

Abstract
Protein homeostasis (proteostasis) is essential for cellular and organismal health. Stress, aging, and the chronic expression of misfolded proteins, however, challenge the proteostasis machinery and the vitality of the cell. Enhanced expression of molecular chaperones, regulated by heat shock transcription factor-1 (HSF-1), has been shown to restore proteostasis in a variety of conformational disease models, suggesting a promising therapeutic approach. We describe the results of a ∼900,000 small molecule screen that identified novel classes of small molecule proteostasis regulators (PRs) that induce HSF-1-dependent chaperone expression and restore protein folding in multiple conformational disease models. The beneficial effects to proteome stability are mediated by HSF-1, DAF-16/FOXO, SKN-1/Nrf-2, and the chaperone machinery through mechanisms that are distinct from current known small molecule activators of the HSR. We suggest that modulation of the proteostasis network by PRs represents a promising therapeutic approach for the treatment of a variety of protein conformational diseases.
Keywords: heat shock, high-throughput screening, small molecules, protein misfolding
Introduction
Proteostasis regulates the functional properties of the proteome to minimize the damage of misfolded and aggregated proteins through a network of pathways for protein synthesis, folding, trafficking and degradation1,2. Loss of proteostatic control has been implicated in aging and multiple disorders of protein misfolding including metabolic diseases, cancer, and neurodegenerative diseases1-3. Eukaryotic cells have developed compartment-specific quality control mechanisms for proteome maintenance, to guide protein folding and transport, and to direct faulty proteins for refolding or clearance. Likewise, each compartment induces a cell stress response to detect and restore balance: the heat shock response (HSR) for cytoplasmic folding and misfolding, and the unfolded protein responses (UPR) in the endoplasmic reticulum (UPR-ER)4 and mitochondria (UPR-MT)5. Across these compartments are other stress responses that detect specific classes of protein damage caused by metal stress and antioxidants (ARE)3,6,7.
The cytosolic HSR is governed by a family of heat shock factors (HSFs) of which HSF-1 is essential for proteotoxic stress and regulation of heat shock proteins (Hsps)8. Many Hsps are molecular chaperones that guide the conformation of proteins during biogenesis and prevent misfolding and aggregation that interfere with cellular function9. Induction of the HSR not only prevents protein damage from persisting, but also restores the cell to the pre-stress condition. Despite these essential cellular stress pathways, the chronic expression and accumulation of misfolded, oxidized and aggregated proteins, as occurs in aging and disease, leads to cellular dysfunction when the quality control machineries become compromised10,11. There is increasing evidence that misfolded proteins expressed in diseases of protein conformation are not efficiently counterbalanced by a compensatory induction of cellular stress responses such as the HSR11. Enhancing the activity of HSF-1 and the levels of molecular chaperones by genetic techniques or pharmacological manipulation has been shown to restore proteostasis in several models of disease12-20.
Given the prominent roles of HSF-1 to maintain cellular proteostasis by upregulation of chaperone expression, there has been substantial effort to identify novel small molecule PRs that modulate HSF-1 activity. Several small molecule activators of HSF-1 are known8,21, including proteasome inhibitors and compounds that selectively bind to the chaperone Hsp90, including radicicol, geldanamycin (GA), and 17-AAG. Another plant-derived compound, the triterpenoid celastrol, identified from a consortium screen for molecules with protective effects in models of Huntington's disease and ALS was shown to potently activate the HSR in mammalian cells. Most recently, a yeast-based high-throughput screen for small molecule activators of HSF-1 identified the compound HSF1A that was shown to activate HSF-1 without associated proteotoxicity20.
Despite the potential benefits, as shown in multiple cellular and animal models of diseases of protein conformation, further development of these small molecules will be necessary prior to their use as a therapeutic agent22,23. For example, although GA and other derivatives that inhibit Hsp90 are currently in pre-clinical and clinical development for the effective treatment of a number of different cancers24, their use for diseases associated with protein misfolding may have limited therapeutic potential. Given the imminent challenges facing the care of individuals afflicted with protein conformational diseases, and the lack of effective therapeutics, we propose that further investigation into small molecule PRs that activate HSF-1 is urgent. Here, we describe the discovery and characterization of novel small molecule PRs that, by enhancing HSF-1 activity, restore proteostasis in multiple diseases of protein conformation. We propose that modulation of the proteostasis network by HSF-1 PRs represents a new therapeutic approach for the treatment of both cytosolic and compartment-specific conformation disorders.
Results
Cell-based HTS for Small Molecule Activators of the HSR
We developed a high-throughput screen (HTS) that measures the activation of the HSR in HeLa cells stably transfected with a heat shock-inducible reporter containing the proximal human Hsp70.1 promoter sequence upstream of a luciferase (luc) reporter gene (Fig. 1a)25,26. To assess the sensitivity and robustness of the cell-based assay before undertaking the HTS campaigns, we established dose-response profiles using three positive controls, celastrol, cadmium chloride (CdCl2), and MG132 (Fig. 1b). The derived EC50 values for celastrol and MG132 were ∼3 and 5 μM respectively, in agreement with previous reports21. Optimization and miniaturization of the HTS formats were carried out independently at the Scripps Research Institute Molecular Screening Center (SRIMSC, Florida) and at the Southern Research Institute (SRI) (Supplementary Table 1 in the Supplementary Results section). MG132, due to its sigmoidal profile, and CdCl2, because of its ability to strongly induce the reporter (Fig. 1b), were subsequently employed as positive controls at the SRIMSC and Southern Research Institute, respectively (Supplementary Table 2). Assessment of critical variables of the assay was carried out during the miniaturization and optimization steps to achieve 384 and 1,536-well plate formats (Supplementary Table 1). The Z′ values for the miniaturized cell-based assays were ≥0.6, indicative of consistency and reproducibility. The HeLa-luc assay was validated by pre-screening two different libraries, the 1,280-molecule Library of Pharmacologically Active Compounds (LOPAC) from Sigma at the SRIMSC, and a 2,000-compound set of known biologically active compounds from MicroSource Discovery Systems at Southern Research Institute.
Figure 1. Identification of small molecule proteostasis regulators (PRs) by high-throughput screening.

(a) HeLa-luc cells were used to screen compound libraries to identify small molecule PRs. The Hsp70.1pr-luc construct is diagrammed. The sequences of the upstream region of the human Hsp70.1 promoter from +1 to -188 are represented by a line. The locations of transcription factor binding sites are depicted as boxes and their corresponding genetic elements are indicated in the boxes. The transcription factors that bind to these regions are indicated above the boxes. The nucleotide sequence of the HSE is shown and the inverted nGAAn repeats, to which HSF-1 binds, are labeled with arrows and marked 1 through 5. (b) HeLa-luc cells were treated with celastrol, cadmium chloride and MG132 at the indicated concentrations and luciferase activity was measured after 24 h. Each experiment was performed in triplicate. The standard deviation is shown. (c) Confirmed hits (263) were clustered accordingly to their chemical substructure and a total of 7 clusters were identified. The number of hits per cluster is shown.
Identification of Novel Classes of Small Molecule PRs
Two primary screens of 803,587 and 100,000 compounds were performed independently at the SRIMSC and at SRI, respectively. The small molecule libraries consisted of: (i) 607,408 compounds from the Scripps Drug Discovery library and 196,179 compounds from the Molecular Libraries Probe Production Center Network (MLPCN) library, both run at the SRIMSC; and (ii) 100,000 compounds from the National Institute of Neurological Disorders and Stroke (NINDS) library, run at the Southern Research Institute. Assay performance was consistent across plates, with robust Z′ factors and signal-to-background ratio (Supplementary Fig. 1 and Supplementary Table 2). The data obtained from the primary screens was normalized to each positive control set as 100% activation. The screen performed at the SRIMSC identified 759 primary hits, whereas 37 primary hits were identified at SRI (Supplementary Table 2).
False-positive hits were excluded by performing a triplicate run on the primary hits that yielded 263 confirmed compounds (Supplementary Table 2). Although the more stringent conditions elicited by CdCl2 led to a lower number of primary hits, these conditions also identified a smaller fraction of false positives, as demonstrated in the confirmatory secondary assays (Supplementary Table 2). Confirmed hit compounds were reordered as dry powders from commercial vendors (ChemDiv, Enamine, ChemBridge, and Asinex) or re-synthesized for retesting in a full dose response. The active compounds were clustered by structure with a 0.8 Tanimoto27 cutoff followed by manual inspection to merge clusters sharing a common scaffold. This analysis resulted in 233 hits grouped into seven clusters (A through G, Fig. 1c) that did not show structural similarities with the remaining 30 hits. Many compounds contained reactive moieties known to activate the HSR, such as reactive α,β-unsaturated carbonyls28,29, but other series had no obvious reactive groups.
The two primary screens performed with different positive controls, chemical libraries and compound concentrations, independently identified three common scaffolds (C, E and F, Fig. 1c and Table 1), providing cross-validation of the cell-based screen. These active compounds were subsequently tested in the HeLa-luc cells to establish dose-response curves and compound toxicity. EC50 and CC50 values were determined: most hits exhibited a sharp activation profile, with only few compounds displaying sigmoidal dose-response curves. Subsequent studies were initially performed with fourteen representative compounds (A1, A2, A3, B1, B2, B3, C1, C2, D1, E1, F1, F2, G1 and G2, Table 1) belonging to the largest clusters A-G (Fig. 1c). The dose-response activity and toxicity profiles for the selected compounds are shown in Supplementary Figure 2. We refer to these compounds as small molecule proteostasis regulators (PRs).
Table 1.
Chemical structure of the selected small molecules PRs
| Cluster A |
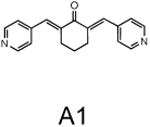
|
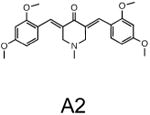
|
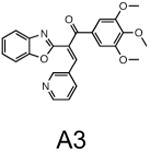
|
| Cluster B |
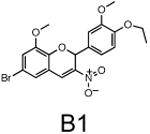
|
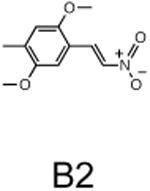
|
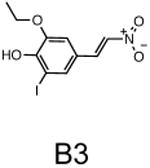
|
| Cluster C |
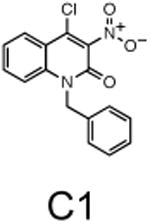
|
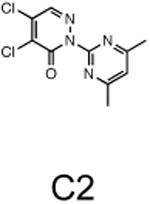
|
|
| Cluster D |
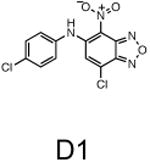
|
||
| Cluster E |
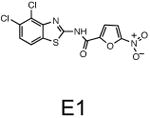
|
||
| Cluster F |
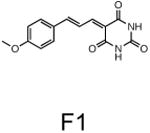
|
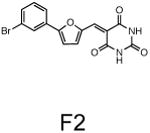
|
|
| Cluster G |
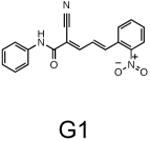
|
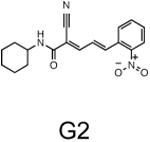
|
Induction of Hsps and Activation of HSF-1 by the PRs
The results of the HTS screen were further validated by direct demonstration that the expression of endogenous Hsp mRNAs and proteins were induced by representative PRs. Exposure of HeLa cells to the PRs induced Hsp70 mRNA levels from 2 to 5-fold (Fig. 2a and Supplementary Fig. 3a). Likewise, western blot analysis (Fig. 2b) showed that the levels of multiple chaperones (Hsp70, Hsp40 and Hsp27) were induced from 2 to 6-fold, similar to the positive controls, respectively (Fig. 2b and Supplementary Fig. 3b).
Figure 2. The small molecule PRs induce Hsp expression by activating HSF-1.

(a) HeLa cells were treated with DMSO, celastrol (Cel, 3 μM), MG132 (MG, 10 μM), CdCl2 (Cd, 50 μM) and selected PRs for 4 h. Similar results were obtained in two independent experiments. Densitometric measurements of Hsp mRNA levels normalized to GAPDH in relation to control DMSO-treated cells were performed using ImageJ software. (b) Western blot analysis of HeLa cells treated with DMSO, celastrol (Cel, 3 μM), MG132 (MG, 10 μM) and selected PRs. Fold induction was calculated as the ratio of normalized Hsp values between a compound-treated sample and the untreated control. Densitometric measurements of Hsp levels normalized to tubulin were performed as in (a). (c) Gel mobility shift assay was performed with a [32P]HSE oligonucleotide and HeLa cell whole cell extracts. DMSO: lanes 2 and 17; celastrol (3 μM): lanes 3 and 18; MG132 (10 μM): lanes 4 and 19; small molecule PRs (10 μM): lanes 5-7 and 20-23. Lanes marked self (lanes 8-12) contained a 200-fold molar excess of unlabeled complementary oligonucleotide. Lanes marked +Ab (lanes 13-15 and 24-29) contained a HSF-1 antibody. (d) HeLa cells were treated with DMSO, celastrol (3 μM) and selected PRs (10 μM) for 30 and 60 min and then chromatin was cross-linked, harvested, and immunoprecipitated with HSF-1 antibody (+Ab). The samples were then analyzed by PCR with primers specific for the Hsp70.1 and the dihydrofolate reductase (DHFR) promoters. The controls include input DNA and a no antibody control (-Ab).
The elevated expression of multiple Hsp genes by the PRs can be most directly explained by induction of the HSR and activation of HSF-1. To address this, we employed electrophoretic gel mobility shift assays (EMSA). Of the fourteen PRs previously tested for chaperone induction (Fig. 2a and b), 7 compounds representative of each chemical cluster were selected for this study (A1, A3, B1, C1, D1, E1, F1). Incubation of HeLa cells with the PRs strongly induced HSF-1 DNA-binding activity (Fig. 2c). The specificity of HSF-1 induction (lanes 3-7 and 18-23) was demonstrated by competition with excess unlabeled HSE oligonucleotide (lanes 8-12) and by anti-HSF-1 antibody supershift experiments (lanes 13-15 and 24, 26-29). The results of EMSA analysis were further confirmed by chromatin immunoprecipitation (ChIP) experiments. The PRs that most strongly induced HSF-1 activation in the gel mobility shift assay (A1, A3, C1, D1 and F1, Fig. 2c) were selected for ChIP analysis and the results showed occupancy of HSF-1 to the endogenous Hsp70.1 (Fig. 2d and Supplementary Fig. 3c), Hsp40 and Hsp27 promoters (Supplementary Fig. 4), but not to the negative control promoter.
From a chemical mechanism perspective, many previously identified small molecule inducers of the HSR have been suggested to activate HSF-1 by causing direct protein thiol oxidation. Compounds A1, A3, C1, D1 and F1 contain cysteine reactive moieties such as unsaturated-carbonyls (Supplementary Table 3). No close analogs of these five compounds that lacked these reactive groups were present in the HTS libraries, and therefore they were not tested. To determine whether the PRs A1, A3, C1, D1 and F1 caused protein thiol oxidation, we tested if their activity could be inhibited by treatment with reducing agents such as N-acetyl cysteine (NAC) and dithiothreitol (DTT). Induction of the HSR by celastrol was inhibited by NAC (2 mM) and DTT (250 μM) treatment, as expected (Supplementary Fig. 5f and l). Only the activity of PR F1 was completely inhibited by NAC and DTT treatment, indicating that F1 may cause oxidative damage by modifying protein cysteine residues (Supplementary Fig. 5e and k). In contrast, the activities of C1 and D1 were only slightly less affected (Supplementary Fig. 5c-d and i-j) and activation of the HSR by A1, and A3 was unaffected (Supplementary Fig. 5a-b and g-h), indicating that these PRs do not act through this mechanism.
PR Activity on Hsp Genes Requires HSF-1 and the HSE
Activation of HSF-1 by the PRs correlates with enhanced expression of chaperone genes, but does not formally demonstrate a requirement for a wild type HSE sequence or a dependence upon HSF-1. To directly address this, we tested the five PRs for their ability to induce luciferase expression in HeLa cells transfected with a mutated HSE sequence fused to a luciferase gene. As expected, cells lacking a wild type HSE failed to induce luciferase expression (Supplementary Fig. 6), indicating that an intact HSE is necessary for activation of the HSR by the PRs. We then treated wild type (WT) and hsf-1 null (hsf-1-/-) mouse embryonic fibroblasts (MEFs) with the PRs and showed that PR-induction of Hsp70 mRNA was detected only in WT cells and not in hsf-1-/- cells (Fig. 3a and Supplementary Fig. 7a). These results provide conclusive evidence that PR induction of chaperone expression is dependent upon activation of HSF-1.
Figure 3. The PRs are HSF-1-dependent.

(a) Wild type (hsf-1+/+) and HSF-1 null (hsf-1-/-) mouse embryonic fibroblasts (MEFs) were treated for 4 h with DMSO vehicle, celastrol (3 μM) or indicated PRs (10 μM). RNA was extracted and reverse-transcribed. PCR reactions were performed on cDNA for the indicated transcripts. GAPDH RNA levels were assayed to determine equal loading. (b, d-g) WT and (c, h-k) hsf-1-/- MEFs were treated for 4 h with either DMSO, MG132 (MG, 1 μM), geldanamycin (GA, 1 μM), tunicamycin (Tm, 1 μM), sulphorahane (Sul, 1 μM) or selected PRs (A3, C1, D1 and F1) at the indicated concentrations. Relative levels of multiple cytoprotective genes were measured by real-time PCR (qPCR) with tubulin serving as a reference gene.
PRs Activate Multiple Proteostasis Network Pathways
We next examined the gene signature of the PRs using a multiplex gene expression analysis to identify additional proteostasis mechanisms regulated by the PRs. We asked whether the PRs could activate other stress responsive proteostasis network (PN) pathways such as the unfolded protein response (UPR) and the antioxidant stress response, in addition to the HSR. Therefore, we monitored the expression of the UPR-inducible gene GRP78/BiP, the antioxidant responsive genes heme oxygenase 1 (HO1) and the regulatory subunit of glutamate-cysteine ligase (GCLM), and the proapoptotic growth arrest- and DNA damage-inducible gene 153 (GADD153, also known as CHOP). WT and hsf-1-/- MEF cells were treated with PRs and the positive controls MG132 (MG) and geldanamycin (GA) that induce the HSR, oxidative stress, and the UPR; tunicamycin (Tm) that induces the UPR; and sulphoraphane (Sul) that activates the antioxidant response (Fig. 3b and c). Untreated (Unt) and DMSO-treated cells served as negative controls (Fig. 3b and c).
The PR stress response signatures were established in WT and hsf-1-/- MEF cells (Fig. 3d-g and h-k). At a range of concentrations of PRs A3, C1, D1, and F1, Hsp70 mRNA levels were induced from 9 to 30-fold in WT MEF cells (Fig. 3d-g). Compound D1 (Fig. 3f) was selective and only induced the expression of Hsp70, whereas A3 and C1 strongly induced Hsp70, in addition to a 3-fold increase in BiP (A3 and C1) and HO1 (A3 only) expression (Fig. 3d and e). Likewise, compound F1 induced multiple responses and strongly induced Hsp70, the oxidative stress response genes (HO1 and GCLM), and a 2.5-fold upregulation of BiP (Fig. 3g).
In performing parallel experiments on hsf-1-/- cells (Fig. 3h-k), we noticed that the level of induction of HO1 was dramatically enhanced from 12 to 130-fold, whereas the expression of GCLM and BiP was comparable to WT MEF cells (Fig. 3h-k). These results suggest that up-regulation of an anti-oxidant stress response compensates for HSF-1 deficiency. At the highest PR concentrations, induction of the cell death pathway (GADD153) was also observed. Our previous experiments employing DTT treatment indicates that PRs A1, A3, C1 and D1 did not activate the HSR by causing oxidative stress, yet we observed potent induction of the antioxidant responsive gene HO1 in absence of HSF-1 (Fig. 3h-k). There may be at least two explanations for this apparent discrepancy. First, if the induction of HO1 by the PRs were due to the generation of oxidative stress, then we would expect a concerted upregulation of the antioxidant GCLM gene, as occurs for compound F1. This, however, is not observed in WT cells. In addition, transcriptional regulation the HO1 gene indicates that expression is regulated by multiple stimuli, and not solely dependent upon oxidative stress30.
PRs Protect Cells Against Severe Stress and Apoptosis
Activation of the HSR and induction of molecular chaperones has been shown to protect cells from the deleterious consequences of protein damage and apoptosis. Therefore we tested if the PRs A1, A3, C1, D1 and F1 displayed cytoprotective properties. Pretreatment with either 42°C heat shock or the PRs A3, D1 and F1 significantly protected cells from cell death induced by a 35 min severe heat shock (Supplementary Fig. 8a). On the contrary, the PRs A1 and C1 did not display any cytoprotective properties and instead increased the fraction of cell death after the 45°C treatment compared to the DMSO control.
We next determined if the PRs protected against apoptotic cell death induced by oxidative stress. Assessment of cellular apoptosis and necrosis was performed by staining HeLa cells with Annexin V and propidium iodide (PI). In agreement with the cytoprotection data, treatment with the PRs A3, D1 and F1 led to a two-fold protection from H2O2–induced apoptosis, as indicated by the reduced number of Annexin V-stained cells compared to the untreated cells (Supplementary Fig. 8b). On the contrary, cells pretreated with PRs A1 and C1 exhibited both an apoptotic (A1 and C1) and necrotic pattern (C1), (Supplementary Fig. 8b).
Proteostasis Restoration in Models of Conformational Disease
In addition to its well-established role in maintaining cytoplasmic proteostasis, HSF-1 has also been recently shown to ameliorate ER stress31. We therefore asked whether the PRs, by enhancing chaperone expression, would reduce protein misfolding in diseases in which expression of mutant proteins accumulates in either the cytoplasm or the ER.
As a representative cytosolic model, we examined the effects of the PRs on Huntingtin aggregation in PC12 cells, conditionally expressing human Huntingtin (htt) exon 1 containing an expansion of 74 glutamines, fused to green fluorescent protein (GFP) (httQ74-GFP)32. In these cells, httQ74-GFP aggregates are detected after 48 hours of induction. PC12 cells were treated with the PRs and visually monitored for aggregate formation. Incubation with A1, D1 and F1 caused a reduction (from 2 to 3-fold) of httQ74-GFP protein aggregates without altering the levels of httQ74-GFP protein (Fig. 4a and b and Supplementary Fig. 9a-f), whereas PRs A3 and C1 had no effect, although Hsp70 levels were induced (Fig. 3d and e).
Figure 4. The PRs restore proteostasis in cell-based models of cytoplasmic and compartment-specific conformational diseases.

(a) PC12 cells expressing httQ74-GFP were treated either with DMSO (panel I), geldanamycin (GA, 200 nM, panel II) or with selected PRs (panels III-V). The representative fluorescence pattern of httQ74-GFP after 72 h of induction is shown. Scale bar: 10 μm. (b) Quantification of results shown in panel (a). Cells containing aggregates were counted and are shown as a percentage of the total number of cells counted. The data shown are derived from three independent experiments. (c) CFBE41o- YFP cells were treated with 0.1% DMSO (black), the positive controls 5 μM SAHA (purple), 10 μM Corrector 4a (Corr4a) (grey) and the PRs A3 (dark blue), C1 (royal blue) and F1 (cyan) at 10 μM for 24 h. Fluorescence quenching is indicative of restored ΔF508-CFTR trafficking (mean ± s.e.m.; n = 3). Color-coded asterisks indicate statistically significant differences from DMSO control at the 30 s time point. (d) CFBE41o- cells were treated with 0.1% DMSO, SAHA (S, 1μM) and selected PRs at the indicated concentrations for 24 h. ΔF508-CFTR trafficking was analyzed by monitoring the band B and C glycoforms (fold increase relative to DMSO band B ± s.e.m.; n = 3) at the various concentrations of PRs. The level of Hsp70 was also monitored by western blot as an indicator of HSF-1 activation. 15 μg of protein were loaded and equal loading was confirmed by staining the membrane with Ponceau S.
We next investigated the PRs on a cellular model of cystic fibrosis. This model corresponds to a human bronchial epithelial cell line (CFBE41o-) stably co-expressing the ΔF508 mutation of the CFTR and a halide-sensing mutant of YFP (H148Q/I152L-YFP). ΔF508-CFTR is defective both in trafficking from the endoplasmic reticulum to the plasma membrane and in channel gating33. Correction of ΔF508-CFTR trafficking by the PRs would lead to increased level of active protein at the cell surface, affording increased flow of extracellular halides into the cell, resulting in reduction of YFP fluorescence intensity.
The same PRs tested in the Huntington's disease cell model were also tested in ΔF508-CFTR expressing cells and positive results were obtained for A3, C1 and F1 (Fig. 4c and Supplementary Fig. 10a). The extent of YFP quenching detected was comparable to that seen with corrector 4a, a commonly used positive control for this assay (Fig. 4c). It is worth noting that compound F1 represents the first characterized small molecule capable of enhancing the correct folding of proteins expressed in two different cellular compartments.
To further confirm that the PRs A3, C1 and F1 rescued ΔF508-CFTR trafficking, we monitored the processing of ΔF508-CFTR. Treatment of CFBE41o- cells expressing ΔF508-CFTR with the PRs generated higher molecular mass forms of ΔF508-CFTR, consistent with full glycosylation (Fig. 4d-f and Supplementary Figs. 10b and 11), indicating that the PRs partially rescued the cell-surface expression and maturation of ΔF508-CFTR. In addition, we show that rescue of ΔF508-CFTR trafficking by the PRs coincides with Hsp70 upregulation, suggesting that the PR action depends on HSF-1 and the induction of molecular chaperones.
Suppression of Aggregation and Toxicity in C. elegans
We asked whether the efficacy of the PRs to reduce protein aggregation could be also observed in a C. elegans model for expression of expanded polyglutamines (35 glutamines fused to YFP, Q35∷YFP) in body wall muscles, that displays age-dependent aggregation and toxicity34. This model exhibits many characteristics of polyglutamine diseases, such as Huntington's disease, and has been a valuable tool in the identification of genetic and chemical modifiers of aggregation and toxicity35,36.
Age-synchronized Q35 animals were treated with the PRs and the effects on aggregation and toxicity scored at 6 days of age. We used 17-AAG as a positive control inducer of the HSR that induces chaperone expression and reduces polyQ aggregation16,18. Treatment of C. elegans with 17-AAG showed a marked reduction in protein aggregates and toxicity (Fig. 5a-c) by inducing the HSR (Fig. 5d). Treatment with PRs A1, D1 and F1 suppressed Q35 aggregation (Fig. 5a and b) without affecting overall levels of Q35 protein (Supplementary Fig. 9g and h). Suppression of polyQ aggregation also ameliorated aggregation-associated toxicity. Expression of Q35 in the body wall muscles reduces motility by 50% relative to WT, and PR treatment of Q35 animals restored (80%-100%) motility to near WT (Fig. 5c). These results reveal that the PR-induced suppression of aggregation also prevented polyQ toxicity.
Figure 5. The PRs reduce aggregation/toxicity in C. elegans models of diseases associated with polyQ expansions.

(a) C. elegans expressing YFP-tagged Q35 protein were treated with either DMSO (panel I) or PRs (panels III-V) at different concentrations (1, 5, 10 and 15 μM) for 4 days. 17-AAG was used as positive control (50 μM, panel II). Fluorescence microscopy images show the PRs that reduced Q35 aggregation at a concentration of 10 μM in 6-day old animals. Panels VI-X show higher magnification images of the boxed areas on the top panels. Scale bar: 0.1 mm. (b) PRs suppress Q35 aggregation as shown by the quantification of fluorescent foci in 6-day old animals, relative to DMSO control. (c) Rescue from polyQ-associated toxicity was determined by comparing the motility of Q35 animals treated with either DMSO alone or the candidate PRs compounds (10 μM) to that of WT animals in DMSO. 17-AAG (50 μM) was used as positive control. Standard error of the mean is shown. (t-test ***p-value<0.001). (d) The PRs up-regulate mRNA expression of cytosolic chaperones (HSP-70 family members and small Hsps) at the concentrations needed to suppress aggregation and toxicity. Real-time qPCR was performed on samples extracted from animals treated with either DMSO, 17-AAG (50 μM), or PRs (10 μM). Standard deviation is shown.
We then confirmed that the effect of the PRs on polyQ aggregation and toxicity was associated with expression of molecular chaperones and induction of the HSR (Fig. 5d). Chaperone expression was HSF-1-dependent (Fig. 6a-c) and moreover downregulation of HSF-1 by RNAi abrogated the PR-induced protection against Q35 aggregation (Fig. 6d).
Figure 6. Chaperone expression and reduction in polyQ aggregation in C. elegans is HSF-1-dependent.

(a-c) For each of the PRs, chaperone up-regulation is HSF-1 dependent. Wild type (control) and HSF-1 mutant (hsf-1(sy441)) animals were treated with each of the PRs or the positive control 17-AAG, and real-time qPCR was performed to show that both HSP-70 (C12C8.1, F44E5.4) and small Hsps (hsp-16.1) induction does not occur in the hsf-1 mutant background. For compounds A1 and D1, DAF-16 also contributes to chaperone up-regulation, as SKN-1 does for F1. Standard deviation is shown. (d) Suppression of Q35 aggregation by the PRs (shown as % of fluorescent foci) is HSF-1 dependent and is not observed when hsf-1 is down regulated by RNAi. (e) Animals carrying temperature sensitive mutations in muscle proteins UNC-52 (perlecan, stiff paralysis) and UNC-45 (myosin assembly, egg laying defect) were incubated with the PRs. At the restrictive temperature of 25°C, F1 suppressed the muscle dysfunction phenotypes, indicating improved folding of UNC-52 and UNC-45. (f) Stress related genes up-regulated by each of the PRs relative to control (DMSO): hsp-4 (ER HSP-70), sod-1 (oxidative stress), daf-21 and ZC395.10 (HSP-90 and co-chaperone) and ubq-2 (ubiquitin). Standard deviation is shown.
PRs Stabilize the Folding of Metastable Proteins
To begin examining the basis for PR induction of the HSR, we asked whether these small molecules caused protein damage and therefore activated a HSR, or alternatively that the PRs activated specific homeostasis regulators to induce chaperone expression. To address this, we used C. elegans strains harboring temperature-sensitive (TS) mutations in specific endogenous muscle proteins, including the basement-membrane protein perlecan UNC-52, and a myosin-assembly protein UNC-45. These conditional mutations do not interfere with folding and function at the permissive temperature (15°C), but cause a complete loss of function at the restrictive temperature (25°C), resulting in distinctive muscle dysfunction (Fig.6e, DMSO 15°C and 25°C). These metastable proteins therefore serve as folding sensors that monitor changes in cellular proteostasis10. To determine whether the PRs induce protein misfolding, we incubated both unc-52(e669su250) and unc-45(e286) animals with PRs A1, D1 and F1 (10 μM) at 15°C. We reasoned that enhancing misfolding would unmask the TS phenotypes at the permissive condition, which was not observed. Alternatively, to determine whether the PRs enhanced the folding environment, we incubated animals with PRs, transferred them to 25°C, and scored for TS phenotypes 2 days later (Fig. 6e). While PR A1 caused an intermediate 35% suppression of the unc-45(e286) TS phenotype, the most potent effect was observed with F1, that suppressed both unc-52(e669su250) and unc-45(e286) phenotypes by 80% and 90% respectively (Fig. 6e). These results suggest that the PRs do not activate the HSR by interfering with cellular protein folding in general, but rather promote folding of metastable proteins.
PRs Depends on the Folding and Quality Control Machinery
The striking improvement in protein homeostasis, following treatment with the PRs, prompted us to investigate the requirements for other regulatory components of the PN such as DAF-16 (FOXO ortholog, insulin/IGF-1-mediated signaling transcription factor) and SKN-1 (oxidative stress response transcription factor). We simultaneously treated Q35 animals with the PRs and RNAi to knockdown the stress regulators DAF-16 and SKN-1. We examined the effects on induction of hsp-70 (C12C8.1, F44E5.4) and small HSP hsp-16.1 and show that A1 and D1-mediated chaperone expression requires both HSF-1 and DAF-16 (Fig. 6a and b), whereas F1 activity is dependent upon HSF-1 and SKN-1 (Fig. 6c).
We explored the role of these stress responses on PR-regulated folding by monitoring the expression of downstream targets of SKN-1 and DAF-16, and other protein folding components including HSP-90 and co-chaperone genes, ubiquitin, and components of the ER HSP-70 (UPR). Compounds D1 and F1 elicited induction of sod-1 (SKN-1 target), hsp-4 (UPR-ER) and hsp-90 and its co-chaperone (ZC395.1) (Fig. 6f). Meanwhile, A1 induced ER hsp-70 levels, and ubiquitin protein ubq-2 (Fig. 6f and Supplementary Fig. 12). We examined the expression of a number of oxidative stress, UPR, mitochondrial-UPR and lifespan/aging regulators (Supplementary Fig. 12), and observed that only genes encoding folding and chaperone components were affected (Fig. 6f). These results indicate that the pathways that are activated to enhance the folding environment depend on HSF-1, DAF-16 and SKN-1 and the chaperone and quality control machinery.
PRs are not Inhibitors of the Proteasome or Hsp90
Having demonstrated that the PRs themselves do not cause protein misfolding, we examined whether the PRs activated the HSR via inhibition of the proteasome or Hsp90 function. For these experiments, we selected the PRs A1, A3 and F1 because A1 and F1 suppressed aggregation in cellular and C. elegans models of conformational diseases and stabilized folding of TS mutant proteins, and A3 and F1 improved the folding stability of mutant CFTR.
Exposure of HeLa cells to MG132 and lactacystin for 3 and 6 hours (Fig. 7a and Supplementary Fig. 13a), reduced proteasome activity to 20 percent relative to DMSO-treated cells (Fig. 7a) and increased the levels of polyubiquitinated substrates (Fig. 7b). By comparison, treatment with the PRs A1, A3 and F1 neither inhibited proteasome activity nor increased polyubiquitinated proteins (Fig. 7a and b).
Figure 7. The PRs are not proteasome or Hsp90 inhibitors.

(a) HeLa cells were incubated with either DMSO, MG132 (10 μM), lactacystin (lactacys., 6 μM) and the PRs A1, A3 and F1 (10 μM) for 6 h. Proteasome-associated chymotrypsin activity was assessed using the fluorogenic substrate Suc-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (suc-LLVY-AMC) as described in Materials and Methods. (b) HeLa cells were either left untreated or treated with DMSO, the proteasome inhibitor MG132 (10 μM), or the PRs A1, A3 and F1 (10 μM) for 16 h. Whole cell extracts of HeLa cells were separated by SDS-PAGE, transferred to membranes, stained with Ponceau S to visualize total protein and probed using a rabbit polyclonal antibody to detect ubiquitin. (c) HeLa cells were treated with either DMSO, 17-AAG (2 μM), MG132 (10 μM), lactacystin (lactacys., 6 μM) or the PRs A1, A3 and F1 (10 μM) for 24 hr. Protein levels of various Hsp90 client proteins (Cdk-4, Raf-1 and Akt) in equal amounts of whole-cell lysates were assessed by western blot analysis. GAPDH was used as loading control. Densitometric measurements of Hsp90 client protein levels normalized to GAPDH in relation to control DMSO-treated cells were performed using ImageJ software. (d) Refolding of chemically-denatured firefly luciferase was assessed in RRL containing 2 mM ATP in the presence of either DMSO (○), 17-AAG (2 μM, ■) or the PR A1 (10 μM, ×). Luciferase activities are expressed as percent of the native enzyme control. The result shown is representative of three experiments.
We next monitored the effects of the PRs on Hsp90 by assessing Hsp90 client protein degradation37. The levels of three well characterized Hsp90 client proteins, Cdk4, Raf-1, and Akt, were quantified by western blot analysis of PR-treated HeLa cells. Relative to the DMSO-treated control cells, a reduction in the levels of the Hsp90 client kinases was observed with the PR A1 (10 μM) after 6 hours of treatment (Supplementary Fig. 13b), whereas treatment with the other two PRs had no effect. The clearance of these Hsp90 clients was even more dramatic after 16 (Supplementary Fig. 13c) and 24 h (Fig. 7c and Supplementary Fig. 7b) of PR treatment, and was comparable to the inhibitory effects of 17-AAG on Hsp90 function.
Since classical Hsp90 inhibitors such as geldanamycin, 17-AAG and radicicol bind competitively to the ATP site of Hsp90, we investigated whether the PRs A1, A3 and F1 had a similar mode of action. We therefore tested whether the PRs could compete in vitro with geldanamycin for binding to the ATP binding pocket of Hsp90. Whereas the positive control 17-AAG competed effectively with geldanamycin, none of the three PRs disrupted the interaction between geldanamycin and Hsp90 even at concentrations in excess of 100-fold (Supplementary Fig. 13d).
Although the competition assay may suggest that the PRs A1 is not an inhibitor of Hsp90 activity, it is also possible that the compound acts through a different mechanism compared to known Hsp90 inhibitors. To further confirm that the previous results were not due to a direct inhibition of Hsp90, we performed a chaperone-dependent protein refolding assay. Refolding of denatured luciferase was monitored in rabbit reticulocyte lysates (RRL) by measuring luciferase activity. In the presence of DMSO, luciferase recovered 40-45% activity, whereas 17-AAG (2 μM) caused only about a 25% recovery (Fig. 7d). Incubation of RRL with the PR A1 (10 μM) did not inhibit luciferase refolding (Fig. 7d). These results indicate that A1 is not a direct inhibitor of Hsp90 activity. Although PR A1 did not directly inhibit Hsp90 activity, we offer possible explanations for the increase in client protein degradation by PR A1. A1 could disrupt the interaction between Hsp90 and its co-chaperone Cdc37 implicated in shuttling kinase clients to Hsp90, or A1 could inhibit Hsp90 activity by binding to a site different than the ATP binding pocket.
Discussion
In this study we describe the results of a large-scale small molecule screen in human cells for HSF-1-dependent activators of chaperone expression. We identified 263 PRs that chemically induce the HSR and result in the activation of HSF-1 and elevated expression of multiple chaperone gene families. The PRs described here represent novel chemical series and, by comparison to previously identified small molecule activators of the HSR, do not cause protein misfolding, proteasome inhibition, or Hsp90 inhibition. A further understanding of these PRs and their ability to activate the HSR and restore protein folding in multiple disease models offers new opportunities and strategies for small molecule chaperone therapeutics for protein conformational diseases with novel specificities and reduced toxicity.
An intriguing observation is that the PRs exhibit complex stress response signatures. In addition to inducing HSF-1 and the expression of multiple cytoplasmic chaperones, we have observed the induction of other major components of the PN including the UPR and the antioxidant response genes. Our PR strategy is based on the proposition that small molecules can mimic the molecular signals recognized by the cell associated with a proteostatic imbalance (Supplementary Scheme 1). This activation of stress-signaling pathways, in turn restores the stability and functionality of the proteome. The ability of these PRs to activate one or more stress response pathways suggests a therapeutic approach that employs the cell biological response to damaged proteins to protect cells against chronic disease. By this approach, we are employing our growing understanding of stress biology to promote the health of the cell. In doing so, we are using compounds to enhance the properties of biological pathways that are already employed by the cell to manage proteostasis, even when challenged by stress and disease. We suggest that this systems and network approach represents an alternative for drug discovery as we harness the protective abilities of cellular stress responses to protect the cell against the multitude of deficiencies that occur during chronic proteotoxicity and stress.
The central role for HSF-1 in maintaining and restoring proteostasis makes this transcription factor and the HSR an attractive target for therapeutic intervention in conformational diseases. The observation that diverse chemical types have in common the ability to induce the HSR, despite their broad activities, supports our proposal that HSF-1 is a stress network hub that integrates multiple stress signaling pathways to coordinate regulatory responses to maintain proteostasis in health, aging, and disease. Of the 263 hits identified in this study that activate HSF-1, we have focused our attention on the 7 major clusters represented by the chemical series: β-aryl-α,β-unsaturated-carbonyls (cluster A), β–nitrostyrenes (cluster B), β-Cl-α,β-unsaturated-carbonyls (cluster C), nitrobenzofurazans (cluster D), nitrofuranylamides (cluster E), unsaturated barbituric acids (cluster F), and 2-cyanopentadienamide (cluster G). These chemical series exhibit a broad range of pharmacological indications and diverse mechanisms of action and, to our knowledge, have not been previously linked to proteostasis and the HSR. For example, compounds in cluster A are chalcone and curcumin analogues and have antibacterial, antioxidant and cancer chemopreventive activities38,39. These compounds are known to inhibit NF-κb and modulate the Keap1-Nfr2 complex40,41. Nitrobenzofurans (cluster E) have antitubercular activity, and nitrofuran antibiotics, i.e. nitrofurantoin, are currently used as second-line agents for urinary tract infections. The nitroimidazole antibiotics are structurally related; for example, metronidazole, is a widely used antibiotic for treatment of anaerobic bacterial and protozoan infections42. Compounds belonging to cluster F are barbiturate analogs associated with anti-inflammatory side effects; in particular thiobarbiturates reduce activation of NF-κb. Thiopental, but not the oxy-analogue pentobarbital, is the only barbiturate suggested to activate the HSR and this property has been attributed to thiopental reactivity with protein thiols43. Of interest, barbiturate analogues have been previously reported as potentiators of defective ΔF508-CFTR channel gating44. Our results reveal unexplored mechanisms by which these chemical classes exert their beneficial effects, and suggest new pathways involved in activation of HSF-1. We propose that the ability of barbiturate analogs to rescue mutant CFTR defective channel gating can now be linked to the activation of the HSR and/or of the UPR. Likewise, the neuroprotective effects attributed to curcumin45, a chalcone analog, may be due to the induction of chaperone expression46. Taken together with the data presented here, we propose that compounds of the same chemical classes identified in our HTS can be reclassified as PRs.
We describe a first generation series of tool compounds that, by activating HSF-1 and other cell-protective stress responses, demonstrate efficacy in cellular and animal models of protein conformational diseases. Although we have shown that the PRs are effective in multiple misfolding disorders, i.e. cystic fibrosis and Huntington's diseases, these molecules may have a broader efficacy. For example, PRs that restore proteostasis by simultaneously inducing the HSR and the UPR should be able to enhance the folding, trafficking and activity of mutant enzymes in a variety of diseases, including lysosomal storage diseases (i.e. Tay-Sachs, Gaucher and Pombe's diseases) and retinitis pigmentosa, that require both ER and cytoplasmic proteostasis47-50. Likewise, PRs selective to the HSR may be beneficial for diseases in which the expression of the affected protein is primarily cytoplasmic and nuclear, as in ALS and in the multiple forms of spinal cerebellar ataxia. Considering that the pathogenesis of many diseases, such as Alzheimer's, Parkinson's, ALS and cystic fibrosis disorders, is also associated with oxidative stress, the activation of the ARE pathway in conjunction with the HSR may be highly beneficial. In support of this, we show that the small molecule PR F1, that simultaneously induced both stress-protective pathways, was the only PR that restored proteostasis in distinct cellular compartments.
In conclusion, we propose that the adjustment of the proteostasis network by small molecule PRs of the HSR provides a previously unexploited and potentially powerful approach to obtaining proteome balance in both loss- and gain-of-function diseases by providing a superior corrective environment based on the principle of proteome balance. Besides the usefulness for potential therapeutic development, small-molecule HSR inducers can be used as pharmacological tools for further dissecting the multi-step activation pathway of HSF-1. We believe that a better understanding of the regulation of HSF-1 activation pathway and of its signaling mechanisms could lead to the discovery of compounds that exhibit stress signatures that are HSF-1 selective or activate multiple stress pathways that can be effective in the control of diseases of protein conformation.
Methods
Cell-based High-throughput Assays
HeLa-luc cells were used to screen three compound libraries consisting of 903,663 structurally diverse small molecules. A library of 100,000 compounds (NINDS library, http://www.ninds.nih.gov/funding/areas/technology_development/HTS_Facility.htm) was screened at the Southern Research Institute and two libraries of 607,408 and 196,179 compounds (Scripps Drug Discovery library and MLPCN library, http://mli.nih.gov/mli/mlpcn/) were screened at the SRIMSC. Detailed protocol and analysis are described in Supplementary Methods.
Reverse Transcription-PCR
RNA was purified using the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. After the reverse transcription reaction, PCR was performed using PCR primers specific for Hsp70 and GAPDH. The human Hsp70 primers were: 5′-AGAGCCGAGCCGACAGAG-3′ (forward) and 5′-CACCTTGCCGTGTTGGAA-3′ (reverse); the mouse Hsp70 primers were: 5′-CACCAGCACGTTCCCCA-3′ (forward) and 5′-CGCCCTGCGCCTTTAAG-3′ (reverse); the human GAPDH primers were: 5′-GTCGGAGTCAACGGATT-3′ (forward) and 5′-AAGCTTCCCGTTCTCAG-3′ (reverse); the mouse GAPDH primers were: 5′-TGCACCACCAACTGCTTAG-3′ (forward) and 5′-GGATGCAGGGATGATGTTC-3 (reverse). PCR products were amplified with Taq polymerase (Promega, Madison, WI) by using standard cycling conditions.
Western Blot Analysis
Analysis of chaperone expression was carried out using HeLa cells that were treated with the indicated compounds for 8 h. Cells were lysed in a buffer containing 20 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid; pH 7.9), 25% (vol/vol) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol and 2 mg/ml of complete protease inhibitor cocktail (Roche, Switzerland) for 30 min on ice. 15 μg of whole cell extracts were run on 7.5% SDS-PAGE gels and transferred to nitrocellulose. Primary antibody incubations were for 12 hours at 4°C in 10% BSA. The following primary antibodies were used: a rabbit polyclonal HSF-1 #4725, a mouse monoclonal Hsp70 antibody (4g4, Affinity Bioreagents Inc., Golden, CO), a mouse monoclonal Hsp40 (αHdj-1 clone 25)25, a mouse monoclonal Hsp27 (MA3-0015, Affinity Bioreagents, Inc., Golden, CO). All primary antibodies were used at a dilution of 1:10,000, except for the Hsp27 antibody, which was diluted 1:500. The anti-β-tubulin antibody (Sigma, St. Louis, MO) was diluted 1:5,000 and used to verify equal protein loading. The secondary antibody was an Alexa Fluor 680 goat anti mouse IgG diluted 1:5,000 (Invitrogen, Carlsbad, CA). Western analysis was performed with the Odyssey system (Li-COR, Lincoln, NE).
Gel Mobility Shift Analysis (EMSA)
Detailed protocol is described in Supplementary Methods.
Chromatin Immunoprecipitation Assays (ChIP)
ChIP was performed essentially as described previously25. Detailed protocol is described in Supplementary Methods.
Effect of NAC and DTT on the PR Activity
HeLa-luc cells were seeded in a white 96-well plate at a density of 10,000 cells/well. Cells were pretreated with either 2 mM NAC or 250 μM DTT for 1 h before addition of the positive control celastrol (Cel; 2.5 and 5 μM) or the selected small molecule PRs (A1, A3, C1, D1 and F1; 2.5, 5 and 10 μM). Cells were incubated with compounds for 24 h before luminescence signal acquisition. DMSO-treated cells were used as negative control.
Multiplex Gene Expression Analysis
WT and hsf-1-/- MEFs were treated with serially diluted compounds in a 7-point dose dependent manner. Cell lysates were pooled with mouse 8-gene multiplex probe sets and with 8 different sets of magnetic capture beads (Luminex Technology, Austin, TX) in a 100 μl/well volume. Fold changes in gene expression were obtained for each gene per well by normalizing the raw data first to the DMSO control and then to the TATA-box binding protein (TBP) housekeeping gene. Detailed protocol is described in Supplementary Methods.
Protein Aggregation Analysis in PC12 Cells
PC12 cells expressing httQ74-GFP were seeded in tissue culture treated 96-well plates as 7,500 cells/well and induced with doxycyclin (1 μg/ml). Compounds A1, A3, C1, D1, and F1 were used at concentrations ranging from 0.75 to 25 μM. GA (200 nM) was used as positive control. Final DMSO concentration was 0.5%. Fluorescence images were taken with a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss, Germany) at 20× magnification and images deconvoluted with the Axiovision software. For quantification of fluorescence microscopy analysis, approximately 500 cells were counted for each treatment.
ΔF508-CFTR YFP Quenching Assay
Human bronchial epithelial cells (CFBE41o-) stably expressing ΔF508-CFTR as well as H148Q/I152L-YFP (CFBE41o- -YFP) were treated with the indicated concentration of compounds in complete growth media and incubated at 37°C, 5% CO2 for 24 h. Cells were subsequently stimulated with a final concentration of 10 μM forskolin (fsk) and 50 μM genistein (gen) for 15 min prior to addition of PBS + NaI (replacement of NaCl with 137 mM NaI). Fluorescence was monitored every second for a total of 30 seconds (3 seconds prior to addition of NaI and 27 seconds after addition of NaI). Detailed protocol and analysis are described in Supplementary Methods.
ΔF508-CFTR Transport Assay
CFBE41o- cells stably expressing ΔF508-CFTR were treated with the indicated concentration of compound in complete growth media and maintained at 37°C, 5% CO2 for 24 hrs. Cells were lysed on ice and the supernatant was collected for analysis. Equal amount of total protein (15 μg) was separated by SDS-PAGE (8% gel) and transferred to nitrocellulose. The blot was probed overnight at 4°C for CFTR (3G11 rat monoclonal antibody at 1:500 dilution) and indicated chaperone proteins. Detailed protocol is described in Supplementary Methods.
C. elegans Assays for Aggregation and Motility Defects
The treatment with chemical compounds was performed in a 96-well plate format, in liquid culture. The animals were scored for changes in aggregation (number of fluorescent foci) using the stereomicroscope Leica MZ16FA equipped for epifluorescence (Leica Microsystems, Switzerland). For the motility assay, animals' movement was digitally recorded using a Leica M205 FA microscope with a Hamamatsu digital camera C10600-10B (Orca-R2, Leica Microsystems, Switzerland), and the Hamamatsu Simple PCI Imaging software. Detailed protocol is described in Supplementary Methods.
Supplementary Material
Acknowledgments
We acknowledge Dr. Joseph Maddry, the SRI and the NINDS for support in performing the primary screen; Peter Chase and Pierre Baillargeon of Scripps Florida for executing the SRIMSC screening activities; Sue Fox, James West, Sandy Westerheide, Jason Moran, Monica Beam and Kai Orton for technical assistance; Dr. Jesper S. Pedersen for help developing the worm tracker system; the Morimoto laboratory, in particular Drs. Tali Gidalevitz, Janine Kirstein, Cindy Voisine and Anan Yu for helpful comments. PC12 httQ74-GFP cells were kindly provided by Dr. David Rubinsztein. Purified Hsp90β was a generous gift of Dr. Ahmed Chadli. We thank Dr. Aaron Ciechanover for the rabbit polyclonal anti-ubiquitin antibody. The CFBE41o- -YFP cells were a generous gift from Dr. Luis Galietta. This work was supported by the National Institutes of Health Training Grant in Signal Transduction and Cancer T32 CA70085 and the National Institutes of Health Training Grant in Drug Discovery in Age Related Diseases T32 AG000260 (to B.C.), Portuguese PhD fellowship from Fundação para a Ciência e Tecnologia (SFRH/BD/28461/2006) (to M.C.S.), National Institutes of Health grants HL 079442, GM42336 and DK785483 (to W.E.B.), a fellowships from the Canadian Institutes for Health Research (CIHR) (to D.M.H.), the National Institutes of Health Molecular Library Screening Center Network MH084512 (to F.M., S.S. and P.H.) and National Institutes of Health grants GM038109, GM081192, AG026647, and NS047331 (to R.I.M.).
Footnotes
Author Contributions: Designed the research plan: B.C. and R.I.M. Designed and performed the research: B.C., M.C.S., F.M., M.A.C., D.M.H., S.K. and S.A.S. Analyzed and interpreted the data: B.C., M.C.S., F.M., D.M.H., S.A.S., P.H., B.D.T., D.G., W.E.B. and R.I.M. Wrote the manuscript: B.C. and R.I.M. All authors reviewed the manuscript.
Competing Financial Interest Statement: B.D.T., D.G., and S.K. are employees of Proteostasis Therapeutics Inc. (Cambridge, MA) that is developing small molecule therapeutics for protein misfolding diseases. W.E.B. is a co-founder, shareholder and a paid consultant for Proteostasis Therapeutics Inc. R.I.M. is founder, shareholder, and paid consultant for Proteostasis Therapeutics Inc.
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–38. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 5.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–55. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 6.Lichtlen P, Schaffner W. Putting its fingers on stressful situations: the heavy metal-regulatory transcription factor MTF-1. Bioessays. 2001;23:1010–7. doi: 10.1002/bies.1146. [DOI] [PubMed] [Google Scholar]
- 7.Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–55. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–91. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 10.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–4. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 11.Gidalevitz T, Kikis EA, Morimoto RI. A cellular perspective on conformational disease: the role of genetic background and proteostasis networks. Curr Opin Struct Biol. 2010;20:23–32. doi: 10.1016/j.sbi.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sittler A, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet. 2001;10:1307–15. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 13.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto M, et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem. 2005;280:34908–16. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- 15.Vacher C, Garcia-Oroz L, Rubinsztein DC. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum Mol Genet. 2005;14:3425–33. doi: 10.1093/hmg/ddi372. [DOI] [PubMed] [Google Scholar]
- 16.Waza M, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–95. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med. 2007;85:1421–8. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujikake N, et al. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–97. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–5. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 20.Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- 22.Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol. 2008;8:370–4. doi: 10.1016/j.coph.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solit DB, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–7. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westerheide SD, et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–60. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 26.Williams GT, Morimoto RI. Maximal stress-induced transcription from the human HSP70 promoter requires interactions with the basal promoter elements independent of rotational alignment. Mol Cell Biol. 1990;10:3125–36. doi: 10.1128/mcb.10.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butina D. Unsupervised data base clustering based on Daylight's fingerprint and Tanimoto similarity: a fast and automated way to cluster small and large data sets. Journal of Chemical Information and Computer Sciences. 1999;39:747–750. [Google Scholar]
- 28.Amici C, Sistonen L, Santoro MG, Morimoto RI. Antiproliferative prostaglandins activate heat shock transcription factor. Proc Natl Acad Sci U S A. 1992;89:6227–31. doi: 10.1073/pnas.89.14.6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trott A, et al. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol Biol Cell. 2008;19:1104–12. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, et al. Pharmacologic induction of heme oxygenase-1. Antioxid Redox Signal. 2007;9:2227–39. doi: 10.1089/ars.2007.1783. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Chang A. Heat shock response relieves ER stress. EMBO J. 2008;27:1049–59. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyttenbach A, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet. 2001;10:1829–45. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- 33.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–26. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 34.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–22. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nollen EA, et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–8. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia SM, Casanueva MO, Silva MC, Amaral MD, Morimoto RI. Neuronal signaling modulates protein homeostasis in Caenorhabditis elegans post-synaptic muscle cells. Genes Dev. 2007;21:3006–16. doi: 10.1101/gad.1575307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 38.Stringer JR, Bowman MD, Weisblum B, Blackwell HE. Improved Small-Molecule Macroarray Platform for the Rapid Synthesis and Discovery of Antibacterial Chalcones. ACS Comb Sci. 2011 doi: 10.1021/co100053p. [DOI] [PubMed] [Google Scholar]
- 39.Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int Immunopharmacol. 2010 doi: 10.1016/j.intimp.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci U S A. 2001;98:3404–9. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dinkova-Kostova AT, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tangallapally RP, et al. Synthesis and evaluation of nitrofuranylamides as novel antituberculosis agents. J Med Chem. 2004;47:5276–83. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 43.Roesslein M, et al. Thiopental protects human T lymphocytes from apoptosis in vitro via the expression of heat shock protein 70. J Pharmacol Exp Ther. 2008;325:217–25. doi: 10.1124/jpet.107.133108. [DOI] [PubMed] [Google Scholar]
- 44.Yang H, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–85. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 45.Yang F, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 46.Teiten MH, Reuter S, Schmucker S, Dicato M, Diederich M. Induction of heat shock response by curcumin in human leukemia cells. Cancer Lett. 2009;279:145–54. doi: 10.1016/j.canlet.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 47.Mu TW, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–81. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–79. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorbatyuk MS, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–6. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tam LC, et al. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90) Hum Mol Genet. 2010;19:4421–36. doi: 10.1093/hmg/ddq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.