked with MOM reagent (Vector) and then blocking solution (10% Normal goat serum in PBS). One of the two sections on each slide was permeabilized with 0.25% Triton-X100 in the blocking solution. After washing, the liquid barrier was removed so that the permeabilized and non-permeabilized sections were stained side-by-side on the same slide to minimize staining variations. An anti-Kv4.2 antibody which recognizes an extracellular epitope (NeuroMab) was applied 1:200 overnight in blocking solution, followed by 1:200 secondary Alexa488-conjugated goat-anti-mouse. Images were taken on a LSM510 confocal fluorescence microscope using identical microscope settings for all slices by an observer blinded to genotype/seizure status of the slice. The lens aperture was set so that the focal length was larger than the section. A single image was measured to avoid photobleaching during the acquisition of confocal stacks. The thalamus was traced out based on the boundaries described by (Paxinos & Franklin 2008), and the image was focused so that the mean pixel intensity was maximized for each section (i.e., the focal z-axis encompassed the entire section) before the final image was acquired. Because side-by-side serial sections were used, the area traced for each permeabilized/non-premeabilized pair was as close to identical as possible. The mean pixel intensity was then recorded for the side-by-side permeabilized and unpermeabilized sections. The ratio of surface-to-total Kv4.2 expressed as percent surface expression was obtained by dividing the intensity of the non-permeabilized slice by the intensity of the adjacent permeabilized slice. For the in vitro treatments, 3 independent experiments (with 3 independent staining batches) were normalized to each other by setting the untreated percent surface expression value for each genotype to 1.
Statistical analysis
Comparisons between groups used two-tailed, unpaired student's t test, or ANOVA. Two-way ANOVA was used for comparisons involving both genotype and seizure status as independent variables, followed by Bonferroni's multiple comparison post-hoc test. p < 0.05 was considered significant, error bars represent SEM.
Results
Seizures induce rapid homeostatic plasticity of thalamocortical neuron excitability that is partially blocked in mLGI1 transgenic mice
We hypothesized that pilocarpine-induced seizures would induce rapid homeostatic changes in thalamic relay neurons. We recorded the responses of ventromedial thalamic relay neurons in acute slices from controls or mice sacrificed two hours following seizure induction. Only mice that experienced stage 4 or 5 seizures were studied. The driver inputs received by first order thalamocortical neurons, including ascending sensory inputs and possibly cortical layer V thalamic inputs, are typically composed of a single strong axonal input. Therefore action potentials within these single axons transmit as a brief, phasic, excitatory postsynaptic current. We modeled this brief phasic input by injecting short pulses of current which mimic an excitatory postsynaptic current (EPSC) into the neuronal soma while bypassing other seizure-induced changes of that native excitatory synapses. We were motivated to test this stimulus protocol because in our preliminary studies, wild-type mice showed very minimal changes in firing rate to a tonic 1 second current injection, but strong suppression of hyperpolarization-induced rebound burst firing in thalamocortical relay neurons (Kasten and Anderson, unpublished). Wild-type thalamocortical neurons that experienced a seizure in vivo displayed a marked decrease of phasic firing in response to current injections (Fig. 1).
Fig. 1.

Seizures strongly suppress phasic firing responses to EPSC-like currents. (a) Representative whole-cell, current-clamp tracings of the response of ventromedial thalamic relay neurons to EPSC-like currents of increasing intensity from control or mLGI1 transgenic mouse brain slices, either at baseline or 2 hours after a pilocarpine-induced seizure. (b) Cumulative probability plot showing reduced probability of firing in post-seizure slices. (c) Threshold current amplitude to fire was markedly increased in post-seizure slices. Two-way ANOVA reveals a significant effect of genotype (F(1,27)=5.74, p = 0.024), a significant effect of seizure (F(1,27)=52.39, p < 0.0001), and a significant interaction (F(1,27)=6.17, p = 0.019). * p < 0.05 ** p < 0.01 *** p < 0.001 Bonferroni's Multiple Comparison Post-Hoc test. n = 7-8.
Neurons were current-clamped at -70mV and increasing amounts of EPSC-like current were injected to simulate synaptic inputs (Fig. 1a). Control wild-type neurons generated action potentials (APs) in response to increasing EPSC-like current injections, while post-seizure wild-type neurons required a much larger current injections to induce the firing response (Control 337.5 ± 42.0, post-seizure, 800 ± 62.7, p < 0.001, t test) (Fig. 1a and b). Thus, seizures induce a rapid homeostatic down-regulation of phasic firing in thalamocortical neurons.
LGI1 protein is associated with human epilepsy and is heavily expressed in thalamus (Schulte et al. 2006). Mice expressing a human epilepsy-associated truncated dominant-negative mutant form of LGI1 are prone to chemically-kindled seizure with repeated subconvulsive doses of GABAergic blockers (Zhou et al. 2009). We speculated that this might be due, at least in part, to reduced anti-epileptic homeostasis. EPSC-like currents were injected into neurons from mLGI1 transgenic mice. Under control conditions, mLGI neurons generated action potentials (APs) in response to increasing current amplitudes at similar thresholds to controls (Fig. 1a and b). Mutant LGI1 transgene partially prevented the seizure-induced suppression of ES coupling seen in wild-type neurons (Fig. 1b). Firing threshold more than doubled from 337.5 ± 42.0 to 800 ± 62.7 in wild-type neurons, but increased more moderately from 342 ± 20.2 to 550 ± 65.8 in mLGI1 neurons following the seizure. Two-way ANOVA reveals a significant effect of genotype (F(1,27)=5.74, p = 0.024), a significant effect of seizure (F(1,27)=52.39, p < 0.0001), and a significant interaction (F(1,27)=6.17, p = 0.019) (Fig. 1c).
For comparison, we injected 1 second current pulses to assess tonic firing. Following the seizure, wild-type neurons required higher-amplitude current injections to fire APs than non-seizure wild-type neurons; however the effect was more modest than for phasic firing (control 27.5 ± 3.7 pA; seizure 43.3 ± 13.1 pA)(Fig. 2a and b). Also, unlike the findings for phasic firing, seizures also increased the threshold to fire in response to a 1 second pulse in mLGI1 neurons with no difference between control and mLGI1 neurons after seizure (Fig 2c). 2-way ANOVA reveals a significant effect of seizure status (F(1,25)=6.88, p = 0.015), but no effect of genotype (F(1,25)=0.16, p = 0.78), and no seizure-genotype interaction (F(1,25)=0.08, p = 0.68). Active and passive membrane properties remained constant before and after seizure (see Table 1).
Fig. 2.

Seizures moderately suppress firing to prolonged 1s current injections. (a) Representative whole cell current clamp tracings to 1 s current injections. (b) Cumulative probability plot of firing in both wildtype and mLGI1 neurons with or without a seizure. (c) Threshold current to fire significantly (2-way ANOVA) increases following a seizure (F(1,25)=6.88, p = 0.015), with no effect of genotype (F(1,25)=0.16, p = 0.78), and no seizure-genotype interaction(F(1,25)=0.08, p = 0.68).
Table 1.
Active and passive membrane properties of ventromedial thalamic relay neurons of wild-type (Wt) and mLGI1 transgenic mice at baseline and following a seizure. All statistics used two-way ANOVA; no significant differences were found (n=7-15).
| Wt | Wt + seizure | mLGI1 | mLGI1 + seizure | |
|---|---|---|---|---|
| Resting potential (mV) | -61.1 ± 1.4 | -63.8 ± 1.5 | -60.9 ± 0.9 | -60.6 ± 1.9 |
| AP Threshold (mV) | -34.9 ± 2.5 | -36.7 ± 1.6 | -38.1 ± 2.5 | -41.9 ± 2.8 |
| AP Spike Width (ms) | 1.4 ± 0.2 | 1.5 ± 0.2 | 1.1 ± 0.1 | 1.4 ± 0.2 |
| Time to first spike (ms) | 74.4 ± 11.5 | 71 ± 14 | 63.9 ± 16.6 | 63 ± 11.3 |
| Kv4.2 inactivation Tau (ms) | 39.5 ± 4.4 | 39.3 ± 3.1 | 35.0 ± 3.1 | 39.8 ± 5.0 |
Seizures increase thalamic relay neuron A-type K+ current in wild-type, but not mLGI1 transgenic mice
The dampening effects of A-type K+ currents regulate the integration of high-frequency inputs in hippocampus (Ramakers & Storm 2002). This current also shows the typical phasic activation and inactivation properties anticipated to selectively regulate phasic (but not tonic) firing responses. To isolate A-type K+ currents, we used a modified pulse voltage protocol based on Chen et al. (2006) (see methods) and isolated the slow and fast K-currents mathematically (Fig. 3a and b). High concentrations of 4-AP (5mM) blocked the fast but not slow K+ current (Fig 3c), a characteristic of A-type K+ currents.
Fig. 3.

Seizures increase A-type K+ currents in thalamic relay neurons. (a) Demonstration of the voltage protocols used to isolate Kv4.2 channels. (b) Representative current traces from control and mLGI1 neurons either at baseline or post-seizure. Only control-seizure neurons show a large (∼50%) increase in A-type K+ current magnitude. (c) 4-AP (5 mM) inhibits A-type K+ current. (d) Seizures effects on A-type K+ currents in control and mLGI1 transgenic neurons. Two way ANOVA reveals a significant effect of inactivation voltage (F(6,176)=41.36, p < 0.0001) and of group (F(1,176)=30.93, p < 0.0001) and a significant interaction (F(6,176)=2.42, p = 0.028). *** p < 0.001, ** p < 0.01, * p < 0.05 by Bonferroni's Multiple Comparison Post-Hoc test. (e) At -70 mV inactivation voltage, seizures significantly increase A-type K+ current only in wild-types. Two-way ANOVA reveals an effect of seizure (F(1,40)=4.1, p = 0.05), no effect of genotype (F(1,40)=2.6, p = 0.11) and no interaction (F(1,40)=1.5, p = 0.23). * p < 0.05 by Bonferroni's Multiple Comparison test. (f) Inactivation curve (b) normalized to peak current reveals (by two-way ANOVA) no effect of seizure (F(6,176)=0.44, p = 0.50), a significant effect of inactivation voltage (F(6,176)=52.84, p < 0.0001) and no interaction (F(6,176)=0.77, p = 0.99). (g) Half inactivation voltage displays no significant (two-way ANOVA) effect of seizure (F(1,39)=3.33, p = 0.08), genotype (F(1,39)=1.5, p = 0.23), or seizure-genotype interaction (F(1,39)=1.2, p = 0.28). (h) Slow K+ currents are similar in control and post-seizure neurons across multiple activation voltages. Two-way ANOVA: inactivation voltage (F(6,168)=101.7, p < 0.0001), group (F(3,168)=1.4, p = 0.23) interaction (F(1,39)=0.3, p = 0.99). (i) At +30 mV activation voltage, the slow K+ currents are similar: Two-way ANOVA: seizure (F(1,39)=0.7, p = 0.40), genotype (F(1,39)=1.8, p = 0.18), or seizure-genotype interaction (F(1,39)=0.3, p = 0.60). Error bars represent SEM. N = 7-15.
Pilocarpine-induced seizures markedly increased A-type K+ current in control mice (Fig. 3b). The current was significantly increased at inactivation voltages between -70 and -50 mV, inclusive (Fig. 3d and e). However, when the inactivation curve was normalized to peak current, the wild-type and post-seizure curves were statisitically indistinguishable, indicating steady-state voltage-dependent inactivation is similar (Fig. 3f). Curves were fit to individual neuron inactivation curves and the voltage resulting in 50% maximum current was not significantly different between control and post-seizure neurons (Fig. 3g). Further, the inactivation time constant of the A-type current was calculated for each cell, and was identical before and after seizure (Table 1). Slow K+ currents were unaltered following the seizure (Fig. 3h and i). The data suggest a channel carrying A-type K+ current may be rapidly recruited to the plasma membrane following a seizure.
To evaluate whether A-type K+ currents fail to increase in mutant LGI1 transgenic mice after seizures, the pulse protocol was repeated in mutant LGI1 under control or post-seizure conditions. Mutant LGI1 transgenic mice also displayed a fast-inactivating K+ current that was similar to wildtype under baseline conditions (Fig. 3b, d, and e). Post-seizure, mLGI1 neurons failed to show the robust increase in fast-inactivating K+ current seen in wild-type animals (Fig. 3b, d, and e). When the total current was normalized to peak current, there were no differences in relative current between different groups at different inactivation voltages (Fig. 3f). The voltage resulting in 50% maximum current was not significantly different from wild-type in either control or post-seizure mLGI1 neurons (Fig. 3g), and no differences in decay kinetics were observed (Table 1). Finally, slow K currents showed no difference in mLGI1 neurons compared to controls, with or without seizures (Fig. 3h and i).
Seizures rapidly increase surface Kv4.2 in wild-type, but not mLGI1 thalamus
The voltage-gating, kinetics, and 4-AP block of the A-type K+ current (Fig. 3) as well as the strong suppression of phasic relative to tonic firing (Figs. 1 and 2) following a seizure suggested possible involvement of Kv4 channels in the homeostatic response. Kv4.2 is the major A-type K+ channel transcript in thalamus (Serodio & Rudy 1998). Thus, we assayed brain sections for the surface expression of Kv4.2 by comparing the ratio of staining intensity on adjacent sections that were either un-permeabilized or triton-permeabilized. In unpermeabilized sections, the anti-Kv4.2 antibody (which binds to an extracellular epitope on Kv4.2) had access to surface, but not internal channel protein, while in triton-permeabilized sections the antibody labeled total Kv4.2. Thus, the ratio of non-permeabilized staining intensity to total staining intensity represents an approximation of the percentage of Kv4.2 expressed on the cell surface.
We found that two hours following a seizure, the percentage of Kv4.2 channels on the surface of wild-type thalamic neurons significantly increased (76 ± 4.4% vs. 100 ± 6.1%, p = 0.006, n = 8 and 6) (Fig. 4a and b) indicating seizures trigger surface recruitment of Kv4.2. To determine if total Kv4.2 is increased after a seizure, we ran western blots of Kv4.2, and found similar levels of Kv4.2 in the thalamus two hours post seizure (Fig. 4c and d). The results indicate seizures rapidly redistribute Kv4.2 to the surface and suggest this occurs without increasing total protein.
Fig. 4.
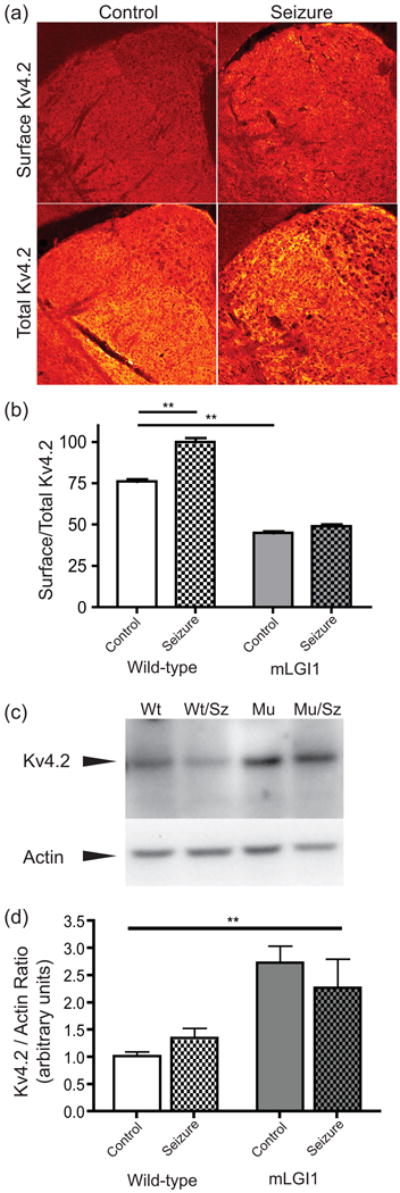
Seizures recruit surface Kv4.2 in wildtype but not mLGI1 transgenic mice. (a) Representative images of permeabilized (bottom, total Kv4,2) or non-permeabilized (top, surface Kv4.2) immunostained thalamus sections from mice given a pilocarpine seizure (right) or controls (left). (b) Quantification of the percent of surface-expressed Kv4.2 based on immunostaining of wild type or muLGI1 mice with or without seizures. Two-way ANOVA: Genotype: F(1,30)=766.3, p < 0.0001, Seizure F(1,30)=89.35, p < 0.0001, Interaction F(1,30)=45.59, p < 0.0001, ** p < 0.001, Bonferroni's Multiple Comparison Post-Hoc test. n = 6-11 adjacent sections from 3-4 animals. (c) Representative western blot showing total Kv4.2 levels in triton-solubilized thalamic lysates. (d) Quantification of western blots. Two-way ANOVA: Genotype: F(1,33)=16.44, p = 0.0003, Seizure F(1,33)=0.039, p = 0.84, Interaction F(1,33)=1.49, p = 0.23, ** p < 0.01, Bonferroni's Multiple Comparison Post-Hoc test.
In mLGI1 transgenic mice, two hours following a seizure, the percentage of Kv4.2 channels on the surface of thalamic neurons was unchanged, indicating a lack of Kv4.2 surface trafficking (45 ± 3.1% vs. 49 ± 3.8%, p = 0.43, n = 11 and 9; Fig. 4a and b). Moreover, the percentage of Kv4.2 at the neuronal surface at baseline was significantly lower when compared to controls (control vs. mLGI1 transgenic: 76 ± 4.4% vs. 45 ± 3.1%, p < 0.0001; Fig. 4b). The finding that surface/total Kv4.2 ratio is decreased, but that total A-type K+ current is unaffected in mLGI1 transgenic compared to wild-type at baseline (Fig. 3a) suggested that total Kv4.2 protein might be increased in mLGI1 transgenic mice to compensate for the reduced fraction of surface protein. This prediction was confirmed by western blot (Fig. 4d). The results indicate epilepsy-associated mLGI1 inhibits Kv4.2 surface recruitment. Under baseline conditions the cell compensates by increasing total Kv4.2 protein, thus normalizing total surface Kv4.2 and the corresponding A-type K+ current. However, following a seizure, the cell is unable to down-regulate its excitability by trafficking this increased pool of cytoplasmic Kv4.2 to the surface.
Direct glutamatergic signaling is sufficient to induce surface Kv4.2 recruitment in wild-type and the in vitro homeostatic response is blocked by mLGI1
To assess whether mLGI1 inhibits seizure-induced homeostatic responses via direct effects at the level of glutamatergic signaling we developed an in vitro assay. Slices were cut from wild-type and mLGI1 transgenic mouse brain as for electrophysiology, and were either left in ACSF or treated with high-KCl ACSF (KCl) or 10 μM glutamate (Glu) to mimic the effects of seizure-like activity in vitro. Surface expression of Kv4.2 was analyzed as above. Both KCl and glutamate increased Kv4.2 channel surface expression in wild-type slices (Fig. 5). By contrast, in brain slices from mLGI1 transgenic mice, Kv4.2 channels failed to redistribute to the surface following KCl or Glu treatment.
Fig. 5.

Glutamate and KCl application recruits surface Kv4.2 trafficking in vitro in wildtype, but not mLGI1 neurons. Slices were incubated in ACSF, ACSF with high KCl, or ACSF with 10 μM glutamate for 20 minutes. % surface expression was normalized to controls within-genotype to compare data among three independent experiments. ANOVA: F5,72=28.95, p < 0.001, * p < 0.01, ** p < 0.001 Bonferroni's Multiple Comparison Post-Hoc test. n = 5-20 pairs of adjacent sections from 3-4 animals.
Discussion
The thalamus relays sensory information to the cerebral cortex, and is intensely activated by seizures in vivo. In order to understand the physiological alterations of thalamocortical neurons triggered by a seizure, we recorded from these neurons in mice after induction of seizures with pilocarpine. We found that acute epileptiform activity strongly inhibited phasic and weakly inhibited tonic firing of thalamocortical neurons. This inhibition of phasic firing was not reversed by blocking the main channels previously implicated in regulating excitability of thalamocortical neurons, SK or Kv1 channels (Kasten et al. 2007) (data not shown), but instead resulted at least in part from an increase of A-type K+ currents associated with the surface recruitment of the A-type K+ channel subunit Kv4.2. Since Kv4.2 channels rapidly activate and then inactivate, enhancement of this particular outward potassium current would be expected to selectively inhibit depolarizing responses to a brief and rapid (EPSC-like) stimulus with little effect on firing to more prolonged stimuli. In mutant LGI1 mice, trafficking of Kv4.2 was impaired both at baseline and following the seizure. While a compensatory increase in total Kv4.2 protein levels normalized total A-type K+ current at baseline, following a seizure, the Kv4.2 surface expression remained at pre-seizure levels, impairing the homeostatic reduction in phasic firing (ES coupling). We propose that this failure to down-regulate neuronal excitability contributes to the seizures associated with LGI1 mutations in mice and humans.
Previous studies have been inconsistent, reporting either an increased or decreased expression of Kv4.2 following neuronal activity or seizures. ERK-mediated phosphorylation of Kv4.2 channels was associated with reduced synaptosomal (but not total) Kv4.2 in rat hippocampus one or three hours following a kainate-induced seizure in rats (Lugo et al. 2008). With longer delays following status epilepticus, others have also reported reduced total Kv4.2 and physiologic effects of A-type K+ currents in hippocampus in vivo (Bernard et al. 2004, Monaghan et al. 2008). Other studies have focused an activity-induced down-regulation of Kv4.2 as a mechanism for hippocampal synaptic plasticity (Chen et al. 2006, Frick et al. 2004, Kim et al. 2007). The pathways leading to Kv4.2 internalization have also been investigated (Kim et al. 2007, Jung et al. 2008). At least some of these studies reporting down-regulation of surface Kv4.2 with activity have been performed in cultured neurons suggesting the possibility that these are developmentally immature synapses. Consistent with this possibility is our observation of a trend towards KCl down-regulation of surface Kv4.2 in vitro in thalamic slices from mLGI1 mice (Fig. 5). Our previous studies have shown that glutamatergic synapses of the hippocampal perforant pathway to the dentate gyrus remain developmentally immature with a high NR2B/NR2A ratio (Zhou et al. 2009). However, glutamate did not produce this same down-regulation. Further studies will be necessary to explain the underlying mechanisms.
Conversely, brief (20 min) activity reportedly increased Kv4.2 surface expression and A-type K+ current in vitro in E18 rat cortical neurons (Shen et al. 2008). Calcium-calmodulin dependent protein kinase II (CAMKIIα) activation also increased Kv4.2 surface recruitment in hippocampal neurons, providing a link between excitatory activity, calcium entry, and Kv4.2 trafficking (Varga et al. 2004). Furthermore, direct SAP97-Kv4.2 interactions facilitated CAMKIIα-dependent up-regulation of Kv4.2 (Gardoni et al. 2007). KChIP isoforms also promote Kv4.2 surface expression, with the KChIP4a isoform effects uniquely facilitated by PKA (Lin et al. 2010). Thus, Kv4.2 surface expression shows bi-directional regulation under different conditions.
Our study focuses on an early, very rapid anti-seizure response in the thalamus in vivo where A-type K+ currents and A-type K+ channel Kv4.2 subunits are recruited to the plasma membrane, presumably inhibiting the transmission of pathologic network activity. This study makes use of a relatively simple, but infrequently used measurement when examining neuron excitability, the injection of short, EPSC-like currents. When more traditional long current injections were used to assess effects on action potential firing, weaker defects were observed that were not inhibited by the mLGI1 transgene. Our study also looked specifically at the ventromedial thalamus, which contains a relatively uniform population of excitatory relay neurons. It is not clear if this rapid neuroprotective response also occurs in other brain regions, however, the seizure-induced increase of surface Kv4.2 appeared to occur across multiple midline nuclei within the thalamus. The broad expression of Kv4.2 mRNA (Jerng et al. 2005) along with other studies showing similar activity-induced increases of Kv4.2 in cortex (Shen et al. 2008) or hippocampus (Varga et al. 2004) suggests Kv4.2 may play a broad role in neuroprotection. However, examples to the contrary also exist (Lugo et al. 2008).
LGI1 mediates developmental pruning of excitatory synapses in the hippocampus. Mutant LGI1 inhibits, while excess wild-type LGI1 magnifies the normal pruning of the dendritic arbor while concurrently lowering NMDA receptor NR2B/NR2A subunit ratio and presynaptic release probability (Zhou et al. 2009, Anderson 2010). Thus, during development, LGI1 acts as a developmental “circuit breaker” to promote the pruning of excess synaptic connections associated with immature neurons and to reduce glutamate synapse excitability to promote the normal developmental maturation of neuronal circuits. We speculate that this novel LGI1-Kv4.2 pathway may act during adulthood as a functional circuit-breaker to decrease neuron excitability and return overactive (seizing) circuits to their baseline state. While the mechanisms for the seizure- or glutamate- induced surface recruitment of Kv4.2 remains unclear, we speculate that they may result from increases of intracellular calcium coupling directly to CAMKIIα (Varga et al. 2004) or indirectly via calcium-dependent adenylate cyclase to PKA (Lin et al. 2010). Mutant LGI1 might inhibit this response by reducing calcium entry via effects on NMDA receptor composition (Zhou et al. 2009) or through effects on these signaling pathways, scaffolding molecules, or accessory channel subunits.
It was previously shown that cortical neurons from Kv4.2 knockout mice have unaffected AP thresholds and similar responses to a prolonged (400ms) current injection(Nerbonne et al. 2008), so alterations in amount of Kv4.2 was not expected to alter these properties as we report (Table 1).
Making a direct causal connection between the increased A-type K+ channel subunit Kv4.2 surface expression, the increased A-type K+ channel current, and the reduced ES coupling is hindered by the lack of an effective toxin for inhibiting Kv4.2 in brain slice preparations. While we demonstrate that 4-AP blocks the A-type K+ currents that we measured (Fig. 3c), the very high (mM) concentrations required would interfere with many other channels (e.g. Kv1 family) also important for ES coupling measurements. The spider toxins, phrixotoxin 1 and 2, are effective on Kv4.2 cDNA expressed in cell culture or Oocytes but have not proven effective on brain slices (Whyment et al. 2011; only 50% reduction in Kv4.2 conductance at 5 μM). However, our immunofluorescence studies showing that both seizures and in vitro glutamate treatments up-regulate Kv4.2 surface expression that is blocked by mLGI1; the biophysical properties of the A-type K+ currents; and importantly, the ES coupling inhibition by mLGI1 strongly supports our hypothesis that Kv4.2 at least partially underlies the rapid homeostatic down-regulation of phasic firing following a seizure.
In conclusion, we have shown that thalamic relay neurons respond to a seizure by trafficking intracellular stores of Kv4.2 to the surface. Mutant LGI1 transgenic mice display altered surface-to-intracellular distribution ratios under baseline conditions, and in response to a seizure fail to traffic increased Kv4.2 to the cell surface. This defect prevents the homeostatic increase in Kv4.2-mediated A-type K+ current seen in wild-type mice that associates with reduced ES coupling, a specific form of neuronal excitability. This lack of Kv4.2 surface trafficking impairs the ability of mLGI1 transgenic mice to homeostatically respond to a seizure by reducing neural excitability, potentially explaining the more severe epileptiform activity and facilitated kindling of seizures present in this model of human epilepsy (Zhou et al. 2009). Thus, we speculate that LGI1 may act via two pathways to create a seizure-prone brain: by interfering with normal glutamatergic circuit pruning and maturation (Zhou et al. 2009) and by disrupting glutamatergic circuit anti-epileptic mechanisms in the adult brain.
Acknowledgments
The authors wish to thank Andrew Varga for initially setting up the methods for Kv4.2 surface immunostaining, Oriana DiStefano and Xuan (Tony) Wang for technical assistance, and Ekkehard Kasper and Yu-Dong Zhou for experimental suggestions. This work was supported in part by the US National Institute of Neurological Disorders and Stroke R21NS070295, R01 NS057444, and K02 NS054674-03 (M.P.A.), Nancy Lurie Marks Family Foundation (M.P.A.), Autism Speaks/US National Alliance for Autism Research (M.P.A.), US National Institute of Mental Health NRSA Fellowship F32 MH087085 (S.E.P.S.), and Beth Israel Deaconess Medical Center.
Footnotes
The authors declare no conflict of interest.
References
- Anderson MP. Arrested glutamatergic synapse development in human partial epilepsy. Epilepsy Curr. 2010;10:153–158. doi: 10.1111/j.1535-7511.2010.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Varghese GI, Purcaro MJ, et al. Cortical and subcortical networks in human secondarily generalized tonic-clonic seizures. Brain. 2009;132:999–1012. doi: 10.1093/brain/awp028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Chabrol E, Navarro V, Provenzano G, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 2010;133:2749–2762. doi: 10.1093/brain/awq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, Shigemoto R, Nicoll RA, Fukata M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A. 2010;107:3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Marcello E, Sala C, Di Luca M, Jeromin A. SAP97 directs the localization of Kv4.2 to spines in hippocampal neurons: regulation by CaMKII. J Biol Chem. 2007;282:28691–28699. doi: 10.1074/jbc.M701899200. [DOI] [PubMed] [Google Scholar]
- Gu W, Brodtkorb E, Steinlein OK. LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol. 2002;52:364–367. doi: 10.1002/ana.10280. [DOI] [PubMed] [Google Scholar]
- Hashiguchi K, Morioka T, Yoshida F, et al. Thalamic hypometabolism on 18FDG-positron emission tomography in medial temporal lobe epilepsy. Neurol Res. 2007;29:215–222. doi: 10.1179/174313206X153851. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Kunjilwar K, Pfaffinger PJ. Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J Physiol. 2005;568:767–788. doi: 10.1113/jphysiol.2005.087858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz C, Nagy F, Watson C, da Silva EA, Muzik O, Chugani DC, Shah J, Chugani HT. Glucose and [11C]flumazenil positron emission tomography abnormalities of thalamic nuclei in temporal lobe epilepsy. Neurology. 1999;53:2037–2045. doi: 10.1212/wnl.53.9.2037. [DOI] [PubMed] [Google Scholar]
- Jung SC, Kim J, Hoffman DA. Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron. 2008;60:657–671. doi: 10.1016/j.neuron.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, Anderson MP. Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J Physiol. 2007;584:565–582. doi: 10.1113/jphysiol.2007.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Wikenheiser AM, Kung F, Hoffman DA. KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol Cell Neurosci. 2010;43:315–325. doi: 10.1016/j.mcn.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Barnwell LF, Ren Y, Lee WL, Johnston LD, Kim R, Hrachovy RA, Sweatt JD, Anderson AE. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J Neurochem. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan MM, Menegola M, Vacher H, Rhodes KJ, Trimmer JS. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience. 2008;156:550–562. doi: 10.1016/j.neuroscience.2008.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–1128. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic Press/Elsevier; Amsterdam; Boston: 2008. [Google Scholar]
- Paz JT, Chavez M, Saillet S, Deniau JM, Charpier S. Activity of ventral medial thalamic neurons during absence seizures and modulation of cortical paroxysms by the nigrothalamic pathway. J Neurosci. 2007;27:929–941. doi: 10.1523/JNEUROSCI.4677-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ramakers GM, Storm JF. A postsynaptic transient K(+) current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:10144–10149. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klocker N, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Serodio P, Rudy B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J Neurophysiol. 1998;79:1081–1091. doi: 10.1152/jn.1998.79.2.1081. [DOI] [PubMed] [Google Scholar]
- Shen B, Zhou K, Yang S, Xu T, Wang Y. The Kv4.2 mediates excitatory activity-dependent regulation of neuronal excitability in rat cortical neurons. J Neurochem. 2008;105:773–783. doi: 10.1111/j.1471-4159.2007.05179.x. [DOI] [PubMed] [Google Scholar]
- Sirerol-Piquer MS, Ayerdi-Izquierdo A, Morante-Redolat JM, Herranz-Perez V, Favell K, Barker PA, Perez-Tur J. The epilepsy gene LGI1 encodes a secreted glycoprotein that binds to the cell surface. Hum Mol Genet. 2006;15:3436–3445. doi: 10.1093/hmg/ddl421. [DOI] [PubMed] [Google Scholar]
- Thomas R, Favell K, Morante-Redolat J, et al. LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010;30:6607–6612. doi: 10.1523/JNEUROSCI.5147-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyvaert L, Chassagnon S, Sadikot A, LeVan P, Dubeau F, Gotman J. Thalamic nuclei activity in idiopathic generalized epilepsy: an EEG-fMRI study. Neurology. 2009;73:2018–2022. doi: 10.1212/WNL.0b013e3181c55d02. [DOI] [PubMed] [Google Scholar]
- Varga AW, Yuan LL, Anderson AE, Schrader LA, Wu GY, Gatchel JR, Johnston D, Sweatt JD. Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J Neurosci. 2004;24:3643–3654. doi: 10.1523/JNEUROSCI.0154-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyment AD, Coderre E, Wilson JM, Renaud LP, O'Hare E, Spanswick D. Electrophysiological, pharmacological and molecular profile of the transient outward rectifying conductance in rat sympathetic preganglionic neurons in vitro. Neuroscience. 2011;178:68–81. doi: 10.1016/j.neuroscience.2010.12.061. [DOI] [PubMed] [Google Scholar]
- Yu YE, Wen L, Silva J, Li Z, Head K, Sossey-Alaoui K, Pao A, Mei L, Cowell JK. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet. 2010;19:1702–1711. doi: 10.1093/hmg/ddq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]