

Abstract
Drug synergy allows a therapeutic effect to be achieved with lower doses of component drugs. Drug synergy can result when drugs target the products of genes that act in parallel pathways (‘specific synergy’). Such cases of drug synergy should tend to correspond to synergistic genetic interaction between the corresponding target genes. Alternatively, ‘promiscuous synergy’ can arise when one drug non-specifically increases the effects of many other drugs, for example, by increased bioavailability. To assess the relative abundance of these drug synergy types, we examined 200 pairs of antifungal drugs in S. cerevisiae. We found 38 antifungal synergies, 37 of which were novel. While 14 cases of drug synergy corresponded to genetic interaction, 92% of the synergies we discovered involved only six frequently synergistic drugs. Although promiscuity of four drugs can be explained under the bioavailability model, the promiscuity of Tacrolimus and Pentamidine was completely unexpected. While many drug synergies correspond to genetic interactions, the majority of drug synergies appear to result from non-specific promiscuous synergy.
Keywords: chemical genetics, drug combinations, drug discovery, genetic interactions
Introduction
Pathogens or tumors that do not respond to single treatments may be amenable to combined drug treatments (Yeh et al, 2006; Azmi et al, 2010). Synergistic drug pairs have special potential for treatment, since they allow a desired effect to be achieved with a lower total dose of administered medicine (Greco et al, 1995). Theoretical and experimental studies have shown that drugs that exhibit synergy for a specific effect are usually not synergistic for side effects (Lehar et al, 2009; Owens et al, 2010). These considerations make synergistic drug pairs ideal candidates for treatment of pathogens or tumors.
As a result of their medicinal potential, synergistic drug pairs are sought both experimentally and theoretically. Large-scale experimental drug synergy screens have found that synergistic drug pairs are rare (4–10%) (Borisy et al, 2003; Zhang et al, 2007; Farha and Brown, 2010). Therefore, methods which predict synergistic drug combinations have been developed, such as network analysis of expression profiles (Nelander et al, 2008), or clustering of chemogenomic profiles of drug perturbations (Jansen et al, 2009). In addition, perturbations with multiple drugs have been used to understand the organization and robustness of biological systems (Lehar et al, 2007).
One model for drug synergy is the parallel pathway inhibition model, which suggests that two drugs will be synergistic if they inhibit two proteins on parallel pathways essential for an observed phenotype (Yeh et al, 2009). To find protein pairs in parallel pathways, we used synergistic genetic interactions, where two combined genetic perturbations result in a phenotype that is more severe than expected (Tong et al, 2004; Mani et al, 2008). Because a drug's action on a target gene product can yield an effect that is similar to a hypomorphic or loss-of-function allele of the gene encoding the drug target (Parsons et al, 2004; Hillenmeyer et al, 2008), two drugs may be synergistic for the growth inhibition phenotype if they target the products of two genes that have a synergistic genetic interaction.
Several examples of genetically consistent synergy have been reported, such as the synergy involving a combination of either two mutations or two drugs that inhibit the DNA polymerase and thymidine kinase of Herpes Simplex Virus (Jacobson et al, 1995). Also, drug synergy between Terbinafine and Rapamycin is known to correspond to a synergistic genetic interaction between the target genes (Lehar et al, 2007).
Other models have been proposed to explain the mechanism of observed synergistic drug interactions (Jia et al, 2009). The bioavailability model suggests that two drugs will be synergistic if one drug's action helps another drug's availability in the target cells (Zimmerman et al, 2007), either by increasing the second drug's entry to the cell or by decreasing the second drug's degradation. For example, it has been reported that 1,8-Cineole enhances the effect of AZT, an anti-HIV drug, by increasing skin permeability (Narishetty and Panchagnula, 2004). Consistently, a number of drugs known to disrupt cell membranes, such as Triclosan in E. coli and ergosterol pathway-targeting drugs in yeast, have been found to exhibit many synergies (Ryder, 1992; Schweizer, 2001).
Combination therapies for which an underlying genetic explanation exists are attractive from a therapeutic standpoint. Such genetically determined drug synergies are expected to be more specific to particular pathways, thus having a reduced potential for unintended consequences. Concordantly, synergy which arises via the bioavailability model comes with the concern that bioavailability may be increased for many drugs beyond the one for which synergy is desirable, including drugs co-administered for other indications. However, it may not always be possible to find an efficacious combination therapy for which the genetic underpinnings are known. Indeed, it is unclear whether correspondence between gene synergies and drug synergies, which we refer to as ‘specific synergy’, is the predominant form of drug synergy.
If some drugs are inherently more likely to participate in synergistic combinations—for example, because they generally increase the bioavailability of other drugs—then the search for synergistic combinations might be accelerated by prioritizing combinations that contain promiscuous synergizers. Because ‘promiscuous synergy’ by definition encompasses many different drugs, it may be relatively common when compared with specific synergy.
Results
We first established an experimental framework for classifying the interaction between two drugs as synergistic, independent, or antagonistic. For each drug pair examined, we examined all possible combinations of seven concentrations for each drug, such that the concentration for each drug ranged from 0 to its minimal inhibitory concentration (MIC). Synergistic or antagonistic interaction can then be classified according to significant deviation from non-interaction behavior under the Loewe additivity model of drug synergy (Greco et al, 1995). The Loewe additivity model is intuitively appealing, in that a drug does not interact with itself under this model. As viewed on a two-dimensional grid, a drug combined with itself will yield linear isophenotypic contours (isobols), curves such that each point along the curve corresponds to a different drug concentration combination that yields the same growth phenotype. However, the contours will be convex when two drugs are synergistic, and concave when they are antagonistic (Figure 1). We used a mathematical model (see Materials and methods) to quantify the shape of isophenotypic contours where values near 0 indicate linearity (independence), negative values indicate convexity (synergy), and positive values indicate concavity (antagonism).
Figure 1.

Experimental set-up for classification of drug interactions. For each drug pair, growth rates were measured for all pairwise combinations of seven drug concentrations, linearly increasing from 0 to the minimal inhibitory concentration (MIC). ‘Isophenotypic’ curves describing drug concentration combinations which yield the same phenotype are shown on the left. Isophenotypic curves are expected to be parallel to the diagonal for independent drug pairs, concave for synergistic drug pairs, and convex for antagonistic drug pairs, according to Loewe additivity. For each drug pair, a drug interaction score (α) quantifying the concavity of the isophenotypic curve was computed, with α=0 defining independence and α taking negative or positive values when drugs are synergistic or antagonistic, respectively. Measurement error in α was assessed by examining the distribution of α for 25 self–self drug combinations, and drug pairs that had significantly smaller or larger α values were classified as synergistic or antagonistic, respectively. The white region shows the margin of variability between self–self interactions (−0.78< α<0.68, 95% confidence interval). Experimental examples of drug interactions are given on the right. In each example, Tacrolimus concentration is increased along the x axis. Drugs used in increasing concentrations along the y axis are Tacrolimus for self–self, Myriocin for independence, Latrunculin B for synergy, and Bromopyruvate for antagonism. For each example, sample isophenotypic curves are depicted in white. Yellow dots mark the longest isophenotypic curve, which was used to classify a drug interaction. See Supplementary Figure 1 for plots of all 200 pairwise drug interaction tests conducted in the course of this study.
To measure experimental variation in this interaction score, we evaluated 25 ‘self–self’ drug combinations that would be expected to yield a score of 0 in the absence of measurement errors. This allowed us to classify drug interactions as independent, significantly synergistic, or significantly antagonistic, depending on where the interaction score falls relative to the observed distribution of self–self interaction scores. We also considered Bliss and Gaddum's interaction models for such binary classifications (Berenbaum, 1989; Greco et al, 1995); however, we found that our model based on Loewe additivity provided the most intuitive and robust classification of drug interactions (see Materials and methods). Importantly, growth rates for self–self drug interaction experiments exhibited very high correlation along the symmetry axis (r=0.98, P<2.2 × 10–16), indicating the reproducibility of our experimental system.
To assess the overlap between synergistic genetic interactions and synergistic drug interactions, we focused on the yeast S. cerevisiae, the organism for which the largest number of synergistic genetic interactions is known. We compiled a catalog of 113 known chemical/target relationships in S. cerevisiae from the literature (Supplementary Table 1). All chemical compounds that yield a growth fitness defect when administered as single agents (referred to in this study as ‘drugs’) were considered, excepting compounds known not to act as inhibitors of their target. We next integrated the resulting drug-target catalog with known synergistic genetic interactions (Baryshnikova et al 2010; Costanzo et al, 2010; Stark et al, 2011; see Materials and methods). This yielded a set of 211 drug pairs predicted to be synergistic according to the parallel pathway inhibition model (Supplementary Table 2). To assess the success of these predictions, 38 of these drug pairs involving a total of 21 drugs were assessed for synergy. Except for three different drugs (DYC, FEN, and HAL) that were each used individually to inhibit Erg2, an ergosterol pathway protein, we used only one drug to inhibit each specific protein corresponding to a gene with a synergistic genetic interaction (see Table I for each drug's target name and function). Nine of these thirty-eight tested drug pairs were also predicted to be synergistic according to the bioavailability model, as they included an ergosterol pathway-targeting drug.
Table 1. List of all drugs used in this study, their abbreviation, highest dose used, target (bold if essential) and target function. Drugs that were found to be promiscuous are highlighted. Literature references are provided in Supplementary Table 1.
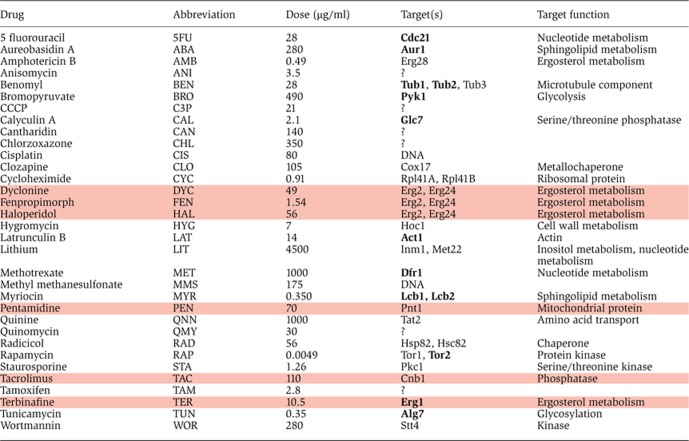
|
We found that 14 of the 38 tested drug pairs predicted to be synergistic using synergistic genetic interactions showed significant synergy (37% accuracy), while 11 of the 38 drug pairs (29%) showed significant antagonism (Figure 2). Although a set of negative control predictions tested within our framework would provide a better validation of the use of synergistic genetic interactions for predicting drug synergies, we initially noted that the prediction accuracy of the parallel pathway inhibition model was much higher than the previously reported background rate of antifungal synergy of 4–10% (Borisy et al, 2003; Farha and Brown, 2010).
Figure 2.

Synergy tests between 38 drug pairs that target proteins encoded by synergistic genes. (A) Drug pairs were combined in 8 × 8 matrices where the concentration of one drug was linearly increased along each axis. The lowest concentration was 0 and the highest concentration was chosen close to the MIC for each drug. Fourteen drug pairs showed significant synergy (marked with S) while eleven were significantly antagonistic (marked with A). Thirteen drug pairs yielded scores that fell within the distribution observed for self–self drug pairs, and were classified as independent (unmarked). (B) Network representation of the drug interactions shown in (A). Edges reflect the interaction type between two drugs and the node pie charts represent the ratio of different types of interactions each drug has in this data set (green: synergy, white: independent; red: antagonism). For drug name abbreviations, see Table I. One example for drug synergy prediction using synergistic genetic interactions: MYR targets Lcb2 and QNN targets Tat2 is also shown. There is a known synergistic genetic interaction between LCB2 and TAT2, which predicts that MYR and QNN will be synergistic according to the parallel pathway inhibition model of drug synergy. However, these drugs were found to be antagonistic.
Of particular note, all of the nine drug pairs that included an ergosterol pathway-targeting drug also exhibited synergy (100% accuracy). Indeed, these nine drug pairs—which represented only 24% of tested pairs—yielded 64% of the synergies among tested pairs, supporting the idea that drugs increasing bioavailability can provide an enhanced rate of synergy (P=1.2 × 10–5, Fisher's exact test). That ergosterol pathway-targeting drugs were enriched for synergy indicated that different drugs may have inherently different background synergy rates. In an effort to assess background synergy rates of different drugs, we tested all drug pairs in a square matrix of 13 drugs (Figure 3). Of these 13 drugs, 10 were partners in successfully predicted synergistic drug pairs in the initial screen (TER, FEN, HAL, DYC, PEN, TAC, LAT, BEN, STA, and RAP; see Table I for abbreviation key). Two drugs were partners in unsuccessful predictions (TUN and CAL) and BRO was added to the list arbitrarily.
Figure 3.

Synergy tests between all pairs among 13 drugs. (A) Drug pairs were combined in 8 × 8 matrices where the concentration of one drug was linearly increased along each axis. Green squares correspond to drug pairs that target proteins encoded by synergistic genes. We found 32 significant synergies (S) and 27 significant antagonisms (A) among the 78 drug–drug interactions tested. Hierarchical clustering of drug interaction score profiles is shown on the left. (B) Network representation of the synergistic and antagonistic drug interactions shown in (A). Edges reflect the interaction type between two drugs and the node pie charts represent the ratio of different types of interactions each drug has in this data set (green: synergy, white: independent; red: antagonism; independence edges are omitted for clarity). Gray box indicates the promiscuous synergizers learned from the drug interaction network in this figure. Yellow circle shows the drugs that target the ergosterol pathway in yeast. For drug name abbreviations, see Table I.
Of the 63 drug pairs in our test space that were not predicted to target synergistic gene products, we were surprised to observe that 21 (33%) showed synergy. The number of synergies each drug exhibited was not uniform (χ2 test, P=0.024), indicating the non-uniformity of background synergy rates for these 13 drugs. This observation suggests that there is no single universal background rate of synergy, which can be used as a basis for comparison of the success for a synergy prediction method.
As an initial attempt to account for background rates of synergy, we considered the baseline synergy rate of each drug tested in our 13 × 13 matrix in turn. For each drug, we examined whether drug pairs involving that drug were more likely to exhibit synergy if they corresponded to a genetic interaction. After correcting for multiple hypothesis testing, we found no evidence for significant enrichment of synergy among drug pairs corresponding to synergistic genetic interactions (using a threshold of 0.05 for adjusted P-values). This was despite the fact that the choice of drugs to fully test within the 13 × 13 matrix was initially biased to include drug pairs corresponding to genetic interaction. Thus, a consideration of baseline rates of synergy for each drug eliminates the significance of our observed correlation between synergistic genetic interaction and drug synergy.
Where drug synergy does correspond to genetic interaction, we wished to further analyze whether the observed drug synergies are the result of synergistic genetic interactions between the drug targets. Therefore, we conducted drug interaction experiments with an S. cerevisiae strain for which the CNB1 gene—encoding the target of TAC (Parsons et al, 2004)—was deleted. CNB1 has a synergistic genetic interaction with ACT1 and PKC1 (targets of STA and LAT, respectively), and we have shown that drug pairs TAC–LAT and TAC–STA are highly synergistic. We hypothesized that if the synergies between these drugs arise from the same mechanism as the synergistic genetic interactions, then these drug synergies should not exist in a cnb1 null mutant. However, we observed both of these drug synergies in cnb1Δ strains. This suggests that the observed synergies of drug pairs TAC–LAT and TAC–STA, successfully predicted on the basis of genetic interaction, are not mediated solely by the CNB1-ACT1 and CNB1-PKC1 synergistic genetic interactions. The observation that TAC exhibits synergies even when its known target has been deleted also suggests that there are other targets of TAC in S. cerevisiae. These results exemplify the idea that the existence of multiple targets for each drug can complicate tests of the correspondence between genetic interaction and drug synergy.
The observation that drugs have different background synergy rates suggested a strategy whereby pairs that involve at least one of these promiscuous synergizers are prioritized for testing. The number of synergies shown by each drug is a proxy for their background synergy rate. However, each of these synergies may be due to the high background synergy rates of the partner drugs. For a more robust analysis of background synergy rates of drugs, we clustered the interaction score profiles of the drugs in the 13 × 13 matrix. Not surprisingly, the three Erg2-targeting drugs were clustered together. Interestingly, these three and the other ergosterol pathway-targeting drug TER were clustered with TAC and PEN, despite the fact that the latter two drugs are not known to disrupt the cell membrane. All pairwise interactions within this set of six drugs were synergistic, except between Erg2-targeting drugs (which may be explained by competition between these drugs at the same target site). We refer to members of this cluster as promiscuous synergizers.
We also carried out a network analysis using the drug interaction network shown in Figure 3B in order to determine the smallest set of drugs that could ‘explain’ all observed drug synergies. When two drugs acted synergistically, we attributed the drug synergy to the drug with the highest level of background synergy. By individually removing the drugs with the highest background synergy rate and recomputing the number of synergies each drug exhibits in the resulting network, we found that the parsimonious classification of six drugs as promiscuous explained all the drug synergies in this data set. This smallest set coincided exactly with the promiscuous synergizers found by clustering analysis. Because the remaining seven drugs in our analysis exhibited relatively few synergies, we refer to these as chaste synergizers.
We next wondered whether promiscuity and chastity of drugs are intrinsic drug properties that would predict drug interaction behavior outside of the 13 × 13 drug matrix in which these properties were learned. Therefore, we further examined three promiscuous (TER, PEN, and TAC) and three chaste drugs (LAT, STA, and BEN), assessing synergy for each of these six against an array of 14 arbitrarily chosen drugs (Figure 4). Fourteen of sixteen synergies observed in this screen included a promiscuous synergizer drug, indicating a significant enrichment (Fisher's exact test, P=8.7 × 10–4) and supporting the idea that promiscuity is an intrinsic property with predictive value. Remarkably, LAT and STA did not show synergy with any of the 14 drugs, supporting the idea that they are intrinsically chaste. We found that BEN showed synergy with two arbitrarily chosen drugs (C3P and QMY) and it clustered with the other three promiscuous drugs in this new data set. While further experiments will be required to confidently characterize BEN as either promiscuous or chaste, these observations support the inherence of promiscuity for TER, PEN, and TAC. These results further bolster the conclusion that background synergy rates of different drugs are highly non-uniform.
Figure 4.

Synergy tests between 6 drugs (3 predicted to be promiscuous synergizers and 3 predicted to be chaste synergizers) and 14 arbitrarily chosen drugs. (A) Drug pairs were combined in 8 × 8 matrices where the concentration of one drug was linearly increased along each axis. Promiscuous synergizers (top three rows) showed 14 synergies (S) while chaste synergizers (bottom three rows) showed 2 synergies. Thirty-seven significant drug antagonisms (A) were observed, predominantly involving chaste synergizers. Green squares correspond to drug pairs that target proteins encoded by synergistic genes. Hierarchical clusterings of drug interaction score profiles are shown on the left and top. (B) Network representation of the synergy and antagonism drug interactions shown in (A). Edges reflect the interaction type between two drugs and the node pie charts represent the ratio of different types of interactions each drug has in this data set (green: synergy, white: independent; red: antagonism; independence edges are omitted for clarity). Gray circle indicates the promiscuous synergizers learned from the drug interaction network in Figure 3B. For drug name abbreviations, see Table I.
We next wondered whether drugs have intrinsic tendencies towards antagonism, as well as synergy. An analysis of the data in the 13 × 13 square matrix of drug interactions does not reject the null hypothesis that background antagonism rates of drugs are uniform (χ2 test, P=0.097). However, we note that BRO, which showed antagonism with every drug against which it was tested, was significantly enriched for antagonisms (P=2.9 × 10–6). Moreover, the synergy and antagonism rates of individual drugs have a significant negative correlation (r=−0.78, P=1.6 × 10–3). Thus, the antagonism background rates of individual drugs may be a useful exclusion criterion for accelerating the search for synergistic drug combinations.
Discussion
In this study, we systematically explored drug combinations to evaluate specific and promiscuous models of drug synergy. We developed a sensitive drug interaction classification assay and used it to find 38 synergistic drug pairs, of which only 1 (Terbinafine–Rapamycin) had been previously reported (Lehar et al, 2007).
To identify cases of specific synergy, predicted by synergistic genetic interaction, we integrated a newly assembled catalog of drug targets with the synergistic genetic interaction network. Although we tested 38 drug pairs corresponding to genetic interactions and found 14 (37%) to be synergistic, it is difficult to assess whether these drug synergies are better explained by genetic interaction or promiscuous synergy. Indeed, after accounting for background rates of synergy for each drug, our results did not show a significant enrichment for drug synergy among drugs targeting the products of genetically interacting genes.
There are many potential explanations for this observation. First, either or both of the drugs may not inhibit their targets completely or at all. Although chemogenomic studies have shown that the deletion of a target gene is often similar to treatment by the drug (Hillenmeyer et al, 2008), and although we excluded drugs that were known not to act as inhibitors, not all of the drugs within our tested set are known to act as inhibitors.
Second, each drug may have multiple targets. Action on additional targets may raise the background rate of drug synergy, making it more difficult to observe a significant enrichment for synergy predicted via genetic interaction. In rare cases, action on these additional targets may even suppress the effects of action on the known targets used to make drug synergy predictions.
Third, some drug targets in our literature-derived catalog may have been erroneously identified. The misidentification of targets would naturally have a profound effect on the apparent overlap between genetic interaction and drug synergy.
Fourth, intrinsic promiscuity of individual drugs may be the dominant factor in predicting drug synergy, again making it difficult to discern significant enrichment for synergy for drug pairs that correspond to genetic interactions. Indeed, our results support the idea that mechanisms other than parallel pathway inhibition of drug synergy are largely responsible for the synergistic drug interactions that are observed in our study.
Another model of drug synergy proposes that drugs may exhibit synergy if a drug's action helps a second drug's availability in the cell (bioavailability model). This model was systematically evaluated in our study, by testing synergy of drug pairs that included drugs that are known to destabilize the cell membrane by targeting the ergosterol pathway in yeast. We have shown that such drug pairs are enriched for drug synergy. We further classified drugs that partake in numerous synergies as ‘promiscuous synergizers’ (Farha and Brown, 2010). Interestingly, two of the drugs we classified as promiscuous synergizers are not known to destabilize the membrane. Further studies will be required to determine whether there are other classes of promiscuous synergizers that do not correspond to the bioavailability model of drug synergy. In fact, after >50 years since the bioavailability model for drug synergy was suggested (Jawetz and Gunnison, 1953), direct experimental evidence for this mechanism has not been produced. However, the analyses presented here strongly suggest that promiscuous synergizers enhance the potency of other drugs. Therefore, promiscuous drugs have a potentially valuable use in accelerating the search for synergistic drug combinations.
Additional models explaining drug synergy have been proposed. The physical interaction model proposes that two drugs physically interact to make a more potent compound. This model, which is largely discredited (Jawetz and Gunnison, 1953), was not evaluated here. The same target model proposes that two drugs will be synergistic if they target different sites on the same protein (Krogstad and Moellering, 1986). Incidentally, it has also been proposed that two drugs will be antagonistic if they target similar sites on a protein (competitive binding model; Krogstad and Moellering, 1986). In this study, we tested all pairwise combinations of three drugs that target the same protein, Erg2. Two of these drug pairs were found to be antagonistic, while none of them exhibited synergy. These observations support the idea that these three Erg2 binding drugs, as predicted by the similarity of their chemical structures, bind to similar pockets on the target protein (Giaever et al, 2004).
Our results show clearly that background synergy rate is drug dependent. This observation is critical for the assessment of any drug synergy prediction method, since it should account for the background synergy rates of all drugs that are in a drug combination.
The wealth of drug interaction results that are obtained in our study allows us to comment on mechanisms of drug antagonism. Two models for drug antagonism have been proposed. The first, referred to as the competitive binding model, was mentioned above with the same target model for drug synergy (Krogstad and Moellering, 1986). The other drug antagonism model suggests that drug antagonism occurs when a drug's action impedes the metabolism of the cell so that the second drug cannot exhibit its optimal effect (metabolic imbalance model; Bollenbach et al, 2009; Xavier and Sander, 2010). Concordantly, we note that Bromopyruvate, which is known to inhibit glycolysis, was antagonistic with all drugs tested, including promiscuous synergizers. Thus, our results hint that background drug antagonism rates are also non-uniform. Notably, a previous study suggested, in agreement with the metabolic imbalance model for drug antagonism, that drugs that target unrelated pathways are more likely to show antagonism (Lehar et al, 2007).
The study presented here provides a systematic exploration of drug synergies in the context of mechanistic models of drug synergy. We found 14 drug pairs to be consistent with specific synergy, that is, for which genetic interaction provides some evidence that the drugs target parallel pathways. Our results also suggest that discovery of synergistic drug pairs can be accelerated by considering the background synergy rate individual drugs. Although the predominant form of drug synergy in our study is promiscuous synergy, it remains possible that genetic interaction will be a good predictor of drug synergy in other contexts. For example, genetic interaction might predict synergies between chaste synergizers. Proper study of this question awaits an expansion in the number of known drug/target relationships and a further-expanded map of genetic interactions.
Materials and methods
Experimental materials and methods
5FU, ANI, BEN, BRO, C3P, CHL, CIS, DYC, FEN, HAL, LIT, MET, MMS, PEN, QNN, TAM, and TER were purchased from Sigma-Aldrich. AMB, CAL, CAN, CLO, CYC, HYG, LAT, MYR, QMY, RAD, RAP, STA, TAC, TUN, and WOR were purchased from AG Scientific. See Table I for abbreviation key. ABA was purchased from Clontech. All drugs were dissolved in DMSO (except Cisplatin which was dissolved in DMF) and kept at −20°C (except Cisplatin and Bromopyruvate which was freshly prepared due to loss of activity upon freezing). All experiments were conducted with S. cerevisiae strain BY4741, purchased from Open Biosystems. Yeast cells were grown in YPD (1% yeast extract, 2% bacto-peptone, 2% glucose) overnight (OD600∼5) and diluted to an OD600 of 0.01 in YPD with the desired drug concentrations controlled for solvent concentrations. Cells were grown for 24 h in 96-well plates in a Tecan F200 microplate reader, with OD595 readings of cell density recorded every 15 min. For each drug–drug interaction assay, we took 6144 cell-density measurements (detailed growth time courses for each of 64 concentration combinations with 96 time points in each time course). Synergy assays were performed for 200 drug pairs in total, so that a total of >1 million measurements were performed during this study. We used the area under growth curve of each condition as a metric of cell growth, after discarding the first 10 measurement points. The entire collection of growth measurement data is available as Supplementary Data Set 1.
Synergy assessment
To assess synergy, we used the Loewe Additivity Model, which defines a drug's interaction with itself as no interaction (for a comparison with other drug synergy models, see below) (Greco et al, 1995). Eight different concentrations of two drugs were combined in a two-dimensional grid, where the lowest concentration for each drug was 0 and the highest concentration for each drug was close to the MIC. The concentration of one drug was linearly increased on each axis (see Table I for the highest dose used for each drug).
The marginal effect of each drug alone was determined by assessing the growth under 0 concentration of the other drug. Additional grid points were imputed (using Akima interpolation; Akima, 1970) and contours were plotted, connecting the marginal growth rates of each drug through the grid along ‘isophenotypic’ curves. The maximum of the lowest marginal growths of each drug was used as a threshold, where any growth lower than this value was not considered (as contours could no longer connect the two marginal axes). The grids were then rescaled from 0 (low growth) to 1 (highest growth) and the longest isophenotypic curve (i.e., the curve described by the greatest number of data points) was selected for assessment. The following model was used to summarize this curve:
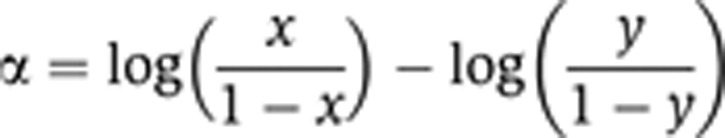 |
where x and y are the normalized drug concentrations and α is a free parameter describing the ‘bend’ of the curve. When α=0, the contour connecting the two marginal growth rates is linear (e.g., describing the case when a drug is tested against itself). When α<0 or α>0, drug synergy or antagonism is concluded, respectively.
To assess measurement error, 25 drugs were experimentally tested for interaction with itself (since by definition, there is no synergy for a drug with itself, any deviations of α from 0 represent measurement errors). We modeled the α values of drug self-interaction experiments as normal (as we did not find sufficient evidence to believe otherwise, KS test, D=0.15, P=0.58), with a mean of 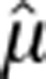 of –0.05 and standard deviation
of –0.05 and standard deviation  of 0.37. Hence, two drugs were considered synergistic or antagonistic when their α value was significantly smaller (α<−0.78) or larger (α>0.68) than the experimental mean of the drug self interaction scores, respectively.
of 0.37. Hence, two drugs were considered synergistic or antagonistic when their α value was significantly smaller (α<−0.78) or larger (α>0.68) than the experimental mean of the drug self interaction scores, respectively.
The synergy levels between drugs were also assessed using Bliss Independence and Gaddum's non-interaction models. For Bliss Independence, parameter β that minimizes the following metric was computed to assess the overall synergy between drugs 1 and 2:
and for Gaddum's non-interaction model, parameter γ that minimizes the following metric was computed:
where f(drug 1[x]) represents the cell growth in drug 1 at concentration x, and β and γ are free parameters describing the deviation from null model surfaces. According to these drug interaction models, β=1 or γ=1 represents non-interaction between two drugs. When these parameters are smaller or larger than 1, synergy or antagonism is concluded, respectively. Parameters β and γ for self–self drug interactions were approximately normally distributed. A comparison of the estimated β values with α scores estimated above showed that these two parameters had a very high correlation (r=0.79, P<10–43); however, γ values showed correlation with neither α nor β parameters. Moreover, according to the estimated γ values for our experiments, none of the tested drug pairs showed synergy or antagonism. While α parameters found only 1 self–self interaction as interacting (P<0.05), the Bliss Model found 2 self–self interactions as interacting (P>0.05). In the light of these observations, we concluded that Bliss Independence and Gaddum's non-interaction models, while useful for finding synergies between specific concentrations of two drugs, does not give robust results for an absolute measure of synergy between two drugs. Hence, we used the Loewe additivity model as our drug synergy-antagonism classifier.
Genetic interaction data
In addition to published sources of genetic interaction data (Baryshnikova et al, 2010; Costanzo et al, 2010; and Stark et al, 2011) we also used 11 unpublished interactions from the Boone and Andrews Labs: AUR1-MET22, ERG2-ACT1, HOC1-TOR2, ACT1-TOR1, ACT1-CNB1, INM1-TOR2, INM1-CNB1, LCB2-TAT2, LCB2-HSP82, LCB2-CNB1 HSC82-PKC1.
Supplementary Material
Raw cell growth measurements for all 200 drug-drug interaction assays. Each file contains a 96x64 matrix of numbers which corresponds to OD595 readings for one drug-drug interaction. Rows correspond to different time points with 15 minutes intervals that span 24 hours. Columns correspond to the 8 x 8 matrix of drug concentration combinations
Supplementary Figure S1, Supplementary Tables S1–2
Acknowledgments
We are indebted to the anonymous reviewers, whose initial comments substantially changed the direction of this study. We thank F Özbay, C Cenik, I Iossifov, M Yıldırım, E Demir, J Mellor, R Deo, and Y Bulut for their valuable comments, and to S Buratowski, N Gray, and C Shamu for providing reagents. M Cokol (MC1) and MT were supported by NIH Fellowships (GM086115 and HG004098, respectively). MC1 was supported by a FP7 Marie Curie IRG Grant (268440) and Scientific and Technological Research Council of Turkey (110S209). HNC was supported by the Agency of Science, Technology and Research, Singapore. CN and GG were supported by the Canadian Institutes of Health (MOP-81340 and MOP-84305). M Costanzo (MC2), AB, CLM, BJA, and CB were supported by a grant from the National Institutes of Health (HG005853-01). FPR was supported by NIH grants (HG003224, HG004756, CA130266, MH087394 and HG004233), a Canadian Institute for Advanced Research Fellowship and a Canada Excellence Research Chair.
Author contributions: MC1 and FPR designed the study. MC1, BM, and YS conducted the experiments. MC1, HNC, BM, ZBW, MEN, GG, and CN constructed the drug-target dataset. MC2, AB, CLM, BJA, and CB contributed genetic interaction data. MC1, HNC, and MT analyzed the data. MC1, ZBW, and FPR wrote the paper.
Footnotes
MC1, HNC, and FPR have filed a US patent application on synergistic antifungal drug combinations discovered in the course of this study.
References
- Akima H (1970) A new method of interpolation and smooth curve fitting based on local procedures. J ACM 17: 589–602 [Google Scholar]
- Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH (2010) Proof of concept: network and systems biology approaches aid in the discovery of potent anticancer drug combinations. Mol Cancer Ther 9: 3137–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, Youn JY, Ou J, San Luis BJ, Bandyopadhyay S, Hibbs M, Hess D, Gingras AC, Bader GD, Troyanskaya OG, Brown GW, Andrews B, Boone C, Myers CL (2010) Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods 7: 1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum MC (1989) What is synergy? Pharmacol Rev 41: 93–141 [PubMed] [Google Scholar]
- Bollenbach T, Quan S, Chait R, Kishony R (2009) Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell 139: 707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER, Serbedzija G, Zimmermann GR, Foley MA, Stockwell BR, Keith CT (2003) Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA 100: 7977–7982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M et al. (2010) The genetic landscape of a cell. Science 327: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farha MA, Brown ED (2010) Chemical probes of Escherichia coli uncovered through chemical-chemical interaction profiling with compounds of known biological activity. Chem Biol 17: 852–862 [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW (2004) Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci USA 101: 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco WR, Bravo G, Parsons JC (1995) The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 47: 331–385 [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320: 362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson J, Kramer M, Rozenberg F, Hu A, Coen DM (1995) Synergistic effects on ganglionic herpes simplex virus infections by mutations or drugs that inhibit the viral polymerase and thymidine kinase. Virology 206: 263–268 [DOI] [PubMed] [Google Scholar]
- Jansen G, Lee AY, Epp E, Fredette A, Surprenant J, Harcus D, Scott M, Tan E, Nishimura T, Whiteway M, Hallett M, Thomas DY (2009) Chemogenomic profiling predicts antifungal synergies. Mol Syst Biol 5: 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawetz E, Gunnison JB (1953) Antibiotic synergism and antagonism; an assessment of the problem. Pharmacol Rev 5: 175–192 [PubMed] [Google Scholar]
- Jia J, Zhu F, Ma X, Cao Z, Li Y, Chen YZ (2009) Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov 8: 111–128 [DOI] [PubMed] [Google Scholar]
- Krogstad DJ, Moellering RCJ (1986) Antibiotics in Laboratory Medicine. Baltimore: Williams and Wilkins [Google Scholar]
- Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, Staunton JE, Jin X, Lee MS, Zimmermann GR, Borisy AA (2009) Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 27: 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehar J, Zimmermann GR, Krueger AS, Molnar RA, Ledell JT, Heilbut AM, Short GF, Giusti LC, Nolan GP, Magid OA, Lee MS, Borisy AA, Stockwell BR, Keith CT (2007) Chemical combination effects predict connectivity in biological systems. Mol Syst Biol 3: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R, St Onge RP, Hartman JL, Giaever G, Roth FP (2008) Defining genetic interaction. Proc Natl Acad Sci USA 105: 3461–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narishetty ST, Panchagnula R (2004) Transdermal delivery of zidovudine: effect of terpenes and their mechanism of action. J Control Release 95: 367–379 [DOI] [PubMed] [Google Scholar]
- Nelander S, Wang W, Nilsson B, She QB, Pratilas C, Rosen N, Gennemark P, Sander C (2008) Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol 4: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens CM, Mawhinney C, Grenier JM, Altmeyer R, Lee MS, Borisy AA, Lehar J, Johansen LM (2010) Chemical combinations elucidate pathway interactions and regulation relevant to Hepatitis C replication. Mol Syst Biol 6: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C (2004) Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 22: 62–69 [DOI] [PubMed] [Google Scholar]
- Ryder NS (1992) Terbinafine: mode of action and properties of the squalene epoxidase inhibition. Br J Dermatol 126 (Suppl 39): 2–7 [DOI] [PubMed] [Google Scholar]
- Schweizer HP (2001) Triclosan: a widely used biocide and its link to antibiotics. FEMS Microbiol Lett 202: 1–7 [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, Reguly T, Rust JM, Winter A, Dolinski K, Tyers M (2011) The BioGRID Interaction Database: 2011 update. Nucleic Acids Res 39: D698–D704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L et al. (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Xavier JB, Sander C (2010) Principle of system balance for drug interactions. N Engl J Med 362: 1339–1340 [DOI] [PubMed] [Google Scholar]
- Yeh P, Tschumi AI, Kishony R (2006) Functional classification of drugs by properties of their pairwise interactions. Nat Genet 38: 489–494 [DOI] [PubMed] [Google Scholar]
- Yeh PJ, Hegreness MJ, Aiden AP, Kishony R (2009) Drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol 7: 460–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yan K, Zhang Y, Huang R, Bian J, Zheng C, Sun H, Chen Z, Sun N, An R, Min F, Zhao W, Zhuo Y, You J, Song Y, Yu Z, Liu Z, Yang K, Gao H, Dai H et al. (2007) High-throughput synergy screening identifies microbial metabolites as combination agents for the treatment of fungal infections. Proc Natl Acad Sci USA 104: 4606–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman GR, Lehar J, Keith CT (2007) Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today 12: 34–42 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw cell growth measurements for all 200 drug-drug interaction assays. Each file contains a 96x64 matrix of numbers which corresponds to OD595 readings for one drug-drug interaction. Rows correspond to different time points with 15 minutes intervals that span 24 hours. Columns correspond to the 8 x 8 matrix of drug concentration combinations
Supplementary Figure S1, Supplementary Tables S1–2