

Summary
This study was performed to elucidate the host cell scaffolding and signaling molecules that Campylobacter jejuni utilizes to invade epithelial cells. We hypothesized that the C. jejuni fibronectin-binding proteins and secreted proteins are required for cell signaling and maximal invasion of host cells. C. jejuni binding to host cells via the CadF and FlpA Fibronectin-binding proteins activated the epidermal growth factor (EGF) pathway, as evidenced by inhibitor studies and immunoprecipitation coupled with immunoblot analysis using antibodies reactive against total and active EGF receptor. Inhibitor studies revealed maximal C. jejuni host cell invasion was dependent upon PI3-Kinase, c-Src, and focal adhesion kinase (FAK), all of which are known to participate in cytoskeletal rearrangements. Knockdown of endogenous Dock180, which is a Rac1-specific guanine nucleotide exchange factor, using siRNA revealed that C. jejuni invasion was significantly reduced compared with cells treated with scrambled siRNA. We further demonstrated that the C. jejuni Cia proteins are, in part, responsible for Rho GTPase Rac1 recruitment and activation, as judged by immunofluorescence microscopy and Rac1 activation. Based on these data, we present a model that illustrates that C. jejuni utilizes a coordinated mechanism involving both adhesins and secreted proteins to promote membrane ruffling and host cell invasion.
Keywords: Bacteria-host cell interactions, cell invasion, cell signaling, integrin, epidermal growth factor, Dock180-ELMO, Rac1, membrane ruffling
Introduction
Campylobacter species are the most common culture-proven cause of bacterial gastroenteritis worldwide, accounting for 400 – 500 million cases of diarrhea each year (Ruiz-Palacios, 2007). Acute campylobacteriosis, which is characterized by fever, severe abdominal cramps, and diarrhea containing blood and leukocytes, is associated with C. jejuni invasion of intestinal cells. Maximal binding of C. jejuni to host cells is dependent on synthesis of two fibronectin (Fn) binding proteins termed CadF and FlpA (Konkel et al., 1997; Monteville et al., 2003; Krause-Gruszczynska et al., 2007; Flanagan et al., 2009; Konkel et al., 2010). In addition, maximal cell invasion requires the secretion of virulence proteins, termed the Campylobacter invasion antigens (Cia), from the bacterium’s flagellar Type III Secretion System (T3SS) (Konkel et al., 1999; Konkel et al., 2004; Malik-Kale et al., 2008; Christensen et al., 2009). The Cia proteins have been proposed to modify host cell regulatory pathways to promote C. jejuni-host cell invasion and intracellular survival.
Integrins are transmembrane glycoprotein receptors composed of heterodimeric α and β subunits that are noncovalently associated with one another (1:1) (Hynes, 1992; Vuori, 1998; Danen et al., 2001; van der Flier et al., 2001). Different α and β subunit combinations dictate the specificity of cell-to-cell and cell-to-extracellular matrix (ECM) component recognition. In addition to binding ECM components, activation of the integrins can trigger host cell signaling pathways. For example, cell-associated Fn binding to α5β1 integrins results in receptor clustering, causing the recruitment of structural and signaling proteins to the cytoplasmic tail of the integrin receptors. Indeed, integrin receptor activation results in the recruitment and activation of Focal Adhesion Kinase (FAK), which initiates a cascade of host signal transduction events that results in actin cytoskeletal rearrangement (Tachibana et al., 1995; Miyamoto et al., 1998).
Focal complexes (FCs) are dynamic protein complexes that connect ECM components, including Fn, to the actin cytoskeleton, thus anchoring the cell to the underlying surface. FCs are comprised of numerous components, including the integrin receptors, FAK, paxillin, and actin. Previous work has revealed that maximal invasion of host cells by C. jejuni is dependent upon at least one component of the FC. For example, C. jejuni infection of epithelial cells results in the transient phosphorylation of paxillin, and the timing of paxillin phosphorylation coincides with a sharp increase in bacterial invasion (Monteville et al., 2003). Interestingly, C. jejuni-induced paxillin phosphorylation is dependent on the C. jejuni Fn-binding protein CadF. C. jejuni internalization also coincides with the activation of the Rho GTPases Rac1 and Cdc42, and dominant negative forms of Rac1 and Cdc42 significantly reduce C. jejuni invasion. As with paxillin, the activation of Rac1 and Cdc42 is dependent on the CadF protein (Monteville et al., 2003; Krause-Gruszczynska et al., 2007). Together these findings indicate C. jejuni adherence to Fn initiates cell signaling and scaffolding proteins, ultimately resulting in host cell invasion. We have defined the cellular components that participate in host cell invasion as the C. jejuni invasion complex (CIC).
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase from the Erb-B receptor kinase family (Citri et al., 2006). EGFR can be stimulated by the binding of extracellular ligand or, in the absence of an extracellular ligand, via integrin signaling. In the case of integrin-dependent EGFR-phosphorylation, activation is dependent upon a multimeric complex of integrins, c-Src, p130Cas, and the EGFR (Cabodi et al., 2004). Once stimulated, EGFR autophosphorylates multiple tyrosine residues on its cytoplasmic C-terminal domain and serves as a scaffold, facilitating the activation of a signaling cascade. The EGF pathway influences mitogenic gene expression and alters components of the cytoskeleton involved with actin organization, focal adhesion formation and resolution, as well as cell-cell adhesion (Thelemann et al., 2005).
The goal of this study was to identify the host cell signaling pathways that C. jejuni exploits for host cell invasion. We present a model of C. jejuni cell invasion whereby the bacterium’s binding to Fn sets the stage for the Cia secreted proteins to activate the Rho GTPase Rac1, resulting in host cell membrane ruffling and bacterial uptake. Evident from our study is that C. jejuni have devised a unique strategy to invade epithelial cells.
Results
C. jejuni invasion triggers the activation of host cell signaling pathways
We determined the number of C. jejuni internalized by INT 407 cells over a 3 hr time course using the gentamicin-protection assay. We included the C. jejuni ciaC mutant and C. jejuni wild-type strain treated with chloramphenicol in this assay as controls. The reason that the C. jejuni ciaC mutant was incorporated is because: 1) this mutant is reduced in host cell invasion compared to a wild-type strain; 2) the invasive phenotype displayed by this mutant is comparable to a Cia-secretion deficient mutant; 3) the invasiveness of this mutant should be similar to the C. jejuni wild-type strain treated with chloramphenicol, as chloramphenicol treatment retards the synthesis of the Cia proteins. The C. jejuni ciaC mutant is deficient in the secretion of one protein with a Mr of 12,164 Da (Christensen et al., 2009). Consistent with previous work, a significant reduction in the number of internalized bacteria was observed for the C. jejuni ciaC mutant when compared to the wild-type strain. Regarding the kinetics of C. jejuni host cell invasion, inoculation of the INT 407 cells with the C. jejuni wild-type strain resulted in a sharp increase in the number of internalized bacteria after a 60 min incubation period (Supplemental Fig. S1). Thereafter, the number of internalized C. jejuni steadily increased over the course of the 3 hr assay. In contrast, treatment of the C. jejuni wild-type strain with chloramphenicol, which is a selective inhibitor of bacterial protein synthesis, retarded the sharp increase in cell invasion.
We assessed the activation of INT 407 cell signaling pathways in response to C. jejuni using the PT-66 α-phosphotyrosine antibody (Fig. 1). INT 407 cells were treated with 100 ng/ml of EGF for a positive control, whereas uninoculated and untreated INT 407 cells were used for a negative control. In contrast to control INT 407 cells, tyrosine phosphorylated proteins were observed in the lysates prepared from the C. jejuni-infected INT 407 cells over the course of the infection assay (i.e., 15 to 90 min). The intensity of the phospho-reactive bands was greatest at 30 and 45 min post-infection, which preceded the sharp increase in the number of internalized bacteria observed at the 60 min time point (see Fig. S1). Genistein, an inhibitor of protein tyrosine kinases, significantly inhibited the uptake of C. jejuni by INT 407 cells (Supplemental Fig. S2). Consistent with published data, genistein-treatment of cells did not alter the invasiveness of Salmonella typhimurium (Wooldridge et al., 1996). Collectively, these results indicate that C. jejuni invasion of INT 407 cells is dependent upon the phosphorylation of tyrosine residues of cellular proteins.
Fig. 1.
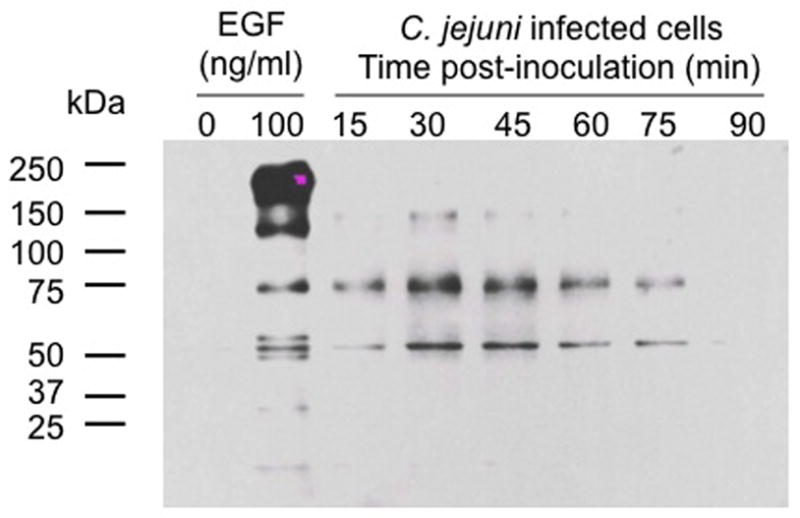
Infection of INT 407 cells with C. jejuni results in an increase in host cell tyrosine phosphorylated proteins. INT 407 cells were infected with C. jejuni for 15 to 90 min. Triton X-100 soluble host cell proteins were extracted, and subjected to SDS-PAGE coupled with immunoblot analysis using the PT-66 α-phosphotyrosine antibody. INT 407 cells treated with 100 ng/ml of EGF served as the positive control, whereas uninoculated INT 407 cells were used as a negative control. Equal amounts of total protein were added to each well of the gel.
C. jejuni binding to fibronectin sets the stage for cell invasion
Immunoblot analysis with the PT-66 α-phosphotyrosine antibody revealed a few bands in the C. jejuni-infected INT 407 cell lysates that had similar molecular masses to bands observed in the lysates of EGF-treated INT 407 cells (~50–75 kDa range). Based on this observation, we tested if C. jejuni invasion to host cells requires the activation of the EGF receptor (EGFR). Both PD168393 and erlotinib, which are specific inhibitors of EGF receptor tyrosine phosphorylation, significantly inhibited C. jejuni cell invasion in a dose-dependent decrease, regardless of the MOI used in an individual experiment (Supplemental Fig. S3, and data not shown). Neither PD168393 nor erlotinib affected the viability of the INT 407 cells or the bacteria (data not shown). We also found that the maximum volume of vehicle (i.e., methanol and dimethyl sulfoxide) used to administer the cellular inhibitors throughout this study had no effect on C. jejuni internalization (Supplemental Fig. S4). To determine if the EGFR is phosphorylated in response to C. jejuni infection, INT 407 cells were inoculated with a C. jejuni wild-type strain and epithelial cell lysates were prepared at different time points following infection. Total EGFR and the amount of phosphorylated EGFR were determined by immunoprecipitation coupled with immunoblotting using specific antibodies. To examine if EGFRs are phosphorylated in response to an integrin-dependent activation signal, we specifically examined whether tyrosine residues 845 and 1068 on the EGFR were phosphorylated. The greatest increase in phosphorylated EGFR was observed in C. jejuni-infected INT 407 cells at 45 min post-inoculation compared to uninoculated cells (Supplemental Fig. S5). This time point correlated with the greatest intensity of the immunoreactive bands observed from C. jejuni-infected INT 407 cells with the PT-66 α-phosphotyrosine antibody (see Fig. 1). In contrast, the C. jejuni cadF flpA mutant did not alter the level of phosphorylated EGFR (Supplemental Fig. S5). A potential caveat of this finding is that the binding of the C. jejuni cadF flpA mutant to INT 407 cells is reduced 42% (P < 0.01) compared to the wild-type strain (data not shown). Taken together, these data indicate that C. jejuni binding to fibronectin (Fn) via the CadF and FlpA proteins sets the stage for phosphorylation of the EGFR via integrin receptor activation.
Host cell proteins are required for efficient C. jejuni invasion
Integrin activation is known to recruit FAK and other cell signaling and scaffolding proteins to focal complexes. Given that C. jejuni causes integrin activation, as evident from the phosphorylation of the EGFR, experiments were performed to examine the participation of other host cell proteins, including FAK, in C. jejuni invasion. Wortmannin and PP2 inhibit PI3-Kinase and c-Src, respectively (Bain et al., 2007), whereas TAE226 is a relatively new inhibitor that specifically suppresses ECM-dependent phosphorylation of FAK at Tyr397 and Tyr861. Incubation of the INT 407 cells and bacteria with wortmannin, PP2, or the TAE226 had no effect on cell viability (data not shown). While treatment of the INT 407 cells with wortmannin, PP2, or TAE226 had no affect on C. jejuni binding to the INT 407 cells (data not shown), each drug significantly inhibited C. jejuni cell invasion in a dose-dependent manner (Fig. 2). Taken together, these results suggest that C. jejuni utilizes components of the focal complex in order to invade epithelial cells.
Fig. 2.
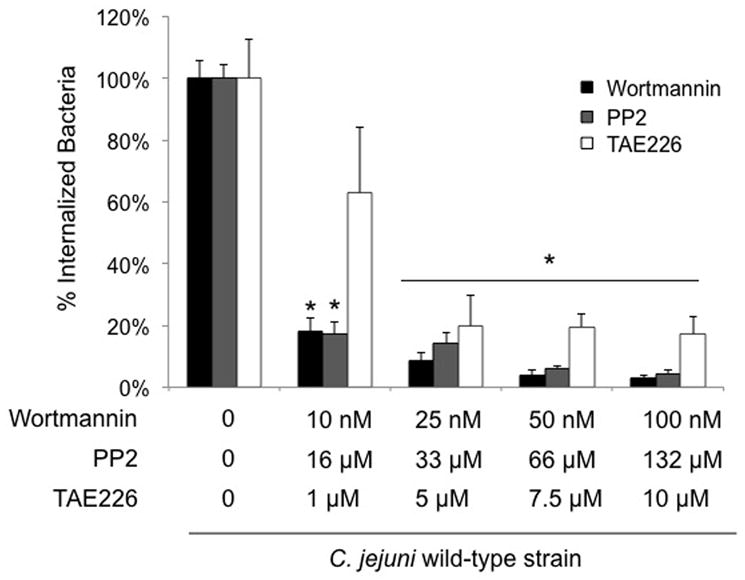
Effect of host cell PI3-Kinase (wortmannin), c-Src (PP2), and FAK (TAE226) inhibitors on C. jejuni invasion. Each inhibitor was tested on a different day. The invasion assay was performed as indicated in the “Experimental Procedures,” whereby the bacteria were incubated with the INT 407 cells for 3 hr prior to gentamicin treatment. Values represent percent of internalized (gentamicin-protected) bacteria ± standard deviation compared to untreated cells. The asterisks indicate significance between untreated and treated samples (P < 0.01), as determined using Student’s t test.
Dock180 contributes to C. jejuni invasion of INT 407 cells
Recruitment and activation of FAK can initiate host signal transduction events leading to the activation of Dock180 (Dedicator of cytokinesis). Dock180 and its binding partner ELMO (Engulfment and Cell Motility) form a bipartite guanine nucleotide exchange factor (GEF), which activates Rac1, leading to lamellipodial extensions. The participation of Dock180 in C. jejuni invasion was assessed using small interfering RNA (siRNA) coupled with the gentamicin protection assay. While the American Type Culture Collection notes the INT 407 cell line was derived via HeLa contamination, we chose to use HeLa cells for this assay because we were able to obtain greater transfection efficiencies with HeLa cells versus INT 407 cells. Consistent with previous work (Konkel et al., 1992; Buelow et al., 2011), C. jejuni were able to adhere to and invade HeLa and INT 407 cells with equal efficiency (not shown). Here we found that treatment of cells with Dock180 siRNA significantly reduced C. jejuni invasion of HeLa cells when compared to cells treated with scrambled siRNA (Fig. 3A). Because Dock180 is a Rac1-specific guanine nucleotide exchange factor, we also examined the amount of activated Rac1 in Dock180 siRNA treated cells by G-LISA™. We found that treatment of cells with Dock180 siRNA decreased the level of C. jejuni induced Rac1 activity to that of uninfected cells (Fig. 3B). The knockdown of Dock180 in siRNA treated cells was determined to be 61% compared to the non-siRNA treated cells as judged by immunoblot analysis coupled with densitometry using a Dock180-specific antibody (Fig. 3C).
Fig. 3.

Dock180 participates in C. jejuni invasion of epithelial cells. HeLa cells were transfected with Dock180 or scrambled (Scr) siRNA. Panels: A) Invasion of the C. jejuni wild-type strain in untreated, Dock180 siRNA-treated, and Scr siRNA-treated cells. Also shown is the number of bacteria internalized for the C. jejuni ciaC mutant. The values represent the mean number of internalized bacteria ± standard deviation. B) Rac1 activation in host cells infected with C. jejuni wild-type strain. Whole cell lysates were processed after 15 minutes of incubation and analyzed for activated Rac1 by G-LISA™. The mean ± standard deviation of total active Rac1 is indicated in Relative Optical Density. The data shown represent at least 10 samples of each condition analyzed in duplicate. The asterisks indicate significance (P < 0.01) in the invasiveness or Rac1 activation of the C. jejuni wild-type strain in untreated versus Dock180-treated cells, as determined using the Student’s t test. C) Whole cell lysates of untreated, Dock180 siRNA-treated, and Scr siRNA-treated INT 407 cells (from left to right) analyzed by immunoblotting with an α-Dock180 antibody.
The C. jejuni CiaC protein potentiates Rac1 recruitment and activity
Based on the fact that the C. jejuni ciaC mutant is significantly less invasive than the cadF flpA double mutant, we formulated the following hypotheses: a) binding of C. jejuni to cell-associated Fn via the CadF and FlpA proteins facilitates the recruitment of host cell signaling and scaffolding proteins to the sites of bacterial attachment; b) integrin activation sets the stage for invasion, but is not required for bacterial uptake; c) the CadF and FlpA proteins participate in efficient delivery of the Cia proteins to host cells; and d) the Cia secreted proteins (e.g., CiaC) play an important role in cell invasion. Because Dock180-ELMO is a Rac1 specific GEF, we specifically examined Rac1 activity in cells inoculated with the C. jejuni wild-type strain and ciaC mutant. We initially performed immunofluorescence microscopy assays to determine if Rac1 is recruited to the sites where C. jejuni is bound to the host cell and performed Rac1 activation assays. Consistent with previous work (Dise et al., 2008), EGF treatment of serum-starved cells promoted Rac1 movement from the cytosol to membrane ruffles whereas Rac1 was dispersed in uninoculated INT 407 cells (Fig. 4). Regarding C. jejuni-infected cells, we observed discrete spots of Rac1 at the sites where C. jejuni were bound (~ 33% co-localization). In contrast, fewer sites of co-localization of Rac1 were observed in INT 407 cells inoculated with the C. jejuni ciaC mutant (~ 13% co-localization)(Fig. 4 and Supplemental Fig. S6). These data indicate that Rac1 is recruited to sites of C. jejuni attachment, and that CiaC is, in part, responsible for Rac1 recruitment.
Fig. 4.

Rac1 is recruited to sites of C. jejuni attachment. The localization of Rac1 in C. jejuni-infected cells was examined by immunofluorescence microscopy as outlined in “Experimental Procedures.” EGF-treated cells served as a positive control and uninoculated (non-EGF treated) INT 407 cells were used as a negative control. EGF treatment of INT 407 cells resulted in Rac1 enriched sites, whereas Rac1 was dispersed in uninoculated INT 407 cells. The cell-associated bacteria are indicated by the circles, whereas the C. jejuni adjacent to sites of accumulated Rac1 are indicated by the circles with the arrowhead. Approximately 33% of the C. jejuni wild-type bacteria were co-localized with Rac1, whereas only 13% of the C. jejuni ciaC mutant bacteria were co-localized with Rac1.
To quantitate the level of Rac1 activation in C. jejuni infected cells, INT 407 cells were inoculated with C. jejuni wild-type strain and the amount of activated Rac1 was determined over a 60 minute time period by G-LISA™. We determined that the greatest level of Rac1 activation occurred 15 minutes following infection (data not shown). We then infected cells with C. jejuni wild type strain, the cadF flpA mutant, or ciaC mutant and determined the relative levels of Rac1 activation 15 minutes following infection. The amount of active (GTP associated) Rac1 significantly increased in INT 407 cells inoculated with the C. jejuni wild-type strain versus uninoculated cells (Fig. 5B). In contrast, inoculation of the INT 407 cells with the C. jejuni cadF flpA mutant resulted in a decrease in active Rac1 and the ciaC mutant resulted in a further decrease in the level of Rac1 activation (Fig. 5A). The relative level of Rac1 activation in both mutant strains correlated strongly with their respective invasion phenotypes (Fig. 5B).
Fig. 5.

Rac1 is activated in INT 407 cells in response to C. jejuni infection. INT 407 cells were inoculated with the C. jejuni wild-type strain, cadF flpA mutant, and ciaC mutant. A) Whole cell lysates were processed after 15 minutes of incubation and analyzed for activated Rac1 by G-LISA™. The mean ± standard deviation of total active Rac1 is indicated in Relative Optical Density. The data shown represent at least 10 samples of each condition analyzed in duplicate. B) C. jejuni invasion of host cells as determined by gentamicin protection assay. Values represent mean ± standard deviation of internalized bacteria/well of a 24-well tissue culture tray. The asterisks indicate a significant difference (P < 0.01) in internalization as compared to the wild-type strain. The asterisks indicate a significance difference (*P < 0.01, **P < 0.05) in Rac1 activity for the wild-type strain versus uninoculated cells and cells inoculated with the C. jejuni ciaC and cadF flpA mutants.
The activation of Rac1 has been reported to result in membrane ruffles (Ridley et al., 1992; Hall, 1998). Therefore, we inoculated INT 407 cells with the C. jejuni wild-type strain and examined the cells by SEM for membrane ruffling. We observed that 74.8% (n = 128 of 171) of the cells infected with the C. jejuni wild-type strain showed intense membrane ruffling (Fig. 6). In contrast, 23.2% (n = 33 of 142) of the cells infected with the C. jejuni ciaC mutant showed membrane ruffling. The difference observed in membrane ruffling of the wild-type strain versus the ciaC mutant was significant (P < 0.01). These findings supported the immunofluorescence microscopy results, which demonstrated that Rac1 was recruited to the sites where the C. jejuni wild-type bacteria were bound more frequently than the ciaC mutant. Additional experiments were performed to examine the effect of cellular inhibitors of C. jejuni invasion on Rac1 activation. Cells that were pretreated with erlotinib, TAE 226, wortmannin, and PP2 prior to C. jejuni inoculation resulted in a significant decrease in Rac1 activation as shown by GLISA™ (Fig. S7). A decrease in membrane ruffling was also observed with the cells treated with the same cellular inhibitors as judged by SEM examination (Fig. S8). Collectively, these data suggest that CiaC contributes to Rac1 membrane recruitment and activation, and that maximal C. jejuni invasion of host cells is dependent Rac1 activity that is induced, in part, by EGFR, FAK, PI3 kinase, and Src activation.
Fig. 6.

Inoculation of INT 407 cells with a C. jejuni wild-type strain induces membrane ruffling. Scanning electron microscopy of INT 407 cells infected with C. jejuni. A) Cell infected with a ciaC mutant; B) Cell infected with a C. jejuni wild-type strain; C) Increased magnification of the image (inset box) shown in Panel A; and D) Increased magnification of the image (inset box) shown in Panel B.
Stimulation of the EGF signaling pathway rescues a C. jejuni ciaC mutant
We noted earlier that C. jejuni binding to host cells could stimulate the EGF pathway, although not to the same extent as observed upon the addition of exogenous EGF (see Fig. 1). Given that over stimulation of the EGF pathway can activate Rac1 (Dise et al., 2008) (Supplemental Fig. S9), we hypothesized that activation of the EGF signaling pathway by the addition of exogenous EGF might restore the invasiveness of a C. jejuni ciaC mutant. Thus, INT 407 cells were pre-treated with varying concentrations of EGF for 30 min, and then inoculated with the C. jejuni wild-type strain and C. jejuni ciaC mutant (Fig. 7). In the absence of EGF-treatment, a significant reduction was observed in the number of bacteria internalized for the C. jejuni ciaC mutant when compared with the C. jejuni wild-type strain. However, treatment of the INT 407 cells with 50 ng/ml of EGF resulted in a significant increase in the number of bacteria internalized for the C. jejuni ciaC mutant. Moreover, the invasiveness of the C. jejuni ciaC mutant improved as the concentration of EGF was increased, demonstrating the rescue effect was dose-dependent. These data demonstrate that ‘hyper-stimulation’ of the EGF pathway, which induces Rac1 activation (Dise et al., 2008), is sufficient to promote the uptake of a non-invasive C. jejuni strain.
Fig. 7.

Stimulation of the EGF signaling pathway rescues the invasiveness of a C. jejuni ciaC mutant. Panels: A) INT 407 cells were incubated with the concentrations of EGF indicated for 30 minutes prior to inoculation with either the C. jejuni wild-type strain or the ciaC mutant. Values represent mean ± standard deviation of internalized bacteria/well of a 24-well tissue culture tray. The asterisks (* P < 0.05, ** P < 0.01) indicate a significant increase in the number of internalized bacteria for cells treated with EGF and inoculated with the ciaC mutant versus untreated cells inoculated with the ciaC mutant. B) Immunoblot probed with the PT-66 α-phosphotyrosine antibody. INT 407 cells were treated with EGF and cell lysates prepared after a 30 min incubation period. The blot was exposed for a short period of time in order to show that the EGFR (Mr = 180 kDa) is activated in a dose-dependent manner in response to increasing concentrations of EGF.
Discussion
The goal of this study was to dissect the mechanism that C. jejuni utilizes for host cell invasion and to identify components of the C. jejuni invasion complex (CIC). Based on the present data, combined with information available in the published literature, we have generated a model whereby maximal invasion of host cells by C. jejuni involves activation of α5β1 integrin receptors. Integrin activation results in the recruitment of additional cellular proteins and activates the EGF signaling pathway. The data indicate that FAK, paxillin, c-Src, PI3-Kinase, Dock180, and Rac1 are key components of the CIC (Fig. 8). Our model supports the proposal that the CadF and FlpA adhesins on the surface of C. jejuni can bind to Fn and result in the activation of α5β1 integrin receptors. Upon integrin activation, the amino terminal tail of FAK can bind to the cytoplasmic tail of the β1 integrin and become activated (Cary et al., 1999; Schlaepfer et al., 1999). Once FAK is activated via autophosphorylation of Tyr397, it becomes associated with the SH2 domain of c-Src (Cary et al., 1999). c-Src phosphorylates other residues of FAK as well as paxillin and p130Cas. The adaptor protein p130Cas recruits CrkII, which binds to Dock180. Dock180 and ELMO act as a bipartite GEF that activates the Rho family monomeric G protein Rac1, ultimately leading to a local restructuring of the actin cytoskeleton (Cote et al., 2007). Our data further support the proposal that the secondary messenger molecule PI3-Kinase (PI3K), which converts PIP2 to PIP3 (Mitra et al., 2005), is involved in the recruitment of proteins to the CIC. Dock180 binds to PIP3, and PIP3 is important for the positioning and the activation of Rac1 at the appropriate sites (Cote et al., 2007). The regulatory p85 subunit of PI3K also binds to FAK at Tyr397. Collectively, our data support a model whereby integrin clustering, which occurs via the tails of the clustered beta-1 subunits, results in the activation of the EGFR. Activation of the EGFR and EGF pathway would, in turn, potentiate the recruitment of the cellular components that comprise the CIC. Recruitment and activation of the CIC ultimately results in C. jejuni uptake where membrane ruffling is triggered in response to Rac1 activation.
Fig. 8.

Model of the host cell EGF signaling pathway and proteins downstream of the EGF receptor that are activated in response to C. jejuni invasion. Indicated in blue are components of the focal complex, (FC) and indicated in orange are other components that likely participate in C. jejuni internalization. C. jejuni binding to Fn results in integrin occupancy and clustering, which in turn, leads to integrin activation. The activation of EGFR and FAK then occurs through from the association with the tails of clustered β1 integrins. Activated FAK autophosphorylates Tyr397, resulting in association of c-Src. c-Src phosphorylates additional residues within FAK as well as other members of the FC, including paxillin and p130Cas. This activation results in the recruitment of CrkII and Dock180, the latter of which associates with ELMO to expose its catalytic domain. The Dock180/ELMO complex activates the Rho family monomeric G protein Rac1, leading to a local restructuring of the actin cytoskeleton.
C. jejuni activates the EGF receptor via crosstalk with integrin receptors
An important aspect of our model is that it is supported by cell biology studies demonstrating that integrin-mediated cell adhesion can induce EGF receptor activation (Moro et al., 2002). We found that PD168393 and erlotinib, which are specific inhibitors of EGF receptor tyrosine kinase activity, significantly inhibited C. jejuni cell invasion in a dose-dependent fashion. Cabodi et al. (Cabodi et al., 2004) reported that phosphorylation of the EGFR can occur in the absence of EGFR ligands. In this instance, the integrins activate c-Src kinase, which in turn results in the phosphorylation of specific residues on the EGFR; this event requires a multimeric complex comprised of the EGFR, integrins, c-Src, and the p130Cas adaptor protein. The tyrosine residues phosphorylated on EGFR in response to integrin-dependent EGFR activation include Tyr-845, Tyr-1068, Tyr-1086, and Tyr-1173. We demonstrate that the EGFR was phosphorylated in C. jejuni inoculated cells using α-EGFR pTyr845 and pTyr1068 specific antibodies, and that this event was dependent on the CadF and FlpA Fn-binding proteins. We also found that PP2, a specific inhibitor of c-Src, significantly inhibited C. jejuni cell invasion in a dose-dependent manner. As mentioned above, c-Src kinase can phosphorylate the EGFR (Cabodi et al., 2004). Finally, others have reported that filipin III treatment of cells results in a reduction in C. jejuni invasion (Wooldridge et al., 1996). Filipin III preferentially sequesters cholesterol from the plasma membrane, resulting in lipid raft disassembly and unclustering of membrane-localized receptors (Rothberg et al., 1990; Schnitzer et al., 1994). Relevant to our model is that the tyrosine phosphorylated species of caveolin-1 is associated with β1 integrin, FAK, and phospho-paxillin (Beardsley et al., 2005), and that crosstalk of the integrins with EGF receptors occurs at caveolae. Caveolin-1 has also been demonstrated to be an important regulator of Fn turnover (endocytosis and degradation) (Sottile et al., 2005). These observations are consistent with the proposal that phosphorylation of the EGFR occurs from crosstalk of the activated and clustered β1 integrin (Cabodi et al., 2004).
FAK, c-Src, paxillin, and PI3-Kinase are components of the C. jejuni invasion complex
Cell biology studies have clearly demonstrated that FAK, c-Src, paxillin, and PI3-Kinase interact (Eide et al., 1995; Schaller et al., 1995; Chen et al., 1996; Zamir et al., 2001; Mitra et al., 2005; Gilcrease, 2007). We show that inhibitors directed against FAK, c-Src, and PI3-Kinase all significantly reduced C. jejuni invasion. Although not addressed herein, paxillin is also involved in C. jejuni invasion. Paxillin is a multi-domain adaptor protein that associates with the tails of β1 integrins and provides a platform for the integration and processing of ECM-initiated signals. Paxillin is critical for the targeting of FAK and c-Src to newly formed focal complexes (Zamir et al., 2001; Gilcrease, 2007). We have previously shown that paxillin is phosphorylated upon C. jejuni infection of epithelial cells. Moreover, the phosphorylation of paxillin upon C. jejuni infection of cells occurs immediately prior to a sharp increase in bacterial cell invasion, and occurs in a CadF-dependent manner (Monteville et al., 2003). The fact that paxillin is phosphorylated in a CadF-dependent manner supports the proposal that the integrin receptors are activated in response to C. jejuni binding to cell-associated Fn.
Rearrangement of the actin cytoskeleton involves Dock180 and Rac1
Activated Rac induces actin polymerization and integrin adhesion complex assembly at the cell periphery, leading to membrane protrusions termed lamellipodia. Activation of Rac in cells is triggered by its interaction with one of multiple Rac-specific guanine nucleotide exchange factors (Rac-GEFs) that convert GDP-Rac into GTP-Rac (Buchsbaum et al., 2002). One Rac-specific GEF is Dock180, which when associated with its binding partner ELMO, promotes nucleotide exchange on Rac. We observed a statistically significant reduction in invasion of the C. jejuni wild-type strain for cells treated with Dock180 siRNA when compared to cells treated with the scrambled control siRNA. Although we expected that the reduction in invasion of the C. jejuni wild-type strain for the cells treated with the Dock180 siRNA should have been greater, treatment of the cells with the Dock180 siRNA only resulted in partial knockdown in Dock180 protein compared to non-siRNA treated cells. Nevertheless, the number of bacteria internalized in cells treated with the Dock180 siRNA and inoculated with the C. jejuni wild-type strain was comparable to that observed with the C. jejuni ciaC mutant used in the assay. Also of note is that the difference in the invasiveness of the C. jejuni ciaC mutant, which was used as a control in this experiment, versus the wild-type strain was not as much as typically observed (see Supplemental Fig. S1). One possible explanation for this result was that it was necessary to use HeLa cells after 48 h of growth, rather than 24 h, for the adherence and internalization assays to allow for the siRNA treatment to knockdown the expression of Dock180. We found that the amount of active (GTP associated) Rac1 was significantly greater in INT 407 cells inoculated with the C. jejuni wild-type strain versus uninoculated cells (Figure 5). Also relevant is that the 553508 InSolution™ Rac1 Inhibitor (EMD, Gibbstown), which specifically interferes with the Rac-specific GEFs Trio and Tiam1, did not alter the amount of Rac1 activated in cells inoculated with the C. jejuni wild-type strain as judged by G-LISA™ (data not shown). Unfortunately, an inhibitor of Dock180 is not currently available to researchers. Finally, we found that the activation of Rac1 was partially dependent on the C. jejuni secreted protein CiaC. Collectively, these data suggest that the Dock180-ELMO-Rac1 complex participates in actin cytoskeletal rearrangement and C. jejuni uptake. Nevertheless, it is possible that other GEFs contribute to the activation of Rac1 and C. jejuni internalization given that multiple cellular pathways are stimulated by this pathogen.
Role of microfilaments (actin polymerization) and microtubules in C. jejuni invasion of host cells
It was first reported that C. jejuni invade cells in a microtubule (MT)-dependent, microfilament (MF)-independent fashion in 1993 (Oelschlaeger et al., 1993; Hu et al., 1999; Kopecko et al., 2001). However, in a subsequent study, it was reported that internalization of C. jejuni was sensitive to both microfilament and microtubule depolymerizing agents (Monteville et al., 2003). Monteville et al. (Monteville et al., 2003) also performed experiments to determine the combined effects of drugs that target MFs (cytochalasin D) and MTs (nocodazole) on C. jejuni internalization, and found that the number of C. jejuni that invaded the epithelial cells was comparable to that obtained when cytochalasin D was used alone. The specific role of MTs or MTs and MFs in this process has not been elucidated. One possibility is that the MTs and MFs are acting cooperatively in recycling integrin receptors. More specifically, the α5β1 Fn receptor is constantly endocytosed and recycled to the plasma membrane to refresh the receptors that can bind ligand (Pellinen et al., 2006). This process of endocytosis occurs in an actin-dependent mechanism, after which the vesicles are transported along the microtubules to recycling endosomes (Balasubramanian et al., 2007). These data, coupled with the previously published data, support a model whereby the MTs and MFs act to regulate the caveolar membrane system, encompassing integrin activation and recycling, which in turn could affect C. jejuni cell invasion. Also of note is that membrane ruffles, which were observed by SEM examination on INT 407 cells in response to infection with C. jejuni, are actin-dependent (Raftopoulou and Hall, 2003). Our finding that membrane ruffling and Rho GTPases are involved in C. jejuni cell invasion is supported by previous work (Krause-Gruszczynska et al., 2007).
Concluding Statements
We present the most comprehensive model to date for C. jejuni stimulation of host cell signaling and cell invasion. Our data support the hypothesis that C. jejuni binding to Fn results in the activation of the integrin receptors, which ultimately results in the stimulation of signaling pathways and actin cytoskeletal rearrangement. Noteworthy is that in the absence of Cia protein secretion and delivery, C. jejuni invasion is reduced, indicating that the Cia proteins play a role in fully activating the cellular signaling pathways or in stabilizing components of the CIC. Evident from this study is that C. jejuni have devised a unique strategy of inducing membrane ruffling in order to invade epithelial cells. Studies are currently underway to further dissect the bacterial and host cell molecules involved in C. jejuni-host cell interactions.
Experimental Procedures
Bacterial strains and growth conditions
All of the bacteria used in this study (i.e., the C. jejuni F38011 wild-type clinical strain, C. jejuni cadF flpA mutant (Kanr Tetr), C. jejuni ciaC mutant (Tetr), Salmonella enterica serovar Typhimurium (S. typhimurium) SL1344, and Escherichia coli XL1-Blue MRF’ (Tetr)] were cultured as described elsewhere (Konkel et al., 1992).
Tissue Culture
INT 407 human intestinal epithelial cells (ATCC CCL6; American Type Culture Collection, Manassas, VA) and HeLa cells (ATCC CCL2; American Type Culture Collection, Manassas, VA) were cultured as described previously (Konkel et al., 1992).
Adherence and Internalization Assays
Adherence and internalization assays were performed as described earlier (Christensen et al., 2009). All assays were performed at a multiplicity of infection (MOI) ranging between 50 and 500, and repeated a minimum of 3 times to ensure reproducibility. The reported values represent the mean counts ± standard deviations derived from triplicate wells. To test our model of C. jejuni invasion, INT 407 cells were pre-incubated for 30 min in MEM-1% FBS with EGF (BD Biosciences, Bedford MA), genistein (Enzo Life Sciences, Farmingdale, NY), PD168393 (EMD Biosciences, San Diego, CA), erlotinib (Woburn, MA), wortmannin (Assay Designs, Ann Arbor, MI), PP2 (Sigma-Aldrich, St Louis, MO), or TAE226. The FAK inhibitor, TAE226, was generously provided by Novartis Pharma AG (Switzerland). Following the pre-incubation step, binding and internalization assays were performed. To assess INT 407 cell viability following inhibitor treatment, the cells were rinsed twice with PBS, stained with 0.5% trypan blue for 5 min, and visualized with an inverted microscope.
SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis
SDS-PAGE and immunoblot analysis were performed using standard protocols (Christensen et al., 2009). Epithelial cells were inoculated with C. jejuni grown for 18 h. At the time intervals indicated, the epithelial cells were lysed at 4°C in 1 ml of lysis buffer [10 mM Hepes, 10% glycerol, 50 mM sodium fluoride, 150 mM sodium chloride, 1% Triton X-100, 20 μg/ml DNAse (Sigma, D5025), Protease Inhibitor Cocktail (Sigma, P2714) and 1 mM sodium orthovanadate]. Supernatants were harvested by centrifugation at 15,000 × g at 4°C and analyzed by SDS-PAGE coupled with immunoblot analyses. The protein concentration of each supernatant was determined by the bicinchoninic acid (BCA, Pierce, Rockford, IL) protein assay and normalized prior to SDS-PAGE. The following antibodies were used for immunoblot analysis: α EGFR (Santa Cruz, sc-03), α Phospho-EGFR (Cell Signaling, pEGFR Tyr845 and Tyr1068), α Phospho-Tyrosine (Sigma, PT-66), α Rac1 (NewEast Biosciences, 21003) and α Dock180 (Santa Cruz, sc-6043). The secondary antibodies used in this study were: α rabbit IgG (Sigma, A6154), α goat IgG (Sigma, A5420), and α mouse IgG (Sigma, A4416). Blots were developed using a FujiFilm LAS-4000 and Western Lightning chemiluminescence (PerkinElmer, Boston, MA) according to the manufacturer’s directions. Densitometry was performed using Fujifilm Multi Gauge software. The intensity of each band was obtained by subtracting the average background from adjacent areas in each lane from the total level of the appropriate molecular weight. Each of these values was then normalized to the cells only negative control.
Contribution of EGFR, Dock180, and Rac1 in C. jejuni host cell invasion
To determine whether EGFR is activated in response to C. jejuni, lysates were generated from INT 407 cells infected with C. jejuni. The lysates were prepared in IP lysis buffer [25 mM Tris, 1 mM EDTA 50 mM sodium fluoride, 150mM sodium chloride, 5% glycerol, 1% Triton X-100, Protease Inhibitor Cocktail (Sigma, P2714) and 1mM sodium orthovanadate]. The cell lysates were centrifuged at 15,000 × g at 4°C, and supernatant collected. The supernatant was transferred to a microcentrifuge tube and 30 μl Protein A/G agarose beads (Santa Cruz sc-2003) and α EGFR (Santa Cruz, mAb 528 sc-120) antibodies were added. The mixtures were incubated overnight at 4°C with tumbling. Following incubation the samples were rinsed three times with wash buffer (20 mM Hepes, 150 mM sodium chloride, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 0.1% Triton X-100, and 10% glycerol) by centrifugation at 2000 × g for 5 min at 4°C. After the final rinse, the pellet was resuspended in 1 X sample loading buffer and analyzed by SDS-PAGE.
To determine if Dock180 participates in C. jejuni invasion, HeLa cells were transfected with siRNA using lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions. Dock180 siRNA (sc-35207) and scrambled control siRNA (sc-37007) were both acquired from Santa Cruz, and applied to the cells 24 hr before assay. A standard binding and adherence assay was then performed. Knockdown of endogenous proteins was confirmed by immunoblot with antibodies for specific host proteins.
To determine the role of Rac1 in C. jejuni invasion, two ml of a bacterial suspension was added to each well of a six-well tissue culture tray with a semi-confluent monolayer of INT 407 cells. The cultures were incubated at 37°C in a 5% CO2 incubator for 15 min, after which the samples were processed by adding cell lysis buffer. The amount of activated Rac1 in the cell lysates was determined with the G-LISA™ Rac1 Activation Assay (Cytoskeleton, Denver, CO) according to the manufacturer’s instructions. The amount of activated Rac1 was also determined from cells treated with 50 μM of 53508 InSolution™ Rac1 Inhibitor (EMD, Gibbstown) and inoculated with the C. jejuni wild-type strain.
Immunofluorescence microscopy
INT 407 cells were incubated with C. jejuni wild-type strain for 15 minutes at 37°C in a 5% CO2 incubator prior to fixing. C. jejuni were stained with a 1° goat α-CadF antibody and a 2° swine α-goat IgG (H+L)-FITC labeled antibody (Boehringer Mannheim, Indianapolis, IN). Rac1 was stained with a rabbit α-Rac1 polyclonal antibody (New East Biosciences, Malvern, PA) and a Texas Red dye-conjugated donkey α-rabbit (Jackson Immunoresearch Labs, West Groves, PA). The coverslips were mounted with Vectashield (Vector Laboratories Inc., Burlingame, CA) containing DAPI (4′,6-diamidino-2-phenylindole) and visualized using the Nikon Eclipse TE2000 inverted epifluoresence microscope. Merged images were generated by overlaying three individual channel images (red, green, and blue) into a single three-color image. A minimum of 50 cell-associated C. jejuni were analyzed for co-localization with Rac1.
Scanning electron microscopy
The C. jejuni used for these assays were harvested from agar plates after 18 hr of growth. The INT 407 cells were grown on coverslips placed in wells on a 24-well tissue culture tray. Following the inoculation of the INT 407 cells with C. jejuni, the tissue culture trays were incubated for 15 min in a CO2 incubator. Following the incubation period, the cells were washed one time with PBS and incubated overnight at 4°C in a solution of 2% glutaraldehyde and 2% paraformaldehyde in a 0.1 M cacodylate buffer. The cells were washed with 0.1 M cacodylate buffer 3 times, and were incubated at 4°C overnight with 2% osmium tertraoxide. After this incubation, the cells were rinsed three times with dH2O, incubated for 15 min with a solution of 0.05% tannic acid, rinsed 3 more times with dH2O, and then taken through a series of ethanol dehydrations (30%, 50%, 70%, 95%, and 100%(3×)). Following ethanol dehydration, the samples were critical point dried using a Samdri-PVT-3D critical point drier (Tousimis, Rockville, MA). The coverslips were mounted and platinum/palladium (pt/pd) coated using a Cressington Sputter Coater 208 HR (Cressington Scientific Instruments Ltd., Watford, UK) to a thickness of 2 nm. The cells were observed using a FEI Quanta 200F field emissions scanning electron microscope (FEI, Hillsboro, OR). Quantification of membrane ruffling was done by two independent observers and tabulated. Only cells with clear boundaries were counted and ruffling positive cells were scored as those that had more membrane ruffles than the cells only negative control as seen in Fig 6.
Statistical Analysis
Statistical significance between samples was determined using Student’s t test, and values of P < 0.05 and P < 0.01 are reported.
Supplementary Material
Supplemental Fig. S1. Temporal kinetics of C. jejuni internalization by INT 407 cells. Values represent the percent of internalized bacteria/well of a 24-well tissue culture tray relative to the invasion of the C. jejuni wild-type stain at 3 hr. Shown are a C. jejuni F38011 wild-type strain (solid line, black), C. jejuni F38011 wild-type strain treated with 1024 μg/ml of chloramphenicol (dashed line), and the C. jejuni ciaC mutant (solid line, gray). The error bars represent the standard deviation of the mean of percent internalized bacteria.
Supplemental Fig. S2. Treatment of INT 407 cells with genistein, a global inhibitor of protein-tyrosine kinases, significantly inhibits C. jejuni invasion. The INT 407 cells pretreated with 250 μM of inhibitor 30 minutes prior to inoculation with C. jejuni and S. typhimurium. The bacteria were incubated with the INT 407 cells for 3 hr prior to gentamicin treatment. Values represent number of internalized (gentamicin-protected) bacteria/well of 24-well tissue culture tray and are given as means of triplicate determinations ± standard deviations. The asterisk indicates significance (P < 0.01) between untreated and genistein-treated samples using the Student’s t test.
Supplemental Fig. S3. Effect of EGFR inhibitors on C. jejuni binding and invasion of INT 407 cells. The INT 407 cells were pretreated with the indicated concentrations of inhibitor 30 minutes prior to inoculation with C. jejuni. The bacteria were incubated with the INT 407 cells for 3 hr prior to gentamicin treatment. Values represent number of percent of adherent bacteria that invaded the INT 407 cells. Significance between samples was determined using Student’s t test. * P < 0.01.
Supplemental Fig. S4. Treatment of INT 407 cells with vehicle has no effect on internalization of a C. jejuni wild-type strain. INT 407 cells were treated with the maximum volume of solvent [i.e., dimethyl sulfoxide (DMSO) or methanol (MeOH)] used for delivery of inhibitors to host cells and inoculated with C. jejuni wild-type strain. Values represent the mean ± standard deviation of internalized bacteria/well of a 24 well tissue culture plate. There was no statistical difference in C. jejuni binding to the INT 407 cells (not shown) (DMSO, P = 0.85, MeOH, P = 0.55) or internalization (DMSO, P = 0.29, MeOH, P = 0.61) observed for either vehicle.
Supplemental Fig. S5. C. jejuni induces EGFR activation in a CadF FlpA dependent manner. INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni cadF flpA mutant. Panels: A) Proteins were immunoprecipitated from whole cell lysates with antibody reactive against total EGFR (α-EGFR MAb528). Blots were probed with an antibody that reacts against the active EGFR (pEGFR Tyr845 and Tyr1068) and an antibody that reacts against EGFR. B) Densitometric analysis of the level of EGFR activation of INT 407 cells inoculated with the wild-type isolate and cadF flpA mutant. The asterisks indicate a significance difference (*P < 0.01, **P < 0.05) in EGFR activation between cells inoculated with the wild-type isolate versus the cadF flpA mutant. The average background level in each lane was subtracted from the band of appropriate molecular weight and normalized to the cells only negative control. Values represent a fold change in activation over basal levels. Each condition was tested in triplicate on multiple days.
Supplemental Fig. S6. Additional evidence that Rac1 is recruited to sites of C. jejuni attachment. The localization of Rac1 in C. jejuni-infected cells was examined by immunofluorescence microscopy as outlined in “Experimental Procedures.” The cell-associated bacteria are indicated by the circles, whereas the C. jejuni adjacent to sites of accumulated Rac1 are indicated by the circles with the arrowhead. In contrast to the C. jejuni wild-type strain, fewer sites of Rac1 co-localization were observed in INT 407 cells inoculated with the C. jejuni ciaC mutant. The circles highlight the bound bacteria.
Supplemental Fig. S7. INT 407 cells inoculated with C. jejuni are deficient in Rac1 activation when pretreated with specific inhibitors of EGFR, FAK, PI3 kinase, or Src. Rac1 activation in cells inoculated with C. jejuni wild type strain following pretreatment with either 20 μM erlotinib, 5 μM TAE 226, 50 nM wortmannin, or 33 μM PP2 for 30 minutes. Values represent at least 5 samples analyzed by GLISA™ in duplicate. The asterisks indicate a significance difference (*P < 0.01, **P < 0.05) in Rac1 activation between untreated cells inoculated with the wild-type isolate versus each treatment group.
Supplemental Fig. S8. INT 407 cells treated with inhibitors of EGFR, FAK, PI3 kinase or Src are deficient in membrane ruffling in response to C. jejuni. Scanning electron microscopy of INT 407 cells inoculated with C. jejuni wild type strain after pretreatment with the indicated inhibitors. Cell ruffling was enumerated and each drug treatment was found to significantly reduce ruffling when compared to the wild type strain (P < 0.01).
Supplemental Fig. S9. INT 407 cells are responsive to the addition of EGF. Panels: A) Scanning electron microscopy (SEM) image of an INT 407 cell treated with 100 ng/ml of EGF for 2 minutes; B) SEM image of a non-treated INT 407 cell; C) Quantification of Rac1 activation by G-LISA™ in cells incubated for 2 minutes with 100 ng/ml of EGF versus untreated cells. The mean ± standard deviation of total active Rac1 is indicated in Relative Bioluminescence.
Acknowledgments
We thank Dr. Ray Reeves, Dr. Charles L. Larson, and Jason M. Neal-McKinney and for critical review of the manuscript. We also thank Dr. Mary Hunzicker-Dunn for helpful discussions. Finally, we thank Derrick Samuelson for the examination of C. jejuni-inoculated epithelial cells by scanning electron microscopy.
This study was supported from funds provided by the School of Molecular Biosciences at Washington State University. Tyson Eucker was supported by Award Number T32GM008336 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian N, Scott DW, Castle JD, Casanova JE, Schwartz MA. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat Cell Biol. 2007;9:1381–1391. doi: 10.1038/ncb1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley A, Fang K, Mertz H, Castranova V, Friend S, Liu J. Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J Biol Chem. 2005;280:3541–3547. doi: 10.1074/jbc.M409040200. [DOI] [PubMed] [Google Scholar]
- Buchsbaum RJ, Connolly BA, Feig LA. Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol Cell Biol. 2002;22:4073–4085. doi: 10.1128/MCB.22.12.4073-4085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelow DR, Christensen JE, Neal-McKinney JM, Konkel ME. Campylobacter jejuni survival within human epithelial cells is enhanced by the secreted protein Cial. Mol Microbiol. 2011 doi: 10.1111/j.1365-2958.2011.07645.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, Moro L, Bergatto E, Boeri Erba E, Di Stefano P, Turco E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans. 2004;32:438–442. doi: 10.1042/BST0320438. [DOI] [PubMed] [Google Scholar]
- Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999;4:D102–113. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Christensen JE, Pacheco SA, Konkel ME. Identification of a Campylobacter jejuni-secreted protein required for maximal invasion of host cells. Mol Microbiol. 2009;73:650–662. doi: 10.1111/j.1365-2958.2009.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. EGF ERBB signalling: towards the systems level. Nature Reviews Molecular Cell Biology. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17:383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen EH, Yamada KM. Fibronectin, integrins, and growth control. J Cell Physiol. 2001;189:1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2008;294:G276–285. doi: 10.1152/ajpgi.00340.2007. [DOI] [PubMed] [Google Scholar]
- Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan RC, Neal-McKinney JM, Dhillon AS, Miller WG, Konkel ME. Examination of Campylobacter jejuni putative adhesins leads to the identification of a new protein, designated FlpA, required for chicken colonization. Infect Immun. 2009 doi: 10.1128/IAI.01266-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilcrease MZ. Integrin signaling in epithelial cells. Cancer Lett. 2007;247:1–25. doi: 10.1016/j.canlet.2006.03.031. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hu L, Kopecko DJ. Campylobacter jejuni 81–176 associates with microtubules and dynein during invasion of human intestinal cells. Infect Immun. 1999;67:4171–4182. doi: 10.1128/iai.67.8.4171-4182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Corwin MD, Joens LA, Cieplak W. Factors that influence the interaction of Campylobacter jejuni with cultured mammalian cells. J Med Microbiol. 1992;37:30–37. doi: 10.1099/00222615-37-1-30. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Garvis SG, Tipton SL, Anderson DE, Jr, Cieplak W., Jr Identification and molecular cloning of a gene encoding a fibronectin-binding protein (CadF) from Campylobacter jejuni. Mol Microbiol. 1997;24:953–963. doi: 10.1046/j.1365-2958.1997.4031771.x. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Kim BJ, Rivera-Amill V, Garvis SG. Bacterial secreted proteins are required for the internalization of Campylobacter jejuni into cultured mammalian cells. Mol Microbiol. 1999;32:691–701. doi: 10.1046/j.1365-2958.1999.01376.x. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Klena JD, Rivera-Amill V, Monteville MR, Biswas D, Raphael B, Mickelson J. Secretion of virulence proteins from Campylobacter jejuni is dependent on a functional flagellar export apparatus. J Bacteriol. 2004;186:3296–3303. doi: 10.1128/JB.186.11.3296-3303.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Larson CL, Flanagan RC. Campylobacter jejuni FlpA binds fibronectin and is required for maximal host cell adherence. J Bacteriol. 2010;192:68–76. doi: 10.1128/JB.00969-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecko DJ, Hu L, Zaal KJ. Campylobacter jejuni--microtubule-dependent invasion. Trends Microbiol. 2001;9:389–396. doi: 10.1016/s0966-842x(01)02107-2. [DOI] [PubMed] [Google Scholar]
- Krause-Gruszczynska M, Rohde M, Hartig R, Genth H, Schmidt G, Keo T, et al. Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell Microbiol. 2007;9:2431–2444. doi: 10.1111/j.1462-5822.2007.00971.x. [DOI] [PubMed] [Google Scholar]
- Malik-Kale P, Parker CT, Konkel ME. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J Bacteriol. 2008;190:2286–2297. doi: 10.1128/JB.01736-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Katz BZ, Lafrenie RM, Yamada KM. Fibronectin and integrins in cell adhesion, signaling, and morphogenesis. Ann N Y Acad Sci. 1998;857:119–129. doi: 10.1111/j.1749-6632.1998.tb10112.x. [DOI] [PubMed] [Google Scholar]
- Monteville MR, Yoon JE, Konkel ME. Maximal adherence and invasion of INT 407 cells by Campylobacter jejuni requires the CadF outer-membrane protein and microfilament reorganization. Microbiology. 2003;149:153–165. doi: 10.1099/mic.0.25820-0. [DOI] [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Boeri Erba E, Smeriglio M, et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- Oelschlaeger TA, Guerry P, Kopecko DJ. Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc Natl Acad Sci U S A. 1993;90:6884–6888. doi: 10.1073/pnas.90.14.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellinen T, Ivaska J. Integrin traffic. J Cell Sci. 2006;119:3723–3731. doi: 10.1242/jcs.03216. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Developmental Biology. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rothberg KG, Ying YS, Kamen BA, Anderson RG. Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J Cell Biol. 1990;111:2931–2938. doi: 10.1083/jcb.111.6.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Palacios GM. The health burden of Campylobacter infection and the impact of antimicrobial resistance: playing chicken. Clin Infect Dis. 2007;44:701–703. doi: 10.1086/509936. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Chandler J. Fibronectin matrix turnover occurs through a caveolin-1-dependent process. Mol Biol Cell. 2005;16:757–768. doi: 10.1091/mbc.E04-08-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K, Sato T, D’Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J Exp Med. 1995;182:1089–1099. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelemann A, Petti F, Griffin G, Iwata K, Hunt T, Settinari T, et al. Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol Cell Proteomics. 2005;4:356–376. doi: 10.1074/mcp.M400118-MCP200. [DOI] [PubMed] [Google Scholar]
- van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285–298. doi: 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- Vuori K. Integrin signaling: tyrosine phosphorylation events in focal adhesions. J Membr Biol. 1998;165:191–199. doi: 10.1007/s002329900433. [DOI] [PubMed] [Google Scholar]
- Wooldridge KG, Williams PH, Ketley JM. Host signal transduction and endocytosis of Campylobacter jejuni. Microb Pathog. 1996;21:299–305. doi: 10.1006/mpat.1996.0063. [DOI] [PubMed] [Google Scholar]
- Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1. Temporal kinetics of C. jejuni internalization by INT 407 cells. Values represent the percent of internalized bacteria/well of a 24-well tissue culture tray relative to the invasion of the C. jejuni wild-type stain at 3 hr. Shown are a C. jejuni F38011 wild-type strain (solid line, black), C. jejuni F38011 wild-type strain treated with 1024 μg/ml of chloramphenicol (dashed line), and the C. jejuni ciaC mutant (solid line, gray). The error bars represent the standard deviation of the mean of percent internalized bacteria.
Supplemental Fig. S2. Treatment of INT 407 cells with genistein, a global inhibitor of protein-tyrosine kinases, significantly inhibits C. jejuni invasion. The INT 407 cells pretreated with 250 μM of inhibitor 30 minutes prior to inoculation with C. jejuni and S. typhimurium. The bacteria were incubated with the INT 407 cells for 3 hr prior to gentamicin treatment. Values represent number of internalized (gentamicin-protected) bacteria/well of 24-well tissue culture tray and are given as means of triplicate determinations ± standard deviations. The asterisk indicates significance (P < 0.01) between untreated and genistein-treated samples using the Student’s t test.
Supplemental Fig. S3. Effect of EGFR inhibitors on C. jejuni binding and invasion of INT 407 cells. The INT 407 cells were pretreated with the indicated concentrations of inhibitor 30 minutes prior to inoculation with C. jejuni. The bacteria were incubated with the INT 407 cells for 3 hr prior to gentamicin treatment. Values represent number of percent of adherent bacteria that invaded the INT 407 cells. Significance between samples was determined using Student’s t test. * P < 0.01.
Supplemental Fig. S4. Treatment of INT 407 cells with vehicle has no effect on internalization of a C. jejuni wild-type strain. INT 407 cells were treated with the maximum volume of solvent [i.e., dimethyl sulfoxide (DMSO) or methanol (MeOH)] used for delivery of inhibitors to host cells and inoculated with C. jejuni wild-type strain. Values represent the mean ± standard deviation of internalized bacteria/well of a 24 well tissue culture plate. There was no statistical difference in C. jejuni binding to the INT 407 cells (not shown) (DMSO, P = 0.85, MeOH, P = 0.55) or internalization (DMSO, P = 0.29, MeOH, P = 0.61) observed for either vehicle.
Supplemental Fig. S5. C. jejuni induces EGFR activation in a CadF FlpA dependent manner. INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni cadF flpA mutant. Panels: A) Proteins were immunoprecipitated from whole cell lysates with antibody reactive against total EGFR (α-EGFR MAb528). Blots were probed with an antibody that reacts against the active EGFR (pEGFR Tyr845 and Tyr1068) and an antibody that reacts against EGFR. B) Densitometric analysis of the level of EGFR activation of INT 407 cells inoculated with the wild-type isolate and cadF flpA mutant. The asterisks indicate a significance difference (*P < 0.01, **P < 0.05) in EGFR activation between cells inoculated with the wild-type isolate versus the cadF flpA mutant. The average background level in each lane was subtracted from the band of appropriate molecular weight and normalized to the cells only negative control. Values represent a fold change in activation over basal levels. Each condition was tested in triplicate on multiple days.
Supplemental Fig. S6. Additional evidence that Rac1 is recruited to sites of C. jejuni attachment. The localization of Rac1 in C. jejuni-infected cells was examined by immunofluorescence microscopy as outlined in “Experimental Procedures.” The cell-associated bacteria are indicated by the circles, whereas the C. jejuni adjacent to sites of accumulated Rac1 are indicated by the circles with the arrowhead. In contrast to the C. jejuni wild-type strain, fewer sites of Rac1 co-localization were observed in INT 407 cells inoculated with the C. jejuni ciaC mutant. The circles highlight the bound bacteria.
Supplemental Fig. S7. INT 407 cells inoculated with C. jejuni are deficient in Rac1 activation when pretreated with specific inhibitors of EGFR, FAK, PI3 kinase, or Src. Rac1 activation in cells inoculated with C. jejuni wild type strain following pretreatment with either 20 μM erlotinib, 5 μM TAE 226, 50 nM wortmannin, or 33 μM PP2 for 30 minutes. Values represent at least 5 samples analyzed by GLISA™ in duplicate. The asterisks indicate a significance difference (*P < 0.01, **P < 0.05) in Rac1 activation between untreated cells inoculated with the wild-type isolate versus each treatment group.
Supplemental Fig. S8. INT 407 cells treated with inhibitors of EGFR, FAK, PI3 kinase or Src are deficient in membrane ruffling in response to C. jejuni. Scanning electron microscopy of INT 407 cells inoculated with C. jejuni wild type strain after pretreatment with the indicated inhibitors. Cell ruffling was enumerated and each drug treatment was found to significantly reduce ruffling when compared to the wild type strain (P < 0.01).
Supplemental Fig. S9. INT 407 cells are responsive to the addition of EGF. Panels: A) Scanning electron microscopy (SEM) image of an INT 407 cell treated with 100 ng/ml of EGF for 2 minutes; B) SEM image of a non-treated INT 407 cell; C) Quantification of Rac1 activation by G-LISA™ in cells incubated for 2 minutes with 100 ng/ml of EGF versus untreated cells. The mean ± standard deviation of total active Rac1 is indicated in Relative Bioluminescence.