

Abstract
Molecular modeling of agonist binding to the human A2A adenosine receptor (AR) was assessed and extended in light of crystallographic structures. Heterocyclic adenine nitrogens of co-crystallized agonist overlayed corresponding positions of the heterocyclic base of a bound triazolotriazine antagonist, and ribose moiety was coordinated in a hydrophilic region, as previously predicted based on modeling using the inactive receptor. Automatic agonist docking of 20 known potent nucleoside agonists to agonist-bound A2AAR crystallographic structures predicted new stabilizing protein interactions, to provide a structural basis for previous empirical structure activity relationships consistent with previous mutagenesis results. We predicted binding of novel C2 terminal amino acid conjugates of A2AAR agonist CGS21680 and used these models to interpret effects on binding affinity of newly-synthesized agonists. D-Amino acid conjugates were generally more potent than L- stereoisomers, and free terminal carboxylates more potent than corresponding methyl esters. Amino acid moieties were coordinated close to extracellular loops 2 and 3. Thus, molecular modeling is useful in probing ligand recognition and rational design of GPCR–targeting compounds with specific pharmacological profiles.
Keywords: G protein-coupled receptor, nucleosides, purines, radioligand binding, docking, X-ray crystallography
Introduction
Despite the great importance in human biology and clinical applications of G protein-coupled receptors (GPCRs),1–3 crystallography has only recently started to overcome technological barriers to yield the first high resolution structures for this membrane protein family.4–8 While initially a number of GPCR structures were resolved in an inactive state stabilized by antagonist or inverse agonist, four GPCRs have also been resolved recently in the active state: bovine opsin,9–12 β2- and β1-adrenergic receptors,13–15 and human A2A adenosine receptor (AR).16,17 Among the conformational changes induced by agonists are common movements and rotations of transmembrane domains (TMs) 3, 5, 6 and 7. These helical rearrangements enlarge a crevice in the intracellular interface of the receptor, facilitating G protein binding and activation.
The crystal structures of the human A2AAR have been solved in complexes with several different agonists using two very different approaches to stabilize the active state of the receptor. In the first case, A2AAR was stabilized by extensive interactions with a bulky (~780 Da), conformationally selective agonist 2-N-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl-N6-(2,2-diphenylethyl)-5′-N-ethylcarboxamidoadenosine-2-carboxamide 1 (UK-432,097) and crystallization in lipidic cubic phase (LCP).16,18 In the second case, A2AAR was thermostabilized by four point mutations in the receptor, allowing its crystallization in complex with the much smaller agonists adenosine 4 and its derivative 5′-N-ethylcarboxamidoadenosine 14 (NECA).17 Despite the differences in complex composition and crystal packing, all these A2AAR structures display very similar activation-related changes on the intracellular side, while additional differences specific for the binding of the bulky agonist 1 to the A2AAR are located at the extracellular surface, mainly within the extracellular loops (ELs) 2 and 3.
There is still significant interest in predicting binding modes of agonists based on the more prevalent inactive state structures. This approach has resulted in several studies over the past few years targeting agonists of the β2 adrenergic receptor8 and A2AAR,19,20 as reviewed by Katritch et al.21 Now, with experimental structures of agonist complexes of the human A2AAR available, it is possible to evaluate the quality of these previous models of agonist binding and to discern which approaches are more likely to accurately predict the binding modes of other known agonists (3 – 22, Table 1). The binding modes of other agonists not yet crystallized in complex with the A2AAR are of current interest, such as 1-[6-amino-9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]purin-2-yl]-N-methylpyrazole-4-carboxamide 10 (CVT-3146, regadenoson, Lexiscan), a short-acting adenosine A2AAR agonist already approved as a diagnostic coronary vasodilator.22
Table 1.
Structures and affinities of agonists of the A2AAR used in docking to the crystallographic structure of the agonist-bound receptor. Unless noted, X = O; Y = N; R', R" = H.
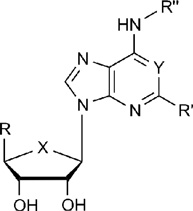 | ||||
|---|---|---|---|---|
| Compound | R | R’ | R’’ | Ki or IC50, nMa (A2A) |
| 1 | CONHC2H5 |  |
 |
4 |
|
3 (X = S) |
H |  |
H | 7.19 |
| 4 | CH2OH | H | H | ~20 |
| 5 | CH2OH |  |
H | 5.7 |
| 6 | CH2OH |  |
H | 58.5 |
| 7 | CH2OH |  |
H | 3.8 |
| 8 | CH2OH | 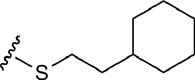 |
H | 372 |
| 9 | CH2OH | 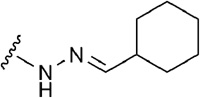 |
H | 270 |
| 10 | CH2OH |  |
H | 290 |
| 11 | CH2OH | H |  |
49.9 |
| 12 | CH2OH | H | 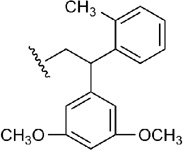 |
168 |
| 13 | CH2OH |  |
 |
20.1 |
| 14 | CONHC2H5 | H | H | 2.2 |
| 15 | CONHC2H5 | 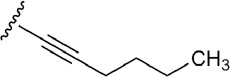 |
H | 6.4 |
| 16 | CONHC2H5 | 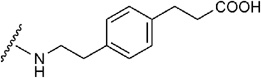 |
H | 27 |
| 17 | CONHC2H5 | 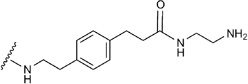 |
H | 5.73 |
| 18 | CONHC2H5 | 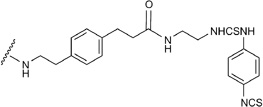 |
H | 7.1 |
| 19 | CONHC2H5 |  |
H | 0.5 |
|
20 (X = CH2, Y = CH) |
CONHC2H5 | H | 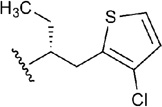 |
56 |
| 21 | 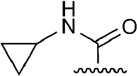 |
 |
H | n.d. |
| 22 | 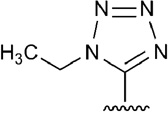 |
 |
H | 2.3 |
The objectives of this study were three-fold: 1) Because much of the docking of biologically relevant GPCRs will still require use of the inactive basal conformational state of a given receptor, we have evaluated the accuracy of previously predicted interactions of A2AAR agonists19,20 in light of the complexes recently crystallized; 2) Docking predictions were made for a wide range of known A2AAR agonists by extending the structures of agonist complexes crystallized. This helps to interpret already elucidated structure activity relationships (SARs) in this chemical series in terms of predicted interactions with the A2AAR receptor; 3) We predicted docking of novel C2 terminal amino acid conjugates of A2AAR agonist 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine 16 (CGS21680) and used these models to interpret effects on the measured binding affinity of the newly-synthesized agonists.
Results
Evaluation of nucleoside binding to the A2AAR: Predicted model based on inactive state vs. crystallographic structure
Because of the activation-related conformational changes, receptor modeling of an agonist binding to a template representing the inactive or basal state of a GPCR is especially challenging and may require additional validation.23 To assess the quality of such docking, we have compared the docking of adenosine derivatives in our previous study19 using the inactive, basal conformation of the A2AAR4 (PDB code: 3EML) with the newly reported X-ray structure of an agonist-bound receptor (PDB code: 3QAK).16 The agonist-bound structure has many of the features of activated GPCRs, including characteristic helical movements and microswitches on the intracellular side.24 The active state of the receptor was also supported by observed pharmacological behavior in modulation of nucleoside binding by sodium ions.16
Previously, various residues located mostly in TMs 3, 5, 6, and 7 of the A2AAR were predicted by modeling to be involved in agonist recognition (Table S1).19 In this model, Thr88 (3.36), Phe168 (EL2), Asn181 (5.42), His250 (6.52), Asn253 (6.55), Ser277 (7.42), and His278 (7.43) are located in proximity to the potent, nonselective agonist 14 and are involved in ligand interactions (the numbers in parentheses correspond to the Ballesteros-Weinstein indexing system).25 The importance for agonist binding of residues located in the TM regions and in EL2 was supported by site-directed mutagenesis data obtained for the A2AAR,26 and also for closely related A1 and A3AR subtypes.27,28 In agreement with the indications from molecular modeling and pharmacological studies, all of these critical residues are located in proximity to the adenosine core of the agonist ligand 1 in the recent crystallographic structure of the A2AAR with 1 bound (designated here 1-A2AAR).16 The previous docking orientation19 of both agonist ligands, adenosine 4 and 5′-uronamide 14, in the inactive state of the human A2AAR4 was nearly identical to the binding mode of the adenosine moiety of the agonist 1 in the TM region as determined by X-ray crystallography (Figure 1).
Figure 1.

A. The newly determined structure of the A2AAR is shown surrounding its synthetic agonist 1.16 The helices (color sequence from red to blue) and the slim connecting loops represent the receptor protein, winding back and forth through the cell membrane. The central ribose moiety (red) of the agonist binds in a hydrophilic region and is critical for activation of the receptor, while the adenine heterocycle (blue) binds in a hydrophobic region. The top (tan-colored) C2 and N6 substituents of the agonist, facing the outside of the cell, effectively fill the remaining spaces in the binding site and stabilize the receptor in order to obtain a crystallized structure. B. Similar view of the agonist docking model of Ivanov et al. using the inactive A2AAR structure.4,19 The potent nonselective agonist 14 is present in the binding site. C. Superposition of the agonist 1 (carbons colored in green) in 1-A2AAR (represented as ribbon colored in green), the antagonist 2 (carbons colored in orange) in 2-A2AAR (represented as ribbon colored in orange), and the predicted docked pose of 14 as in Ivanov et al. (carbons colored in cyan).
A specific mode of overlay of both small agonists 4 and 14 with the position of antagonist 4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol 2 (ZM241,385) inside the inactive A2AAR (designated here 2-A2AAR) was predicted,4,19 and this correspondence was compared with the position of 1 in the co-crystallized structure.16 The modes of overlay and interactions with specific amino acid residues in the X-ray structure 1-A2AAR and in the predicted model are shown in Supporting Information. The adenine moiety of the agonists and the triazolotriazine fragment of 2 have very similar positions inside the binding site and are involved in similar interactions with the receptor, i.e. a π-π stacking interaction with Phe168 (EL2) and H-bonding of the exocyclic amine with Asn253 (6.55). The position of the carboxylate of Glu169 (EL2) has shifted in the 1-A2AAR X-ray structure from the exocyclic amine of adenine in docked agonists 4 and 14 to the urea group of the extended C2 side chain of 1. This side chain shift is considered to occur only after the binding of agonists with bulky substituents at the N6, such as 1, and was not expected in the earlier modeling predictions, possibly because no N6 and C2 substitutions were examined. In the recent structure of 14 bound to a thermostabilized A2AAR, Glu169 (EL2) does not shift away from the (unsubstituted) exocyclic amine.17 The adenine C2-position of the modeled agonists 4 and 14 was oriented toward the extracellular part of the receptor, as found in the 1-A2AAR structure and later confirmed in the crystal structures of the A2AAR in complex with 4 (designated here 4-A2AAR) and 14 (designated here 14-A2AAR). The ribose moiety of 4 and 14 docked to 2-A2AAR was predicted to be in proximity to hydrophilic residues in TM3 and TM7, i.e. Thr88, Ser277, and His278. The docking poses of 4 and 14 were able to predict the interactions of the ribose ring with residues in TM3, TM6, and TM7 but could not predict the backbone changes of the TMs upon binding with agonists, as shown in the agonist-bound crystal structures.
Evaluation of binding of known active nucleosides at the A2AAR: Predictions based on agonist-bound receptor complexes
Re-docking of co-crystallized agonist 1
To investigate the ability of molecular docking to reproduce an experimentally observed ligand binding mode, the potent A2AAR agonist 1, formerly in clinical trials by Pfizer for chronic obstructive pulmonary disease,29 was re-docked to the crystal structure of the 1-A2AAR. Different automated docking approaches with various protocols and distance constraints were tested in order to choose the docking methods that were capable of retrieving a docking pose for 1 with a root mean square deviation (RMSD) less than 2Å from the published crystal structure.16 The peculiar molecular properties of 1 and the shape and electrostatic characteristics of the binding cavity of the active receptor, with a big portion of the ligand exposed to the solvent, make automated docking challenging. Compound 1 breaks three of Lipinski’s rules of five30 for the druglikeness with 7 H-bond donors, 11 H-bond acceptors, and a molecular weight of 778 daltons. The numerous rotatable bonds in 1 define a high conformational freedom of the ligand, and usually a compound’s high flexibility negatively affects the accuracy of automated docking.31 Moreover, the shape of the receptor’s binding pocket in the 1-A2AAR structure shows a large portion of the co-crystallized ligand in the extracellular media. Some automated docking protocols can perform poorly in retrieving accurate poses for partially buried ligands.31 Those issues might account for some of the difficulties in reproducing the co-crystallized orientation of 1 in the binding site of the 1-A2AAR structure with standard automated docking procedures such as Glide32 or MOE.33
Unexpectedly, the Glide docking was unable to find a docking pose for 1 with an RMSD lower than 2Å compared with the corresponding crystal structure, even after the use of distance restraints to force the formation of the key H-bond interactions in the binding site. One docking protocol among the tested ones able to retrieve an accurate pose of 1 closer than an RMSD of 2Å from the experimental conformation was the MOE docking with the pharmacophore placement method, where a pharmacophore model was used to guide the docking process. Pharmacophore-based docking is a useful way to include the known ligand-receptor interactions in the docking procedure. In the pharmacophore query used in this docking study (refer to the methodology paragraph for more details), four pharmacophore features were used: the projections of the N6 H-bond donor and N7 H-bond acceptor of the adenine ring on Asn253 (6.55), and the projections of the OH groups at 2’ and 3’ position of the ribose ring on His278 (7.43) and Ser277 (7.42) respectively (Figure S2). These ligand-receptor interactions are considered to be critical for the binding of 1 in the binding pocket of the A2AAR and are expected to be conserved for all the adenosine-like agonists. The most favorably MOE-docked pose of compound 1 in the binding pocket of 1-A2AAR had an RMSD of 0.42Å from the X-ray conformation. The second and the third best poses had a RMSD of 1.29Å and 1.63Å, respectively, and showed the adenosine core and the N6-diphenylethyl moiety correctly positioned in the binding site, but with the terminal end of the C2-substituent in a slightly different orientation (Figure S2). It should be noted that the majority of the docking solutions had an RMSD greater than 2Å due to the misplacement of the long and highly flexible chain at the C2-position of 1 even if the adenosine core of the compound was correctly positioned in the binding pocket.
After extensive comparison of Glide and MOE, we also found that re-docking of 1 using the ICM-Dock module of Molsoft34 reproduced the position of the agonist in the X-ray structure with a RMSD value within 1Å.
Molecular docking of diverse known agonists to agonist-bound A2AAR crystal structures
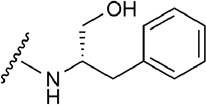
A diverse set of nucleosides that are potent and/or selective agonists to the A2AAR was assembled (Table 1).22,29,35–45 These 20 adenosine derivatives contain modifications of the ribose moiety, a nucleobase substitution, or combinations thereof. They include clinical candidates, such as 2-[2-(4-chlorophenyl)ethoxy]adenosine 6 (MRE-0094, sonedenoson),56 2-((cyclohexylmethylene)-hydrazino)adenosine 9 (WRC-0470, binodenoson),57 13 (UK-371,104),36 [trans-4-{3-[6-amino-9-(N-ethyl-β-D-ribofuranosyluronamide)-9H–purin-2-yl]prop-2-ynyl}cyclohexanecarboxylic acid methyl ester 19 (ATL-146e, apadenoson),39 [1S-[1a,2b,3b,4a(S*)]]-4-[7-[[2-(3-chloro-2-thienyl)-1-methylpropyl]amino]-3H-imidazo[4,5-b]pyridyl-3-yl]cyclopentane carboxamide 20 (AMP-579),44 4-{3-[6-amino-9-(5-cyclopropylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]prop-2-ynyl}piperidine-1-carboxylic acid methyl ester 21 (ATL-313),39 and (2R,3R,4S,5R)-2-(6-amino-2-{[(1S)-2-hydroxy-1-(phenylmethyl)ethyl]amino}-9H-purin-9-yl)-5-(2-ethyl-2H-tetrazol-5-yl)tetrahydro-3,4-furandiol 22 (GW 328267X, which also binds to the A3AR),45,58 and the approved clinical diagnostic agent 10. Various agonists used as research tools and molecular probes of the A2AAR (such as the the amine congener 17 and its irreversibly binding isothiocyanate derivative 18)43 are also included. Various 2-hexynyl derivatives of adenosine, e.g. 5 and 15, were found to maintain high affinity at the A2AAR.40 The 2-hexynyl 5’-truncated agonist 3, a 4’-thioadenosine, maintains affinity and selectivity at the A2AAR.38 Compounds 11 and 12, containing the same N6-(2,2-diphenylethyl) group as 1, were among the first A2AAR agonists identified.42 Several other 2-ether, i.e. the bulky naphthyl derivative 7,41,50 or 2-thioether derivatives, i.e. cyclohexylethyl derivative 8 that was converted to a prodrug form,37 were included. Each of these agonists was sequentially docked to the 1-A2AAR or 14-A2AAR crystal structure. A variety of substituents at the 5’, C2, and N6 positions was chosen to allow the characterization of the different subpockets where those substituents locate in the binding cavity of the receptor and to highlight the relevance of receptor residues important for the binding and activity of those compounds.
An automated docking approach was used to place the molecules in the rigid binding cavities of 1-A2AAR or 14-A2AAR after removal of the co-crystallized ligand, using, as for compound 1, different techniques and protocols in order to find a placement able to reproduce the common interactions with the adenosine moiety shown by 1 in the crystal structure. The indispensable chemical features needed by an agonist to activate the A2AAR are shared by all the agonists in our list: the aromatic nature of the nucleobase for the π–π stacking with Phe168 (EL2) and the stabilizing interactions with Leu249 (6.51) and Ile274 (7.39), the H-bond acceptor nitrogen atom at N7 and the exocyclic amino group of the adenine core to interact with Asn253 (6.55), and the two hydroxyl groups at 2’- and 3’-positions to interact with Ser277 (7.42) and His278 (7.43). These molecular characteristics of the nucleoside agonists are crucial for the ligand recognition at the receptor site and are considered essential for the molecules’ biological activity.
Docking poses with the correct critical interactions with Phe168, Asn253, Ser277 and His278 in the binding cavity of the receptor were so retrieved and further optimized by means of a Monte Carlo Multiple Minimum (MCMM) conformational search of the ligands and the residues with a radius of 4Å from the docked poses in order to simulate the complementary adjustments of bound ligand. The final docking solutions showed structural characteristics of the agonists that might not be necessary for the activation of the receptor, but clearly are crucial for the potency and the selectivity of those compounds.
The native agonist 4 and potent nonselective 5’-uronamide 14 were docked in the rigid binding site of 1-A2AAR and then optimized along with the binding cavity residues located within 4Å from the docked poses. Because both of these agonists were unsubstituted at the N6 and C2 positions, the docking poses of 4 and 14 highlighted the crucial anchoring interactions with the binding site of A2AAR that are expected to be common among all the agonists in our list. Strong H-bond interactions were observed between the carboxamide group of Asn253 (6.55) and the primary N6 amino group and the N7 of the adenine ring of 4 and 14; between Ser277 (7.42) and the 3’-OH group and between His278 (7.43) and the 2’-OH group of the ribose moiety. Favorable van der Waals (vdW) contacts further stabilized the poses of 4 and 14, involving residues Val84 (3.32), Leu85 (3.33), Trp246 (6.48), and Leu249 (6.51) located in close proximity to the ribose moiety, and Ile274 (7.39), Met270 (7.35), Phe168 (EL2), and Met177 (5.38), which embedded the adenine core of the two agonists.
In the optimized binding pose of 14, the 5’ carboxamide group, in common with 1, was firmly locked in the cavity by H-bond interactions with His250 (6.52) on one side and Thr88 (3.36) on the other side, while the attached hydrophobic ethyl group was embedded by favorable vdW interactions with residues Leu85 (3.33), Gln89 (3.37), Ile92 (3.40), Met177 (5.38), Asn181 (5.42), Cys185 (5.46), Val186 (5.47), and Trp246 (6.48) (Figure 2C).
Figure 2.

Docking poses of 4 (panel A and B, with carbon atoms colored in green and orange) and 14 (panel C, with carbons colored in yellow) in the binding site of the agonist-bound 1-A2AAR structure. The key H-bond interactions between the compounds and the residues of the binding pocket are highlighted as dotted lines. Adenosine 4 may assume an orientation of the 5’-OH group that points toward H250, like the docking mode in panel A, or with the 5’-OH group pointing and interacting with T88, docking mode in panel B. The uronamide group of 14 interacts with both H250 and T88. The H-bond interactions between the adenosine moiety of the agonists and the key residues N253, S277, and H278 as well the hydrophobic interactions with F268, L249, I274 are believed to be conserved for all the agonists in our list.
The 5’ substituent of 4 consists of a hydroxymethyl group. A high mobility of the small OH group in the 5’-substitution pocket of 1-A2AAR appeared in the docking results for hydroxymethyl agonists 4–13, with two different orientations of the OH group. The more favorably scored docking orientation of 4 was characterized by an interaction of the 5’-OH group with Thr88 (3.36), where the threonine was acting as H-bond acceptor (Figure 2B). The other most common docking orientation was pointing the 5’-OH group toward His250 (6.52) (Figure 2A). Both docking placements of the 5’-OH group were plausible, since the hydroxyl group could closely resemble the role of the uronamide NH group of the co-crystallized 1, donating the H-bond interaction to Thr88 (3.36), or, at the same time, it could be placed where the uronamide carbonyl oxygen was found in co-crystallized 1, accepting an H-bond interaction from His250 (6.52). After optimization of both representative docking orientations of 4 in the binding pocket of 1-A2AAR, the lowest energy conformations of the ligand were engaged in a H-bond interaction with His250 (6.52) and not Thr88 (3.36). This energy trend was maintained also for the other agonists having a 5’-OH group. For all the agonists on our list with a hydroxyl group at the 5’-position, the two different docking orientations of the OH were considered for the further MCMM optimization. All of these agonists showed the same trend as 4, i.e., the energetically favored conformations showed the 5’-OH group engaged with His250 (6.52). The binding energies between the individual residues in the binding site of 1-A2AAR and the minimized docking poses of 4 were calculated and listed in Table 2. When the 5’-OH of 4 was pointing toward Thr88, the contribution to the binding energy by the proximity of His250 (6.52) was small and predominantly a vdW contribution due to the distance between the residue and the ligand. The energy contribution of Thr88, instead, was still substantial even when the 5’-OH group was engaged with His250, making this orientation the energetically most favored pose after optimization. Furthermore, the predicted energy contributions of each contact residue did not take into account the presence of water molecules which may impact the interaction network. The importance of water has been recently demonstrated by the 4-A2AAR structure,17 where the interaction between the 5’-OH group of the co-crystallized adenosine 4 and the residues His250 (6.52) and Asn181 (5.42) is mediated by a structured water molecule (HOH 2017 in the PDB entry 2YDO17).
Table 2.
Energy contributions to binding at the A2AAR associated with individual amino acid residues in proximity to the co-crystallized ligand in the binding site. Bold font refers to residues that form an H-bond with the ribose moiety.
| Residue | Minimized docked pose A (toward H250), 4 |
Minimized docked pose B (toward T88), 4 |
Minimized docked pose, 14 |
|||
|---|---|---|---|---|---|---|
| Tot En | vdW | Tot En | vdW | Tot En | vdW | |
| V84 | −7.26 | −6.63 | −8.65 | −7.19 | −10.03 | −9.26 |
| L85 | −6.22 | −6.74 | −6.51 | −6.64 | −17.21 | −15.61 |
| T88 | −10.18 | −6.51 | −28.40 | −4.62 | −29.98 | −5.95 |
| F168 | −30.17 | −30.45 | −30.91 | −30.93 | −30.77 | −30.60 |
| E169 | −46.96 | −3.40 | −46.15 | −3.75 | −45.86 | −3.39 |
| M177 | −10.99 | −9.26 | −9.31 | −8.55 | −10.11 | −10.83 |
| W246 | −9.66 | −7.11 | −8.69 | −6.28 | −11.58 | −9.99 |
| L249 | −13.31 | −15.38 | −13.86 | −15.25 | −14.55 | −15.35 |
| H250 | −27.87 | −1.81 | −5.16 | −4.41 | −36.64 | −4.60 |
| N253 | −43.30 | −3.08 | −42.14 | −3.58 | −44.03 | −3.98 |
| M270 | −7.07 | −5.27 | −6.84 | −5.16 | −7.23 | −5.36 |
| I274 | −17.35 | −14.54 | −16.86 | −14.28 | −17.77 | −14.64 |
| S277 | −30.83 | −2.61 | −28.44 | −3.21 | −33.07 | −1.81 |
| H278 | −18.31 | −4.54 | −18.70 | −4.35 | −17.13 | −4.93 |
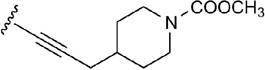
From the docking results in 1-A2AAR and from the 4-A2AAR complex, the small and flexible 5’-OH group of agonists 4–13 could assume different orientations in the 5’ subpocket of A2AAR, and a water molecule could fit as well in the pocket to enhance the contacts of the hydroxyl group with polar residues in the binding cavity. For compounds 21 and 22,39,45 instead, the 5’-substituent was bulky and rigid, and the residues in this 5’-subpocket needed to adjust during the MCMM optimization in order to accommodate the cyclopropylcarboxamido group of 21 and the tetrazole moiety of 22. The movement of the residues in the 5’-subpocket were minimal without any major reorientation of the residues side chains, nevertheless the need of a slightly bigger pocket was evident during the automated docking of compound 21 and 22. The cyclopropyl ring of 21 was embedded by favorable hydrophobic interactions with residue like Leu85 (3.33), Ile92 (3.40) and Trp246 (6.48), while His250 (6.52) and Thr88 (3.36) coordinated the 5’ amide moiety with H-bond interactions. At the same time, hydrophilic residue side chains in the 5’-subpocket were able to stabilized the bulky tetrazole ring of 22. The predicted binding mode of 22 showed H-bond interactions with Thr88 (3.36) and Gln89 (3.37), and Asn181 (5.42) was found in proximity to the 5’ moiety of 22.
Compared to the antagonist-bound A2AAR structure, Glu169 in the EL2 assumed a different and specific rotameric orientation of the side chain in the 1-A2AAR structure. Glu169 is oriented toward the C2-substituent of 1 in order to engage in an H-bond interaction with the urea moiety of the agonist. In 2-A2AAR the carboxyl group of Glu169 is oriented toward the binding site, interacting with the exocyclic amine group of the antagonists and further stabilized by the H-bond contact with His264 in EL3. The shifted orientation of the Glu169 side chain in the 1-A2AAR structure is believed to be very specific for agonists sharing with 1 both a bulky substituent at the N6 position and H-bond donor groups in the C2-substituent. In the case of agonists non-substituted at the N6 position, Glu169 can be expected to assume a 2-like rotameric orientation of the side chain in order to H-bond with the exocyclic amine. The prediction of the rotameric adjustment of Glu169 toward the adenine primary amine has been confirmed by the 14-A2AAR and 4-A2AAR structures, where the rotameric state of Glu169 and the conformation of EL3 are very similar to the ones observed in the 2-A2AAR complex. Here we also considered this assumption for agonists with hydrophobic and bulky substituents at the C2 position (α-naphthylethyl ether derivative 7 among others41). After the docking of 7 in the rigid 1-A2AAR structure, the steric and electrostatic clashes between the ligand naphthyl substituent and Glu169 were preventing the retrieval of low energy docking poses. After MCMM optimization of the docked pose of 7 and the residues in the proximity, a rotation of the Glu169 carboxyl chain occurred, relieving the clashes with the ligand and enhancing the binding interactions with an additional strong H-bond interaction with the exocyclic amine (Figure 3A). The intuitive supposition that, for N6 non-substituted agonists, Glu169 should contribute favorably to the binding energies through an H-bond interaction with the exocyclic amine was emphasized by the prominent energy contribution of Glu169 in anchoring the adenine moiety of the small and non-substituted agonists 4 and 14, as shown in Table 2. All the agonists in our list with a primary amine at the N6 position were docked in the optimized binding site of the 1-A2AAR structure with a 2-like rotamer of Glu169.
Figure 3.

Docking poses of the 7 (panel A, with carbon atoms colored in orange), and 10 (panels B and C, with carbon atoms colored in orange and green, respectively) in the optimized binding site of the 1-A2AAR structure. The key H-bond interactions between the compounds and the residues of the binding pocket are highlighted as dotted lines. The bulky and hydrophobic naphthyl moiety of 7 could fit in the binding site of 1-A2AAR only after the rotation of Glu169 side chain toward the adenine moiety of the ligand, engaging in a strong H-bond interaction with the exocyclic amine group of 7 and relieving the steric clash with the C2 substituent of the agonist. The rotamer flexibility of Glu169 in the 1-A2AAR structure is shown with the two possible binding modes of 10. The carboxyl group of Glu169 could interact with the exocyclic amine group of the ligand (panel B) or with the amide carbonyl group at the C2 substituent of 10 (panel C).
The diagnostic vasodilator 10, an adenosine derivative containing a primary amine at the N6 position and a hydrophilic side chain at the C2 position,22 was docked in both the binding sites of the 1-A2AAR structure before and after the rotameric optimization of Glu169. The contributions to the binding energy of Glu169 to the docked pose of 10 in both binding sites, shown in Table 2, were prominent in both the docked poses. Moreover, no rotation of the Glu169 side chain was observed after optimization by means of MCMM conformational search analysis of the docked poses in the 1-A2AAR structure. The docking poses of compound 10 are shown in Figures 3B and C. The 4-methylamide pyrazole chain of compound 10 was protruding toward the extracellular part similarly to the 2-substituent of 1 in the crystal structure. Interesting, the pyrazole moiety of compound 10 was about the same length as the ethylamide linker of compound 1 with respect to the overlap of the NH group of the methylamide moiety of compound 10 with one of the NH groups of the urea moiety in compound 1. After optimization of the docking pose of 10 and the binding site residues of 1-A2AAR, the carboxyl group of Glu169 was still oriented to interact with the C2-amide of agonist 10. In this predicted pose of compound 10, the carboxyl group of Glu169 acted as H-bond acceptor from the NH group of the same amide, while the hydroxyl group of Tyr271 (7.36) acted as H-bond donor in favor of the carbonyl group of the methylamide chain of the agonist. This double H-bond lock of the C2-chain stabilized the agonist 10 in the active mode inside the binding cavity in a manner similar to 1, although less strongly. Beside the favorable electrostatic interactions, hydrophobic residues in the extracellular part of the pocket were embedding the substituent at the C2 position of the purine moiety, such as Leu267 in EL3 and Leu167 in EL2 that are in proximity of the methyl group of the proximal amide. On the other hand, the optimized binding orientation of 10 in the 1-A2AAR binding site with a 2-like rotamer of Glu169 led to a similar conformation of the ligand with the H-bond interaction between the C2-side chain and the hydroxyl group of Tyr271 (7.36), and no further rotation of Glu169 toward the amino group at the C2-substituent, while the interaction between Glu169 and the purine exocyclic amine of 10 was maintained (Figure 3B). We interpreted this finding as the ambivalent ability of Glu169 to interact with both H-bond donor groups of 10 in order to stabilize the molecule in the binding cavity of the receptor. The rotamer flexibility of Glu169 in the 1-A2AAR structure might be favored by the distance between Glu169 and His264 (EL3) and the consequent lack of the H-bonding interaction between these two residues as shown in the 2-A2AAR complex.
On the other hand, favorable interactions of the rotamer of Glu169 in the 1-A2AAR X-ray structure contributed to the stabilization of the predicted binding modes for those agonists having substitution at the N6 position. A secondary amine at N6 of the adenine core is prevented from interacting with the Glu169 side chain, since the only available hydrogen atom is already engaged in an H-bonding interaction with Asn253 (6.55). Beside compound 1, in our list there are four other agonists that are substituted at the N6 position (Figure 4). Compound 11,42,46 a monosubstituted adenosine derivative that was one of the first agonists to display any degree of A2AAR selectivity, is similar to 1 at N6 but is lacking the C2-substituent. The most favorable binding poses for compound 11 showed the diphenyl moiety oriented similarly to the orientation of the group in the 1-A2AAR X-ray structure. As in the case of 1, the diphenyl moiety of compound 11 had extensive hydrophobic interactions with the receptor. Met270 (7.35) stabilized both the phenyl rings from above with hydrophobic interactions. Tyr271 (7.36) and Ala273 (7.38) were in close proximity to one aromatic ring of agonist 11 offering good hydrophobic interactions, while Thr256 (6.58), Ile252 (6.54) and Ala256 (EL3) were stabilizing the other phenyl ring. Met174 (5.35) was found in close proximity to the ethyl linker between N6 and the phenyl rings (Figure 4).
Figure 4.

Panel A: Docking poses of 11 (with carbon atoms colored in green), 12 (with carbons in orange), and 13 (with carbons colored in cyan) in the binding site of the 1-A2AAR structure. The bulky N6 aromatic rings of the agonists are located in a hydrophobic pocket at the extracellular region of TM6, TM7, EL2, and EL3. Panel B: predicted binding orientation of 20 in the binding site of 1-A2AAR. The key H-bond interactions between the compounds and the residues of the binding pocket are highlighted as dotted lines.
The chirality of the unevenly substituted diphenyl derivative 12 was studied,42,46 and both diastereomers were docked in the binding cavity of the receptor. The best poses for the diastereomer of agonist 12 having an (R) configuration of the N6 group showed an orientation of the phenyl rings similar to the docking pose of 11, with the methyl-substituted ring close to Tyr271 (7.36) and below Met270 (7.35), while the dimethoxy-substituted ring was closer to the EL2 residues. The methyl group of the first ring could be found pointing toward TM6 and TM7, close to Ile252 (6.54) and Ala273 (7.38), and groups on the other ring were found pointing toward the solvent and close to the methyl group of Thr256 (EL3) and Met270 (7.35) (Figure 4A). The (S) diastereomer of 12, instead, showed an orientation of the diphenyl moiety that leaded to a shifted adenine ring weakening the interaction between the adenine ring and Asn253 (6.55) (Figure S3). From these results we suggested that between the two diastereomers of 12 the (R)-diastereomer should be more active than the (S).
Diphenyl derivative 13 is similar to compound 1 with its C2-substituent containing an amide group and a piperidine ring.36 The docking poses of compound 13 into the binding cavity of the receptor showed the orientation of the diphenyl substituent at the N6 position comparable to the orientation assumed by the best poses of compound 11 and 12. The C2-substituent is pointing toward the upper part to orient the piperidine ring between Tyr271 (7.36) and Leu167 (EL2), stabilized by hydrophobic interactions with these two residues and also with Met270 (7.35) and one of the phenyl rings of the N6-diphenyl moiety of the ligand itself.
Carbocyclic derivative 20 shows a different core at the N6 substituent with a chiral chain and a thiophene ring.44 In the best docking pose, the thiophene ring was located below Met270 (7.35) with the edge of the aromatic ring close to Tyr271 (7.35) and the chlorine atom pointing toward the interface between TM6 and TM7 close to Ile252 (6.54) and Ala273 (7.38). The ethyl group attached to the chiral center was instead oriented toward the extracellular part of TM6, reaching Thr256 (6.58) (Figure 4B). An unusual feature of compound 20 is also the substitution of the classical ribose of the adenosine-like agonists with a cyclopentyl ring. Nevertheless, the conformation of the 5-membered ring was similar to the one assumed by the ribose ring of the other agonists in our list, thanks to the strong H-bond interactions between the substituents at 2’, 3’, and 5’ positions and the residues Ser277 (7.42), His278 (7.43), His250 (6.52) and Thr88 (3.36) in the binding pocket.
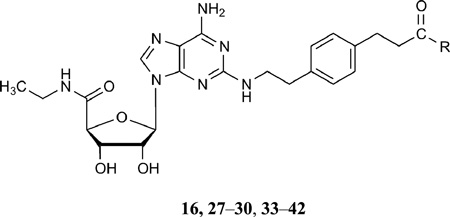
A widely used A2AAR agonist 16 shows a long and flexible chain at the C2-position ending with a carboxyl acid group. Among the docking poses of compound 16 in 1-A2AAR, some had an H-bond interaction between the carboxyl group of the ligand and the hydroxyl group of Tyr271 (7.36), but the lowest energy conformation had the terminal carboxylate group free in the extracellular media. The C2 aromatic ring of 16 was located in the space between Tyr271 (7.36), Leu267 (7.32), and Leu167 (EL2), embedded by favorable hydrophobic interactions with these residues, but more possible orientations of the C2 side chain were found due to the flexibility of the chain and the open solvent exposed space between the upper part of TM7 and EL2.
Also, many conformations of the long and positively charged C2-chain of amine congener 17 were found after the MCMM optimization of the docked complex in 1-A2AAR, and the most energetically favorable orientation was found with the positively charged terminal amino group of 17 interacting with both the carboxyl groups of Glu169 and Asp170 in EL2.
Compound 18 acts at the A2AAR in a peculiar manner, since the terminal aryl isothiocyanate group of the agonist C2 chain is assumed to bind irreversibly to the receptor.43 The possible residues that can react with the isothiocyanate group are lysine or, less likely, cysteine, and since the C2-substituent is protruding toward the extracellular medium, the targeted lysine residues should be found in the extracellular part of the receptor; however, there are only two lysines in the extracellular portion of the A2AAR, Lys150 and Lys153, located in the EL2. Unfortunately, neither is present in the 1-A2AAR X-ray structure, since that portion of EL2 was missing.
We also studied the docking of compounds 16, 17, and 18, all members of the same chemical series of long chain C2 derivatives,43 in the 14-A2AAR structure. The region where the C2-substituent was positioned in the extracellular part of the binding cavity was formed by residues from the upper part of TM7 (e.g. Met270 and Tyr271), EL2, (e.g. Glu169 and Leu167), and EL3 (e.g. His264 and Leu267). This region is very different between the 1-A2AAR and the 14-A2AAR X-ray structures. In the 1-A2AAR structure, EL3 is folded away from the binding site allowing a more open cavity mouth in order to adjust to the bulky and aromatic nature of the N6 substituent of 1. Instead, the smaller and non-substituted agonists fit perfectly in the pocket of the 4-A2AAR and 14-A2AAR structures with the EL3 conformation closer to the binding pocket and the EL2. Another novel structural feature of the A2AAR appearing in the 4-A2AAR and 14-A2AAR structures is the presence of the entire EL2 domain, included residues 149–157 missing in both the 1-A2AAR and 2-A2AAR structures. The closer conformation of EL3 and the complete EL2 domain in the 4-A2AAR and 14-A2AAR structures led to a different shaped opening of the pocket compared to the one observed in 1-A2AAR.
The binding sites of the 4-A2AAR or 14-A2AAR structures are not optimal for docking the adenosine moiety because of the substitution of the binding site residue Gln89 (3.37) with Ala in the thermostabilized constructs. Gln89 is not involved in direct interactions with the agonists, as shown in the 1-A2AAR structure, but its mutation could affect on the binding of ligands and in the activity of the A2AAR (Table S1). Nevertheless, the 4-A2AAR and 14-A2AAR structures, compared to the 1-A2AAR structure, have the advantages of the presence of the density of the whole EL2 domain and the proximity of EL3 to the binding cavity. In the 1-A2AAR, the residues 149–157 are missing, and within this region are the positively charged residues Lys150 and Lys153, which in the 14-A2AAR complex are located in proximity of the binding site above Glu169 and Asp170.
The docking poses of 16, 17, and 18 in the 14-A2AAR structure were overall similar to the ones in the 1-A2AAR complex but with different orientations and different interactions of the C2 substituents. The binding pose of 16 in the 14-A2AAR binding site showed the carboxyl group anchored to Lys153 by an ionic interaction (Figure 5A). The positively charged amino group of compound 17 docked in the 14-A2AAR structure was engaged in an ionic interaction with Glu169, while the amide carbonyl group was anchored to Lys153. Those interactions oriented the long C2 chain of amino congener 17 in the groove formed by residues of EL2 and EL3 at the top of the opening of the receptor binding cavity (Figure 5B).
Figure 5.

Docking poses of 16 (panel A, with carbon atoms colored in green), 17 (panel B, with carbons in orange), and 18 (panel C, with carbons colored in green) in the binding site of the 14-A2AAR structure. The key H-bond interactions between the compounds and the residues of the binding pocket are highlighted as dotted lines. The proximity of the isothiocyanate moiety of 18 to the positively charged amine of Lys150 suggested the involvement of this residue in the irreversible binding of 18 to the receptor.
After an MCMM conformational search of the isothiocyanate derivative in the binding site of 14-A2AAR, the long C2-chain of the affinity label 18 was found in the EL2-EL3 groove with the thiourea anchored to the carboxyl group of Glu169 and the isothiocyanate group in close proximity to the positively charged amino group of Lys150 (Figure 5C), identifying this residue as a possible irreversible anchoring site to the A2AAR by 18 and perhaps other electrophilic affinity labels.
Molecular docking of novel agonists to agonist-bound A2AAR crystal structures and their synthesis and pharmacological characterization
The terminal carboxylate of 16 was selected as the site for modification in new derivatives to be synthesized (Table 3). Amide derivatives were prepared by condensing the α-amino group with various charged or aromatic amino acids in protected form with 16, followed by saponification of the ester protecting groups. The amino acid conjugates were tested in standard binding assays at AR subtypes and a functional assay at the A2AAR. The pharmacological properties of L- and D-stereoisomers were compared. The corresponding methyl esters of the Phe and Trp derivatives, which were synthetic intermediates, were also included in the biological assays.
Table 3.
Binding affinity of a series of adenosine C2 long chain amide derivatives of 16 at three subtypes of human ARs.
 | |||||
|---|---|---|---|---|---|
| Compd | R = | Affinity Ki, nM or (% inhibition)a |
Efficacy (A2A), % of maximalb |
||
| A1 | A2A | A3 | |||
| 16 | OH | 380 | 70 | 570 | 100 |
| 27 | L-Phe-OMe | 1080 ± 210 | 160 ± 50 | 130 ± 40 | 110.9 ± 2.5 |
| 28 | D-Phe-OMe | 1230 ± 180 | 84.3 ± 3.0 | 160 ± 80 | 92.6 ± 12.4 |
| 29 | L-Trp-OMe | 1670 ± 260 | 87.2 ± 3.8 | 140 ± 10 | 104.5 ± 13.6 |
| 30 | D-Trp-OMe | 1610 ± 100 | 130 ± 4 | 250 ± 90 | 118.6 ± 5.7 |
| 33 | L-Asp-OH | 1900 ± 660 | 180 ± 60 | 1460 ± 600 | 94.8 ± 6.7 |
| 34 | D-Asp-OH | 1180 ± 360 | 110 ± 10 | 790 ± 160 | 94.3 ± 7.3 |
| 35 | L-Arg-OH | 1110 ± 30 | 100 ± 4 | 620 ± 250 | 94.5 ± 5.8 |
| 36 | D-Arg-OH | 990 ± 320 | 50 ± 5 | 220 ± 60 | 94.7 ± 6.7 |
| 37 | L-Phe-OH | 640 ± 170 | 63.7 ± 13.1 | 260 ± 140 | 103.3 ± 13.8 |
| 38 | D-Phe-OH | 550 ± 100 | 34.0 ± 3.2 | 140 ± 30 | 93.4 ± 12.4 |
| 39 | L-Trp-OH | 1060 ± 310 | 71.7 ± 16.9 | 200 ± 50 | 95.8 ± 7.4 |
| 40 | D-Trp-OH | 2160 ± 280 | 130 ± 30 | 520 ± 60 | 113.3 ± 5.0 |
| 41 | L-His-OH | 1000 ± 160 | 110 ± 30 | 830 ± 290 | 105.2 ± 4.9 |
| 42 | D-His-OH | 350 ± 40 | 40 ± 4 | 320 ± 150 | 105.9 ± 6.5 |
Using CHO or HEK293 (A2A only) cells stably expressing a human AR (Supporting Information); affinity was expressed as Ki value (n = 3–5) or percent inhibition of radioligand binding at 10 µM. Compounds 31 and 32 are conjugates of L- and D-His-OMe, respectively, and were not tested biologically.
Maximal efficacy (at 10 µM) in an A2AAR functional assay, determined by stimulation of cyclic AMP production in stably transfected CHO cells, expressed as percent (mean ± standard error, n = 3 – 5) in comparison to effect (100%) of full agonist 16 at 10 µM.
D-amino acid conjugates were generally more potent than L- and free terminal carboxylates more potent than the corresponding methyl esters (Table 3). Compounds 38 and 42, D-Phe and D-His conjugates, respectively, were the most potent at the A2AAR with slightly greater affinities than the parent 16. The selectivity for the A2AAR in comparison to the A1AR was generally greater than with respect to the A3AR. The differences in A2AAR affinity depending on the attached amino acid were relatively small, indicating that this region of the nucleoside is not subject to precise constraints imposed on the adenosine moiety deeper in the binding site. The functional assay of stimulation of cAMP production indicated that all of the analogues were full agonists of the A2AAR.
Upon receptor docking, the amino acid moieties were coordinated in a subpocket close to the exofacial surface. This subpocket was previously defined in the binding site of the terminal portion of the C2 chain of 16–18. In the predicted binding modes, the free carboxyl terminal of all the amino acid derivatives was engaged in an ionic interaction with one of the lysine residues in EL2, mostly Lys153, as was the carboxyl group of 16. Doubling the negative charge with aspartic side chains in derivatives 33 and 34 led to another predicted ionic interaction with also Lys150, but not to a gain in binding affinity, possibly because of the repulsive proximity of Glu169, located just below the two lysine residues, which kept the aspartic side chain of 33 and 34 away from the EL2-EL3 groove. We tried the addition of a positive charge with the guanidinium group of an arginine with the aim to create an interaction with Glu169. In the predicted docking modes of 36, the long arginine chain not only interacted with Glu169 but was able to reach to Thr256 (6.58).
We used some aromatic side chains to test the engagement of His264 (EL3) in the binding affinity of these agonist derivatives. The phenylalanine ring, in the docking poses of 37 and 38, was located between the positive charge of Lys150 (EL2) and the aromatic imidazole of His264 gaining favorable interactions and binding affinity. The same involvement of Lys150 and His264 to sandwich the aromatic moiety of the agonist substituent was observed in the predicted binding modes of 40 and 42. The tryptophan indole moiety was, evidently, too bulky to fit properly in the EL2-EL3 groove, particularly for the L-enantiomer 39, while the small imidazole ring of the histidine substituent, studied in both its protonated and non-protonated states, could be easily located between Lys150 and His264 with also an H-bond anchoring to Glu169.
Discussion
We have found that docking of known adenosine agonists to the agonist-bound X-ray crystallographic structures of the A2AAR provides a consistent set of interactions between ligand and receptor. These docking models are closely tied to physically determined structures and are expected to be more accurate than previous agonist binding to homology models based on the inactive state of bovine rhodopsin or other receptors. Nevertheless, many of the agonist binding features determined in prior modeling to inactive states of GPCRs were confirmed in the subsequent crystallographic studies.
It is anticipated that the new structures will guide the structure-based discovery of new GPCR agonists, and specifically AR agonists. We have tested the approach of using the docking of new compounds with a series of amino acid derivatives. The amino acids were appended at the same site, located at the terminal position of a long C2 chain, which was largely exposed to the extracellular space in the crystal structure of the 1-A2AAR complex. Thus, the interaction with the receptor was expected to be less constrained than if similar changes were made in other parts of the molecule closer to the minimal nucleoside pharmacophore.
Adenosine core subpocket of A2AAR
The adenosine pharmacophore of the agonists is bound in a cavity formed among TM3, TM6, and TM7. In the pocket, the adenine ring oriented vertically, anchored by strong π-π stacking interactions with Phe168 (EL2), H-bond interactions with Asn253 (6.55), and favorable vdW contacts with Leu249 (6.51) and Ile274 (7.39). The ribose moiety was located deeper in the pocket, anchored by H-bond interactions with Ser277 (7.42) and His278 (7.43) through the 2’- and 3’-OH, and embedded by hydrophobic residues Val84 (3.32), Leu274 (7.39), and Leu85 (3.33). More H-bond interactions are possible with His250 (6.52) and Thr88 (3.36) through the 5’-substituents of the ligands. The interactions between the agonists and the residues in this subpocket are expected to be conserved for all the agonists sharing the adenosine moiety. The poses of 1 re-docked into the 1-A2AAR structure were retrieved with a docking procedure based on the knowledge of those interactions between the agonist and the receptor, i.e. the H-bond interactions with Asn253 (6.55), Ser277 (7.42), and His278 (7.43). For the agonists in our list, those main interactions between the adenosine core and the residues of this subpocket were all retrieved by our docking approach in 1-A2AAR or 14-A2AAR.
Ribose 5’ subpocket of A2AAR
The biological activity of truncated nucleosides lacking 5’-substitution at the furanose ring, e.g. compound 3, suggests that the occupation of the 5’ subpocket is not essential for the binding to the A2AAR, although the favorable interactions with this pocket of a 5’-uronamide, as in 14, tend to enhance the activity and the selectivity of the agonists for the receptor. From the docking results, the 5’-subpocket could adjust to different substituents, from the small and flexible hydroxyl group to the bulky and rigid tetrazole moiety, in order to create favorable interactions between the residues and the substituents. The pocket of A2AAR accommodating the agonist 5’-substituent was formed by both hydrophilic residues, such as His250 (6.52), Thr88 (3.36), Gln89 (3.37), and Asn181 (5.42), and hydrophobic and bulky residues, like Leu85 (3.33), Trp246 (6.48), Ile92 (3.40), Cys185 (5.46), and Val186 (5.47). The OH-group of agonists 4–13 was small enough to assume different orientations in the pocket, preferring the orientation toward His250 (6.52), and, as shown by the adenosine-bound A2AAR complex,17 the interposition of a water molecule. The ethyl- and cyclopropylcarboxamide groups of agonists 14–21, instead, were firmly locked in the 5’ subpocket by H-bond interactions with His250 (6.52) and Thr88 (3.36) and further stabilized by favorable vdW contacts through their hydrophobic terminal. In particular, the vdW contributions to the binding energy of Leu85 (3.33) was more conspicuous in the case of the ethylcarboxamide substituent than in the case of the smaller OH group, as shown by the energy components in Table 2. The docking results of compound 22, with the bulky and rigid tetrazole moiety, suggested that the 5’ subpocket was spacious and easily adaptable to accept substituents of different size and nature. The 5’ subpocket is a potential site for the development of new substituted agonists in order to analyze the SARs needed to improve the binding and the selectivity of the adenosine derivatives as agonists for the A2AAR (Tosh et al., in preparation).
Adenine N6 subpocket of A2AAR
The region of 1-A2AAR that accommodates the bulky N6-diphenylethyl group of 1 is located between the extracellular termini of two helices (TM6, TM7) and two loops (EL2, EL3) and is formed predominantly by hydrophobic residues. The same subpocket was occupied by the N6 substituents of compounds 11–13, and 20, according to their predicted binding modes. The subpocket that holds the N6 substituent in the A2AAR is quite flexible due to the easy movements of some of the residues in TM7 and EL3. In fact, comparing the X-ray structures of 1-A2AAR and 14-A2AAR or 4-A2AAR, the pocket for the N6 substituent is wide open in the 1-A2AAR but obstructed in the 14-A2AAR structure. The specific rotamer of Met270 (7.35) and the outward folding of EL3 in 1-A2AAR allowed the formation of a subpocket for the N6 substituent of 1. The space created in TM7 between Met270 (7.35) and Ile274 (7.39) by the movement of Met270 side chain toward the extracellular part of the cavity of 1-A2AAR, according to the predicted binding modes of 11–13, and 20, could be occupied by one of the aromatic rings of the N6 substituents, a phenyl ring for 11 and 13, the methylphenyl ring for 12, and the thiophene moiety for 20.
Other hydrophobic residues embedding this aromatic ring were Leu249 (6.51), Ile252 (6.54) and Ala273 (7.38). In the predicted binding poses, the other phenyl ring of the N6 substituent of 11–13 was always pointing toward the solvent in the extracellular media, exiting the binding cavity in a vertical way between Met270 (7.35) and Thr256 (6.58). The side chains of Thr256 (6.58), Glu169 (EL2), Met270 (7.35), Met174 (5.35), and Ile252 (6.54) also contributed with favorable vdW contacts to the stabilization of the N6 substituent. In the predicted binding modes of the N6-substituted agonists, Glu169 in EL2 had to assume a rotamer similar to the one in 1-A2AAR in order to allow the bulky substituents of 11–13, and 20 to adjust in the pocket and to contribute with favorable vdW contacts through the aliphatic part of the side chain, pointing the charged carboxyl group away even in the absence of a polar substituent at the C2 position of the agonists, like in the case of 11, 12, and 20.
Obviously, Met270 (7.35) was a key residue in the binding the N6 substituted agonists, not only contributing with favorable interactions to the recognition of the N6 group, but also with its flexibility that allowed the formation of the subpocket of the substituent. The non-conserved nature of the residue at position 7.35 among the adenosine receptors subtypes could also be a discriminating residue for the selectivity of N6-substituted agonist. The corresponding residues of Met270 in A2AAR are the small Thr270 in A1AR and the bulky Leu264 in A3AR.
Another residue located in the N6 subpocket and involved in the anchoring of the N6 substituent of the A2AAR agonists, but non-conserved among the adenosine receptors, is the Thr256 (6.58), which becomes a bulky leucine in A3AR.
Adenine C2 subpocket of A2AAR
The C2-subpocket of A2AAR is formed between TM7, EL2, and, marginally, TM2. A key residue in the C2-subpocket of A2AAR is Tyr271 (7.36), at the extracellular terminus of TM7. The polar and aromatic nature of Tyr271 could contribute in different ways to the anchoring of the agonists. In the 2-A2AAR structure, the binding contribution from Tyr271 was minimal and predominantly from vdW contacts. In the 1-A2AAR structure, instead, the contribution of Tyr271 to the binding of 1 became prominent due to the strong H-bond interaction between the OH group of the tyrosine and the carbonyl group of the urea moiety in 1. Obviously, the involvement to the ligand binding by Tyr271 was less relevant for small agonists not-substituted at the C2 position, like 4 or 14. From the predicted binding modes of the C2-substituted agonists in 1-A2AAR and 14-A2AAR, the bivalent role of Tyr271, able to adjust to substituents of a different nature, was strategic for the creation of favorable contacts with the ligands. For agonists with hydrophobic C2 side chains, compounds 3 and 5 amongst the others, the main contribution to the stabilization of agonists by Tyr271 was through vdW contacts. For compounds with a H-bond acceptor group on the C2 chain, like in the case of 10 and 13, Tyr271 could form strong H-bond interactions to anchor the molecule in the binding site. Other residues in the C2 subpocket are hydrophobic, like Ile274 (7.39), Ile66 (2.66), Leu167 (EL2), Met270 (7.35), and Leu267 (7.32), favoring hydrophobic chains at the C2 position of the agonists. Longer C2 side chains, like for compounds 17 and 18, once reached the top of the cavity preferred to turn toward TM6 occupying the groove formed between EL2 and EL3, in order to interact with the residues of the ELs, such as Glu169, Lys150 and Ly153.
The binding region on the receptor for long and flexible C2 substituents extended above the principal adenosine binding cavity, following the groove between EL3 and EL2 but mostly exposed to the solvent. Compared to 1-A2AAR, this EL2-EL3 groove in 14-A2AAR is narrow due to the proximity of EL3 to EL2 through the H-bond interaction between His264 and Glu169. The main functional side chains of the receptor exposed to the agonist C2 substituents are the two positively charged lysine residues, Lys150 and Ly153, the negative carboxylate group of Glu169, and the indole ring of His264.
In the predicted binding poses, the carboxyl group of 16 was engaged in an ionic interaction with Lys153, while the long chain of agonist 17 interacted with the Lys153 through its amide carbonyl group and with Glu169 through its charged amine group. The longer and specific side chain of 18 was locked in the EL2-EL3 groove of the 14-A2AAR structure by strong interactions between the thiourea moiety and Glu169 and the possible attack by the thiocyanate group to the amine group of Lys150.
In order to understand if interactions with the residues in the EL2-EL3 groove could bring advantage to the binding affinity of the agonists, we designed some amino acid derivatives of 16 with different functional groups that, from the predicted binding poses, could fit the C2 subpocket. In predicted binding poses of nucleoside derivatives with long C2 side chains the C2 substituents were mostly directed toward the flexible and solvent exposed region at the opening of the binding pocket formed by residues of EL2 and EL3. This region of the receptor is the most variable in amino acid nature within the ARs, possibly granting some selectivity toward specific subtypes of the ARs. Moreover, upon agonist binding this region undergoes many conformational changes as showed by the comparison between the antagonist-bound and the agonist-bound structures of A2AAR, making this region easily adjustable to various substituents.
The D-derivatives of this amino acid series of compounds showed higher binding affinities compared to the L-derivatives. From the predicted poses, the L-side chains were located very close to the EL2 wall, while the D-side chains were more in the middle of the groove. EL3 is more flexible than EL2 and can adjust more easily to the shape of the various ligand substituents, as shown by the outfolding of EL3 in the 1-A2AAR structure. Besides the highly flexible portion with Lys150 and Lys153, present only in the 14-A2AAR and 4-A2AAR structures, EL2 contains a α-helical portion with Phe168 and Glu169 that is quite fixed in the binding cavity of A2AAR and less adjustable to bulky ligand substituents. Among the amino acids derivatives tested, some gain in affinity at the A2AAR was possible with the positive charged side chain of 36 and the aromatic side chains of 38 and 42. In the predicted binding pose of 36 in the 14-A2AAR binding cavity, the arginine side chain was anchored to Glu169 (EL2) and Thr256 (6.58), while the aromatic rings of 38 and 42 were located between Lys150 (EL2) and His264 (EL3). Those docking poses suggested an involvement of residues in the ELs in the coordination of long C2 substituents of nucleoside agonists.
In conclusion, molecular modeling of GPCRs is shown to be a useful technique in probing the determinants of agonist recognition, especially when the modeling takes into account all of the available supporting data (multiple X-ray structures, mutagenesis, and SAR). The position of the adenosine moiety and its overlay with a bound antagonist were well approximated using modeling based on the inactive A2AAR structure, and now the agonist-bound structures provide detailed information. By examining the molecular recognition of 20 known A2AAR agonists, a structural basis for previous empirical SARs consistent with previous mutagenesis results is provided. We are just at the beginning of learning how to effectively utilize the new agonist-bound X-ray structures of GPCRs in drug discovery, but it is clear that the insights gained can be used to guide the rational design of new synthetic analogues. Docking predictions made for these agonists can now be used as an aid in extending the SAR in this chemical series to predict the likelihood in increasing A2AAR affinity. We have prepared a series of amino acid derivatives at the terminal position of a C2 chain that maintain moderate affinity and the putative site of binding of the added moieties associated with EL2 and EL3 has been predicted. This approach can potentially be extended to other regions of the nucleoside structure.
Experimental Section
Chemical synthesis
Briefly, commercially available nucleoside carboxylic acid 16 was coupled to various carboxylate-protected amino acids using either EDC.HCl (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) or HATU (2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium) as coupling agent in anhydrous DMF to obtain the corresponding amino acid coupled methyl ester derivatives (Supporting information). Subsequently, the methyl ester was hydrolyzed using 0.1M aqueous NaOH to give the target carboxylic acid derivatives 33–42.
Purification of the nucleoside derivatives for biological testing was performed by HPLC with a Luna 5 µ RP-C18(2) semipreparative column (250 mm × 10.0 mm; Phenomenex, Torrance, CA) under the following conditions: flow rate of 2 mL/min, 0.5% trifluoroacetic acid in H2O-CH3CN from 100:0 (v/v) to 50:50 (v/v) in 32 min. Analytical purity of compounds was checked using a Hewlett-Packard 1100 HPLC equipped with Zorbax SB-Aq 5 µm analytical column (50 mm × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm. All derivatives tested for biological activity showed >99% purity by HPLC analysis (detection at 254 nm).
For structural confirmation, 1H NMR spectra were recorded at 300 Mz. Chemical shifts are reported in parts per million (ppm) relative to deuterated solvent as the internal standard. ESI-High resolution mass spectroscopic measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine.
Computational methods
Receptor and agonists structures
The crystal structures of the A2A receptor in complex with 1, 4, and 14 were obtained from the Protein Data Bank (PDB entry codes: 3QAK16, 2YDO17, 2YDV17). All the proteins were cleaned from water molecules and ions and hydrogen atoms were added to the receptors and minimized by means of the Protein Preparation Wizard in Maestro31. The T4-lysozyme insertion replacing the third intracellular loop (IL3) in the 3QAK crystal structure was removed. The IL3, which is far from the binding cavity, was not replaced. The protonation states of the histidine residues in the binding cavity were based on hydrogen bonding networks present in the crystal structures. The previously published complexes of the docked agonists 4 and 14 to the antagonist-bound A2AAR crystal structure19 were also used. The structures of the agonists were sketched in Maestro and minimized using the Polak-Ribiere conjugated gradient (PRCG) method with a convergence gradient of 0.001 kJ/mol·Å.
Molecular Docking of agonist 1 to the 1-A2AAR structure
The automated docking of 1 was performed in a rigid binding site of the A2AAR with different docking protocols to test the docking ability to reproduce the co-crystallized orientation. The binding pocket of 1 in the crystal structure of the 1-A2AAR complex was considered for all the docking approaches. The best scored docking poses from each protocol were compared to the co-crystallized orientation of 1 and the root-mean-square deviation (RMSD) was calculated as a measure of docking reliability. Glide31 was used to dock 1 using both the SP (standard precision) and XP (extra precision) procedures, with default parameters or with hydrogen bond constraints. The H-bond constraints in Glide were applied during the generation of the docking grid to enforce the H-bond formation between the adenosine core of 1 and the crucial residues in the binding site, i.e. between the amide group of Asn253 and the N6 amino group and N7 of 1, between the Nε of His278 and the 2’-OH group of the agonist, and between the OH group of Ser277 and the 3’-OH group of 1.
MOE32 docking was performed with Alpha PMI, Alpha Triangle, Proxy Triangle, and Triangle Matcher as docking placement functions. The co-crystallized orientation of 1 in the binding site of 1-A2AAR with a RMSD lower than 2Å was reproduced using the “Pharmacophore consensus” tool implemented in MOE. A receptor-based pharmacophore model was build based on the crucial interactions between the adenosine moiety of 1 and the binding site residues and the pharmacophore model was then used to filter the docking placements in order to retrieve only the docking poses that could reproduce those relevant agonist-protein interactions. The MOE pharmacophore query (figure S2) was defined on the crystal structure of the 1-A2AAR complex, and four pharmacophore features were used: the H-bond acceptor/donor interactions between Asn253(6.55) and the exocyclic amino group and N7 of 1, and the H-bond acceptor interactions between Ser277 and His278 and the 2’-OH and 3’-OH of 1. The radii of the features were not modified.
Agonist 1 was docked to the binding site of the 1-A2AAR structure also with ICM-Dock module in the MolSoft LLC suite,47,48 using mmff charges and the default setup. The binding cavity of the receptor was defined as being within 5 Å of the co-crystallized 1 in the 1-A2AAR structure. The best docking pose of 1 was chosen based on the docking energies and the best RMSD value compared with the X-ray pose of 1.
Molecular docking of agonists 11–13 and 20
The molecular docking of 11–13 and 18 was performed in the binding site of 1-A2AAR with the Glide SP protocol without distances restraints or MOE docking and the pharmacophore query, as described for agonist 1. The best scored docking poses showing the crucial H-bond interactions between the adenosine core of the agonists and the important residues Asn253, Ser277, and His278 of the receptor were retained and subjected to MCMM conformational analysis by means of MacroModel31,47,48 for further structural optimization and energy minimization of the complexes. During the conformational search, full flexibility was granted to the ligand and the residues of the receptor within a radius of 4 Å from the ligand. All the other residues were considered conformationally frozen during the calculations. 1000 steps of MCMM were performed with the MMFFs force field and the water GB/SA model as implicit solvent. Polak-Ribier Conjugate Gradient (PRCG) minimization method with a convergence threshold on the gradient of 0.05 kJ/mol·Å was used.
Molecular Docking of agonists 3–10, 14–19, and 21–22 in the optimized binding site of 1-A2AAR
The automated docking of agonists 3–10, 14–19, and 21–22 was performed in the optimized binding sites of 1-A2AA with a different rotameric state of the Glu169 side chain from the crystal structure. A rotamer of Glu169 with the carboxyl group oriented toward the binding site was chosen after MCMM conformational search of the binding site in order to recreate the H-bond interaction between this residue and the primary N6 exocyclic amine of these agonists, in a similar way as in the 2-A2AAR, 14-A2AAR, and 4-A2AAR crystal structures. The same molecular docking protocols and parameters describe above for agonists 11–13 and 18 were used. After the automated docking the best scored poses of the agonists were optimized by means of MCMM conformational search as described in the previous section.
Molecular Docking of agonists 16–18, and 27–42 in the optimized binding site of 14-A2AAR
The binding site of 14-A2AAR was considered optimal for the docking study of agonists with long C2 substituents due to the presence of the entire EL2 of the receptor, missing in the 2-A2AAR structure. The automated docking and MCMM conformational search for agonists 16–18 and 27–42, characterized by long C2 substituents, were conducted in the binding site of 14-A2AAR using the same protocols described above.
The graphical pictures were made with the Pymol program (Delano Scientific LLC, CA, USA) and MOE.
Pharmacological characterization
Binding assays were performed using membranes of mammalian cells stably expressing recombinant A1AR or A3AR (CHO, Chinese hamster ovary) cells or the A2AAR (HEK-293, human embryonic kidney cells).49–51 as radioligands [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA; 2000 Ci/mmol), [3H]R-N6-phenylisopropyladenosine ([3H]R-PIA, 63 Ci/mmol), [3H]16 (47 Ci/mmol), as described (Supporting information).50,51 IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation.52 Data were expressed as mean ± standard error.
Functional assays of cAMP accumulation were performed using membranes of CHO cells stably expressing recombinant A1AR or A2AAR, as described.53,54 Statistical analysis was performed using Prism 4.0 (GraphPad, San Diego,CA).
Supplementary Material
Figure 6.

Predicted docking orientations of D-amino acid conjugates 36 (panel A, with carbons colored in yellow), 38 (panel B, with carbons in orange), and 42 (panel C, with carbons colored in orange). The guanidium group of 36 was engaged in H-bond interactions with Glu169 (EL2) and Thr256 (6.58). The phenyl ring of 38 and the imidazole ring of 42 were located between the positively charged amino group of Lys150 (EL2) and the aromatic side chain of His264 (EL3).
Chart 1.

Structures of agonist (1) and antagonist (2) AR ligands that were co-crystallized with the human A2AAR.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases and the PSI:Biology program, National Institute of General Medical Sciences U54 GM094618. We thank Dr. Stefano Costanzi (NIDDK) and Dr. Soo-Kyung Kim (California Inst. of Technology) for helpful discussion.
Abbreviations
- AR
adenosine receptor
- CHO
Chinese hamster ovary
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- HPLC
high performance liquid chromatography
- IL
intracellular loop
- MCMM
Monte Carlo Multiple Minimum
- MOE
Molecular Operating Environment
- NECA
5′-N-ethylcarboxamidoadenosine
- RMSD
root mean square deviation
- TM
transmembrane α-helix
- vdW
van der Waals
Footnotes
Supporting information available:
Comparison of the predicted role in agonist recognition of selected residues of the human A2AAR and relation to the agonist binding interactions in the X-ray structure, docking figures for selected compounds, procedures for synthesis and biological assays for novel nucleoside derivatives, and coordinates of complexes with 7 and 19. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 3.Tyndall JD, Sandilya R. GPCR agonists and antagonists in the clinic. Med. Chem. 2005;1:405–421. doi: 10.2174/1573406054368675. [DOI] [PubMed] [Google Scholar]
- 4.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 10.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 11.Standfuss J, Edwards P, D’Antona A, Fransen M, Xie G, Oprian DD, Shertler GFX. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 13.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the β,2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunihara RK, Kobilka BK. Crystal structure of the β,2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a β,1-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveak common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol. 2011 doi: 10.1016/j.sbi.2011.06.007. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov AA, Barak D, Jacobson KA. Evaluation of homology modeling of G protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J. Med. Chem. 2009;52:3284–3292. doi: 10.1021/jm801533x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaakola VP, Lane JR, Lin JY, Katritch V, IJzerman AP, Stevens RC. Ligand binding and subtype selectivity of the human A2A adenosine receptor: identification and characterization of essential amino acid residues. J. Biol. Chem. 2010;285:13032–13044. doi: 10.1074/jbc.M109.096974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katritch V, Abagyan R. GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol. Sci. 2011;32:637–643. doi: 10.1016/j.tips.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zablocki J, Palle V, Blackburn B, Elzein E, Nudelman G, Gothe S, Gao Z, Li Z, Meyer S, Belardinelli L. 2-substituted pi system derivatives of adenosine that are coronary vasodilators acting via the A2A adenosine receptor. Nucleosides Nucleotides Nucleic Acids. 2001;20:343–360. doi: 10.1081/NCN-100002306. [DOI] [PubMed] [Google Scholar]
- 23.Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol. Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009;30:249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 26.Kim J, Wess J, van Rhee M, Schöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2A adenosine receptor. J. Biol. Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olah ME, Jacobson KA, Stiles GL. Role of the second extracellular loop of adenosine receptors in agonist and antagonist binding: Analysis of chimeric A1/A3 adenosine receptors. J. Biol. Chem. 1994;269:24692–24698. [PMC free article] [PubMed] [Google Scholar]
- 28.Gao ZG, Chen A, Barak D, Kim SK, Müller CE, Jacobson KA. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J. Biol. Chem. 2002;277:19056–19063. doi: 10.1074/jbc.M110960200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mantell SJ, Stephenson PT, Monaghan SM, Maw GN, Trevethick MA, Yeadon M, Walker DK, Selby MD, Batchelor DV, Rozze S, Chavaroche H, Lemaitre A, Wright KN, Whitlock L, Stuart EF, Wright PA, Macintyre F. SAR of a series of inhaled A2A agonists and comparison of inhaled pharmacokinetics in a preclinical model with clinical pharmacokinetic data. Bioorg. Med. Chem. Lett. 2009;19:4471–4475. doi: 10.1016/j.bmcl.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 30.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 31.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 32.Prime, version 2.2, Glide, version 5.6, MacroModel, version 9.8, Maestro, version 9.1. New York, NY: Schrodinger, LLC; [Google Scholar]
- 33.Molecular Operating Environment (MOE), version 2009.10. 1255 University St., Suite 1600, Montreal, QC, H3B 3×3 (Canada): Chemical Computing Group Inc; [Google Scholar]
- 34.Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997 Suppl 1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 35.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev. Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnard A, Keir R, Salmon G, Stuart E, Trevethick M, Yeadon M. New insights into the anti-inflammatory effects of adenosine A2A receptor agonists. UK-371,104, a novel adenosine A2A receptor agonist, inhibits acute mediator release in the human neutrophil: comparison with CGS-21,680 and the phosphodiesterase 4 inhibitor, cilomilast. Proc. Physiol. Soc. Life Sci. 2007 [Google Scholar]
- 37.El-Tayeb A, Iqbal J, Behrenswert A, Romio M, Schneider M, Zimmermann H, Schrader J, Müller CE. Nucleoside-5’,-monophosphates as prodrugs of adenosine A2A receptor agonists activated by ecto-5’,-nucleotidase. J. Med. Chem. 2009;52:7669–7677. doi: 10.1021/jm900538v. [DOI] [PubMed] [Google Scholar]
- 38.Hou X, Kim HO, Alexander V, Kim K, Choi S, Park SG, Lee JH, Yoo LS, Gao ZG, Jacobson KA, Jeong LS. Discovery of a new human A2A adenosine receptor agonist, truncated 2-hexynyl 4′,-thioadenosine. ACS Med. Chem. Lett. 2010;1:516–520. doi: 10.1021/ml1001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rieger JM, Brown ML, Sullivan GW, Linden J, Macdonald TL. Design, synthesis, and evaluation of novel A2A adenosine receptor agonists. J. Med. Chem. 2001;44:531–539. doi: 10.1021/jm0003642. [DOI] [PubMed] [Google Scholar]
- 40.Cristalli G, Müler CE. Volpini, R. Recent developments in adenosine A2A receptor ligands. Handb. Exp. Pharmacol. 2009;193:59–98. doi: 10.1007/978-3-540-89615-9_3. [DOI] [PubMed] [Google Scholar]
- 41.Daly JW, Padgett WL, Secunda SI, Thompson RD, Olsson RA. Structure-activity relationships for 2-substituted adenosines at A1 and A2 adenosine receptors. Pharmacology. 1993;46:91–100. doi: 10.1159/000139033. [DOI] [PubMed] [Google Scholar]
- 42.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein J, Gross AS, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: Sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg. Med. Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobson KA, Barrington WW, Pannell LK, Jarvis MF, Ji X-D, Williams M, Hutchison AJ, Stiles GL. Agonist-derived molecular probes for A2-adenosine receptors. J. Mol. Recognition. 1989;2:170–178. doi: 10.1002/jmr.300020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smits GJ, McVey M, Cox BF, Perrone MH, Clark KL. Cardioprotective effects of the novel adenosine A1/A2 receptor agonist AMP 579 in a porcine model of myocardial infarction. J. Pharmacol. Exp. Ther. 1998;286:611–618. [PubMed] [Google Scholar]
- 45.Beattie D, Brearley A, Brown Z, Charlton SJ, Cox B, Fairhurst RA, Fozard JR, Gedeck P, Kirkham P, Meja K, Nanson L, Neef J, Oakman H, Spooner G, Taylor RJ, Turner RJ, West R, Woodward H. Synthesis and evaluation of two series of 4’,-aza-carbocyclic nucleosides as adenosine A2A receptor agonists. Bioorg. Med. Chem. Lett. 2010;20:1219–1224. doi: 10.1016/j.bmcl.2009.11.131. [DOI] [PubMed] [Google Scholar]
- 46.Trivedi BK, Blankley CJ, Bristol JA, Hamilton HW, Patt WC, Kramer WJ, Johnson SA, Bruns RF, Cohen DM, Ryan MJ. N6-Substituted adenosine receptor agonists: potential antihypertensive agents. J. Med. Chem. 1991;34:1043–1049. doi: 10.1021/jm00107a025. [DOI] [PubMed] [Google Scholar]
- 47.Chang G, Guida WC, Still WC. An internal coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1998;111:4379–4386. [Google Scholar]
- 48.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel (1990) An integrated software system for modelling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 49.Klotz K-N, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes –, characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’,s Arch. Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 50.Gao ZG, Mamedova L, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem. Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YC, Ji X-D, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J. Med. Chem. 2000;43:1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 53.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal. Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 54.Post SR, Ostrom RS, Insel PA. Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol. Biol. 2000;126:363–374. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 56.Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem. Pharmacol. 2009;77:1117–1124. doi: 10.1016/j.bcp.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udelson JE, Heller GV, Wackers FJ, Chai A, Hinchman D, Coleman PS, Dilsizian V, DiCarli M, Hachamovitch R, Johnson JR, Barrett RJ, Gibbons RJ. Randomized, controlled dose-ranging study of the selective adenosine A2A receptor agonist binodenoson for pharmacological stress as an adjunct to myocardial perfusion imaging. Circulation. 2004;109:457–464. doi: 10.1161/01.CIR.0000114523.03312.7D. [DOI] [PubMed] [Google Scholar]
- 58.Bevan N, Butchers PR, Cousins R, Coates J, Edgar EV, Morrison V, Sheehan MJ, Reeves J, Wilson DJ. Pharmacological characterisation and inhibitory effects of (2R,3R,4S,5R)-2-(6-amino-2-{[(1S)-2-hydroxy-1-(phenylmethyl)ethyl]amino}-9H–purin-9-yl)-5-(2-ethyl-2H–tetrazol-5-yl)tetrahydro-3,4-furandiol, a novel ligand that demonstrates both adenosine A2A receptor agonist and adenosine A3 receptor antagonist activity. Eur. J. Pharmacol. 2007;564:219–225. doi: 10.1016/j.ejphar.2007.01.094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.