

Abstract
The von Hippel-Lindau (VHL) tumor suppressor gene product is the recognition component of an E3 ubiquitin ligase and is inactivated in patients with VHL disease and in most sporadic clear cell renal carcinomas (RCC). pVHL controls oxygen-responsive gene expression at the transcriptional and post-transcriptional levels. The vascular endothelial growth factor A (VEGFA) mRNA contains AU-rich elements (AREs) in the 3' untranslated region, and mRNA stability or decay is determined through ARE-associated RNA binding factors. We show here that levels of the ARE binding factor, AUF1, are regulated by pVHL and by hypoxia. pVHL and AUF1 stably associate with each other in cells and AUF1 is a ubiquitylation target of pVHL. AUF1 and another RNA binding protein, HuR, bind to VEGFA ARE RNA. Ribonucleoprotein (RNP)-immunoprecipitations showed that pVHL associates indirectly with VEGFA mRNA through AUF1 and/or HuR, and this complex is associated with VEGFA mRNA decay under normoxic conditions. Under hypoxic conditions pVHL is downregulated, while AUF1 and HuR binding to VEGF mRNA is maintained, and this complex is associated with stabilized mRNA. These studies suggest that AUF1 and HuR bind to VEGFA ARE RNA under both normoxic and hypoxic conditions, and that a pVHL-RNP complex determines VEGFA mRNA decay. These studies further implicate the ubiquitin-proteasome system in ARE-mediated RNA degradation.
Keywords: von Hippel-Lindau, hypoxia, VEGF, AUF1, HuR, hnRNP
Introduction
Germline inactivation of the VHL tumor suppressor gene is linked with development of von Hippel-Lindau (VHL) disease, an autosomal dominantly inherited cancer syndrome. VHL patients are predisposed to develop various vascular tumors, including hemangioblastomas of the retinas and central nervous system, clear cell RCC, pancreatic cysts and adenocarcinomas, and adrenal pheochromocytomas (1). The VHL gene is also inactivated in the majority of patients with sporadic clear cell RCC (1). The VHL gene encodes proteins (pVHL) of 25 and 19 kDa through use of alternative translation initiation codons, and both isoforms appear to possess tumor suppressor activity (2). pVHL has α and β structural domains that are critical to its function as the substrate recognition component of a cullin-RING E3 ubiquitin ligase (CRL) (3–5). The N-terminal β domain is associated with target protein recognition, while the α domain contains a SOCS box that interacts with elongin C and links pVHL to the ubiquitin-ligase complex containing elongin B and C and Cul2. Perhaps the best characterized pVHL CRL targets are the α subunits of hypoxia-inducible factor (HIF) (6). Hydroxylation of conserved prolines on HIFα subunits under normoxic (21% O2) conditions provides a substrate recognition motif for pVHL polyubiquitylation, and proteasomal degradation. In hypoxia (1% O2) prolyl hydroxylase activity is inhibited, unmodified HIFα subunits are stabilized, and the hypoxia response is initiated. We recently demonstrated that pVHL levels are suppressed in hypoxia (7), providing an additional mechanism for HIFα upregulation in hypoxia.
HIF activation stimulates transcription of several hundred genes whose products have varied functions, including nutrient uptake, glycolytic metabolism, and neovascularization (8, 9)). However, many of the hypoxia-inducible genes also exhibit control at the level of mRNA stability. Such mRNAs are labile under normal conditions and become stabilized in hypoxia. This regulation is mediated through adenine/uridine-rich elements (AREs) that are typically found in the 3' untranslated regions (UTRs) of these mRNAs, and interactions with ARE-binding proteins determines the stability or decay of the mRNA (reviewed in (10)). Two well characterized are AUF1 and HuR (10). AUF1 (or hnRNP D) represents a family of proteins of 37, 40, 42, and 45 kDa that are produced through alternative splicing of a single pre-mRNA (11). All AUF1 proteins contain two non-identical RNA recognition motifs (RRMs), a consensus RNA-binding domain, and may bind to ARE RNA as heterologous oligomers (12). Sequences flanking the RRMs seem to contribute to ARE binding, including an alanine-rich region near the N-terminus and a short glutamine-rich region near the C terminus. While AUF1 frequently is associated with mRNA destabilizing activity, several studies have indicated that AUF1 stabilizes some mRNAs (reviewed in (13)). The independence and/or interdependence of the various AUF1 isoforms is not yet fully understood. HuR is a 36 kDa protein that is ubiquitously expressed and is a member of the embryonic lethal abnormal vision (ELAV) family of RNA binding proteins. HuR has three RRMs and is generally considered to be involved in the stabilization of ARE-containing mRNAs (10). Overexpression of HuR has been noted in malignancies, including renal cell carcinoma (RCC) (14).
Overexpression of a key angiogenic factor, vascular endothelial growth factor (VEGF) A, has long been appreciated in RCC and VHL-associated tumors tumors. The importance of VEGFA activity in RCC is underscored by the fact that anti-VEGF therapies are now the first-line standard of care for patients with metastatic RCC (reviewed in (15)). VEGFA is a classical hypoxia-inducible gene product. In normoxia basal levels of VEGFA transcription may be controlled by the SP1 general transcription factor (16, 17), but the VEGFA mRNA is unstable in normoxia with a half-life of 15 to 40 minutes (18–20). After a shift to hypoxia an 8- to 30-fold increase in steady state VEGF mRNA levels in various cell lines has been observed (reviewed in (21)). VEGF induction in hypoxia occurs through increased transcription initiation mediated through HIF binding to VEGFA promoter enhancer elements (22), as well as through an increased mRNA half-life of up to 4 h (21). The involvement of several RNA binding proteins in the regulation of VEGFA mRNA levels has been described, including HuR (23), hnRNP L (24, 25), and Tis11b (26).
In this report we examined further the role of pVHL in regulating VEGFA mRNA stability. RCC cell lines with inactivated pVHL exhibit a condition that has been termed pseudohypoxia, high expression levels of classically-defined hypoxia-responsive gene products in the presence of normal oxygen levels. Restoration of pVHL expression in RCC cell lines restores the normal hypoxic response. We show here that the ARE RNA binding protein, AUF1, is regulated by pVHL and by hypoxia, that pVHL and AUF1 directly associate with each other, and pVHL appears to target p45AUF1 and p42AUF1 for ubiquitylation. AUF1 and HuR bind to VEGFA and tumor necrosis factor (TNF) α ARE RNA sequences under both normoxic and hypoxic conditions. In normoxia pVHL is also found in these complexes, and this binding is associated with rapid decay of the ARE-containing RNA. In hypoxia, when pVHL levels are decreased, the continued binding of AUF1 and HuR is associated with stability of the ARE-containing RNA. These results directly link pVHL, and perhaps its ubiquitin-ligase activity, with enhanced degradation of ARE-containing RNA.
Materials and Methods
Antibodies and Reagents
Monoclonal antibodies (mAb) directed against hemagglutinin (HA, HA.11), tubulin, and ubiquitin (P4G7) were from Covance; M2 anti-FLAG mAb was from Sigma; HuR and Cul2 mAb, rabbit polyclonal pVHL antibody (FL181), normal rabbit IgG, and normal mouse IgG were from Santa Cruz Biotechnology; pVHL mAb (Ig32) was from Pharmingen. Rabbit affinity purified AUF1 antibody was previously described (27) or from Upstate Biotechnology or Phoenix Biochemicals. Proteasome inhibitors were from Calbiochem and desferrioxamine was from Sigma.
Cell Culture
Cell lines were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum in a humidified, 5% CO2 incubator at 37°C. A complete description of the cell lines used in these studies is provided in Supplementary Table 1. UOK121 RCC cells are from the Urologic Oncology Branch cell line repository and were provided by Dr. W. Marston Linehan, National Cancer Institute. Human embryonic kidney 293T cells and 786-O RCC cells were obtained from the American Type Culture Collection. 786-O G7F cells and 786-O 157Δ express wild-type Flag-tagged VHL or truncated and inactive pVHL (amino acids 1–157) cDNAs, respectively (7). HA-VHL-expressing 786-O RCC cells (WT7) and control cells (ARZ2 and pRC9) were provided by Dr. William G. Kaelin, Dana-Farber/Harvard Cancer Center. All of the cell lines used during the course of these studies were maintained in continuous culture for 4 months or less.
Hypoxia experiments were performed in a chamber monitored by a BioSpherix C21 dual O2/CO2 controller that was calibrated to 1% O2 levels according to the manufacturer's specifications. In a typical experiment cells were seeded in 100 mm tissue culture dishes and cultured in 5% CO2 in normoxia (room air) for 18 to 24 h before transfer to the hypoxia chamber.
Immunoprecipitation
Cells were harvested at less than 75% confluence in all experiments by scraping in ice-cold phosphate buffered saline (PBS) and centrifugation at 500 × g. Cell were lysed in either Igepal lysis buffer (100 mM NaCl, 0.5% Igepal CA630 (Sigma-Aldrich), 20 mM Tris-HCl, pH 7.0, 5 μM MgCl2, 1 mM sodium orthovanadate, 5 mM levamisole, aprotinin (1 μg/ml), 0.5 mM 4-(2-aminoethyl) benzene sulphonyl fluoride) (all from Sigma), and Complete protease inhibitor cocktail (Roche), or RIPA buffer (150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 20 mM Tris-HCl, pH 7.0, 5 mM MgCl2, and the same inhibitors as above. For immunoprecipitations, 500 μg cell extract was pre-cleared overnight at 4°C with protein G Sepharose (50% v/v; Sigma) that had been pre-blocked with phosphate buffered saline (PBS) containing 20 mg/ml bovine serum albumin, 5 mg/ml heparin, and 0.1 mg/ml yeast tRNA. Immunoprecipitating antibody (1–2 μg) was added to the pre-cleared lysates and incubated at 4°C for two hours to overnight, followed by two hours incubation with pre-blocked protein G Sepharose (50% v/v) on a rotator at 4°C. Beads were washed five times in lysis buffer, samples were resolved in SDS-polyacrylamide gels, and electro-transferred onto PVDF membranes. Western blots were developed using chemiluminescence (ECL, Amersham).
In vitro protein interactions and ubiquitylation assays
Full length or truncated GST-VHL and GST-AUF1 clones in pGEX-2T were prepared by PCR amplification of cDNA clones. cDNAs encoding p45AUF1 or p37AUF1 were used to generate GST-AUF1 clones that either contained both or lacked both exon 2- and exon 7-encoded sequences, respectively. Fusion protein expression in E. coli BL21 with IPTG induction was confirmed by Coomassie Blue staining of SDS-polyacrylamide gels. Radiolabeled proteins were synthesized in vitro in the presence of [35S]-methionine using the TNT coupled reticulocyte lysate system (Promega). GST pull-down assays were performed by pre-binding GST fusion proteins to glutathione-Sepharose (50% v/v; Sigma) in PBS containing 1% NP-40, washing extensively, and then adding radiolabeled in vitro translated protein in PBS+1% NP-40, 1 mg/ml heparin, 5 mM levamisole, 1 mM sodium orthovanadate, and Complete protease inhibitors. Incubations were for 1 hr at 4°C, and the beads were then washed 4 times with PBS containing 500 mM NaCl, 1% NP-40, and 0.5 μg/ml BSA. Samples were resolved by 12% SDS-PAGE, and gels were dried and autoradiographed.
In vitro ubiquitylation assays were performed with p25VHL and AUF1 proteins that were translated in vitro using the TNT coupled reticulocyte lysate system (Promega) in the presence of cold methionine. Five μl each of pVHL and AUF1 were mixed and incubated for at 37°C for 4 h in the presence of an ATP regeneration system, E1 ubiquitin-activating enzyme, UbcH5b E2 ubiquitin-conjugating enzyme (all purchased from Boston Biochemical), 5 μg ubiquitin, and 0.5 μg ubiquitin aldehyde (Sigma) in a total volume of 25 μl. Reactions were stopped by addition of Laemmli SDS-sample buffer and boiling, resolved by 8% SDS-PAGE, and analyzed by western blotting.
RNA-protein interaction assays
VEGFA ARE regions defined by Levy et al (28) (see Supplemental Figure 3A) were amplified by PCR using a VEGFA 3' UTR cDNA clone as template. Each amplicon had a T7 RNA polymerase site at the 5' end, and T7 in vitro transcription reactions were performed in the presence of α-32P-UTP or biotin-11-GTP (Perkin Elmer). Complementary double strand DNA oligonucleotides were synthesized corresponding to the TNFα ARE (5'-AATTATTTATTATTTATTTATTATTTTATTATTTAA-3') or a mutated ARE sequence (5'-AATGATGTACTACTTGTTCATGATGTTCTTCTTGAA-3') (29), were cloned into pGEM4, and run-off transcripts were made with T7 RNA polymerase using linearized plasmid as template. Radiolabeled transcripts (2×105 cpm/reaction) were incubated with 30 μg cytosolic extracts in binding buffer (10 mM HEPES, pH 7.5, 10% glycerol, 5 mM MgCl2, 50 mM KCl, 0.5 mM EGTA, 0.5 mM DTT, 100 μg/ml yeast tRNA, and 5 mg/ml heparin), and incubated at 30°C for 20 min followed by RNase treatment (40 units RNase T1 and 1 μg RNase A) for 15 min at room temperature. Samples were run in 8% non-denaturing polyacrylamide gels in 0.5× TBE buffer, gels were dried, and subjected to autoradiography.
RNP immunoprecipitation and RT-PCR
For RNP-immunoprecipitation, cultures at 75% confluence were washed with cold PBS and extracts were prepared using 0.5 ml/100-mm dish RNP-ip lysis buffer (50 mM HEPES, pH 7.5, 10 mM sodium pyrophosphate, 150 mM NaCl, 1.5 mM MgCl2, 0.5% Igepal CA630, 10% glycerol, 100 mM sodium fluoride, 0.2 mM sodium orthovanadate, 1 mM EGTA, and Complete protease inhibitor cocktail). Insoluble material was removed by centrifugation for 30 min at 20,000×g. Antibodies were added to 1 μg/ml and incubated for 2 h at 4°C on a rotator. Immune complexes were collected with the addition of pre-blocked protein G Sepharose (50% v/v) for an additional 1 h at 4°C on a rotator. Samples were washed five times in RNP-ip lysis buffer and then were extracted with 50 μl Trizol (Invitrogen). The aqueous phase was isolated and ethanol precipitated at −80°C overnight with 1 μg glycogen added as carrier. Ethanol precipitates were centrifuged at 20,000×g for 1 h at 4°C, washed with 70% ethanol, and the pellets were solubilized in 8 μl water for 15 minutes at 65°C. The entire contents of each tube were used for first-strand cDNA synthesis (Invitrogen) according to the manufacturer's protocol. PCR was performed using primers specific for VEGFA or actin as previously described (30) except that 35 amplification cycles were performed. In control RT-PCR reactions first-strand cDNA that was prepared from total cellular RNA was used, and 25 amplification cycles were performed. PCR products were resolved in 2% agarose-1000 (Invitrogen) gels.
Reporter plasmid constructs and transfection
Fragments from the VEGFA 3' UTR were generated by PCR and subcloned into the XbaI restriction site downstream of the firefly luciferase gene translation termination codon in the pGL3-Promoter vector (Promega). A 193 bp fragment, corresponding to VEGFA 3' UTR nucleotides 306 to 498 (relative to the translation termination codon; Genbank submission number Y08736), contains an ARE that was shown previously to confer stability under hypoxic conditions (24, 25, 31). A control, non-ARE containing 193 base pair fragment corresponding to VEGFA nucleotides 113 to 305 was also cloned into the pGL3-Promoter vector. The orientation and sequence of the inserts were verified by DNA sequencing. Cells were transfected in 60 mm tissue culture dishes (LipofectAMINE 2000; Invitrogen). After 24 h, the cultures were washed, trypsinized, and divided onto two 60 mm tissue culture dishes, which were then cultured for an additional 6 h and then either transferred to the hypoxia incubator (1% oxygen) or maintained in room air. After 12 h, the cells were extracted with RNP-ip lysis buffer or with Trizol and analyzed by RNP-ip or RT-PCR as described above. Luciferase-specific PCR primers were: forward 5'-AGATGCACATATCGAGGTGGACAT-3' and reverse 5'-ATCGTATTTGTCAATCAGAGTGCT-3', which amplifies a 786 bp fragment. Aliquots of the cell extracts were also used for luciferase assays, and transfection efficiencies were determined by co-transfection of a plasmid expressing CMV-β-galactosidase.
Gene Silencing
pSilencer-AUF1 and control plasmids (32) were obtained from Dr. M Gorospe, National Institute of Aging, NIH. Verified, pooled HuR siRNA were obtained from Dharmacon. 293T cells were transfected using Lipofectamine 2000, and after 48 h total RNA or RIPA extracts were prepared for quantitative RT-PCR or western blotting analyses, respectively. Primers for (VEGFA, AUF1, HuR, 18S rRNA, and TBP) were obtained from Qiagen, and quantitative PCR analyses were performed with the ABI StepOnePlus Real-Time PCR system with SYBR green detection. Target mRNA expression levels were measured in triplicate and relative quantitation (RQ) values were determined relative to TBP mRNA or 18S rRNA mRNA by the comparative CT (ΔΔCT) method with the ABI StepOne software package. 18S rRNA was used as an internal control in hypoxia experiments, because, unlike TBP, its levels were unaffected by hypoxia treatment.
Results
AUF1 protein levels are regulated by pVHL and hypoxia
In order to understand better regulation of ARE-mediated mRNA stability and decay in normoxia and hypoxia, we examined AUF1 expression in RCC cells and in 293T cells. AUF1 protein levels were lower in pVHL-expressing 786-O G7F RCC cells as compared to 786-O 157Δ RCC cells that express inactive pVHL (Figure 1A). AUF1 is expressed as 4 isoforms of 37, 40, 42, and 45 kDa, and we detected similar regulation of all isoforms. Therefore, we will refer to expression levels of the 4 isoforms collectively as AUF1. When cultured in hypoxia, AUF1 levels were increased in 786-O G7F RCC cells, and AUF1 expression was comparable to that seen in pVHL-negative 786-O 157Δ RCC cells (Figure 1A). AUF1 expression levels in 786-O 157Δ RCC cells were unchanged in hypoxia (Figure 1A), suggesting that AUF1 is not hypoxia-responsive in these cells. We also determined HuR expression levels in these experiments and found that HuR was equally expressed in pVHL-positive or –negative RCC cells and that HuR expression was unaffected by hypoxia (Figure 1A). AUF1 levels also increased within 4 h of addition of the hypoxia-mimetic, desferrioxamine, in 786-O G7F RCC cells, while desferrioxamine treatment had no effect on AUF1 levels in 786-O 157Δ RCC cells (Figure 1B). AUF1 shuttles between the nucleus and the cytosol. Immunofluorescence staining suggested that while desferrioxamine treatment increased total AUF1 levels, AUF1 subcellular localization was not changed (Supplemental Figure 1A). AUF1 expression levels also increased after culture of 293T cells in hypoxia (Figure 1C). As we previously showed (7), p25VHL expression and to a lesser extent p19VHL is suppressed by culture in hypoxia (Supplemental Figure 1B). These results suggest that the hypoxia-induced increase in AUF1 protein levels in 786-O G7F RCC cells or 293T cells may be linked to decreased pVHL levels.
Figure 1.

VHL-, oxygen-, and proteasome-dependent regulation of AUF1 expression in RCC and 293T cells. AUF1 levels in pVHL-positive (786-O G7F) and pVHL-negative (786-O 157Δ) RCC cells that were cultured (A) under normoxic (room air) or hypoxic (1% O2) conditions for 18 h or (B) in the presence of the hypoxia mimetic, desferrioxamine (DFO; 100 μM), for 2 to 18 h. U, untreated cells. C, 293T cells were cultured under normoxic or hypoxic conditions for 18 h. D, 786-O G7F RCC cells were cultured in the absence or the presence of the proteasome inhibitor, MG132 (20 μM), for 2 h. For the experiments in panels A–D, protein extracts were prepared with RIPA buffer, and 40 μg cellular protein was resolved by 12% SDS-PAGE followed by western blotting using the indicated antibodies. Tubulin or Cul2 detection were used as protein loading controls. E, Quantitative real-time RT-PCR analysis of VEGFA and AUF1 mRNAs in 293T cells that were cultured in normoxia or hypoxia for 18 h. The primers used for PCR are specific for VEGF exons 1 and 3 and AUF1 exons 3 and 5, which are contained in all alternatively spliced forms of these mRNAs. Expression levels of the target mRNAs were determined in triplicate and averages are expressed relative to 18S rRNA levels. F, Relative AUF1 mRNA levels in pVHL-positive, WT7, and pVHL-negative, PRC9, RCC cells were determined by quantitative real-time RT-PCR. AUF1 mRNA expression levels were determined relative to TBP mRNA levels, and data are expressed as relative RQ values with WT7=1.
We treated 786-O G7F RCC cells with the proteasome inhibitor, MG132, and found that AUF1 levels increased (Figure 1D), suggesting that the decreased AUF1 protein levels detected in these cells was proteasome-dependent. Similar results were seen after treatment with proteasome inhibitors, MG115 and lactacystin, as well as the calpain inhibitors ALLN and ALLM, which also inhibit proteasome activity (33) (Supplemental Figure 1C). On the other hand, treatment of 786-O 157Δ RCC cells with these same inhibitors had no effect on AUF1 protein levels (Supplemental Figure 1C). Our results suggest that AUF1 protein is downregulated in pVHL-expressing RCC cells as compared to isogenic pVHL-negative RCC cells, and this downregulation is dependent on proteasomal activity.
The data presented in Figure 1A–C suggest that AUF1 is a hypoxia-responsive gene product. However, quantitative RT-PCR analyses showed that total AUF1 mRNA levels were decreased in 293T cells after 6 h and 16 h culture in hypoxia to 65% and 49%, respectively, of the levels of normoxic 293T cells (Figure 1E). We also found that total AUF1 mRNA levels were similar in the pVHL-positive and –negative cell lines that were tested (Figure 1F). Therefore, the increased AUF1 protein levels detected in hypoxia or in pVHL-positive and –negative cells was not likely to be due to differing AUF1 mRNA levels, suggesting that a post-transcriptional mechanism accounts for the pVHL- and oxygen-dependent regulation of AUF1 protein levels.
pVHL and AUF1 associate in cells
We next found that AUF1 and pVHL associate in cells. Either anti-pVHL or anti-AUF1 antibodies co-immunoprecipitated a pVHL-AUF1 complex from cells extracts from 786-O G7F RCC cells (Figure 2A). 786-O G7F cells express both p25 and p19 VHL with a carboxy-terminal Flag epitope tag, and both isoforms were co-immunoprecipitated with AUF1 antiserum. In reciprocal experiments, anti-Flag antibodies co-immunoprecipitated AUF1 isoforms (Figure 2A). Similar results were obtained with HA-VHL-expressing WT7 cells (Figure 2B), providing independent confirmation of the pVHL-AUF1 interaction. We also determined association of endogenous AUF1 and endogenous pVHL in 293T cells. We previously showed that 293T cells express only p19 VHL (7), and p19 VHL-AUF1 complexes were detected in reciprocal co-immunoprecipitations (Figure 2C). Cul2 was present in both pVHL and AUF1 immunoprecipitations (Figure 2B, C), suggesting that the pVHL CRL complex associates with AUF1. Taken together our results indicate that stable pVHL-AUF1 complexes are isolated from pVHL-expressing RCC and 293T cell lines.
Figure 2.

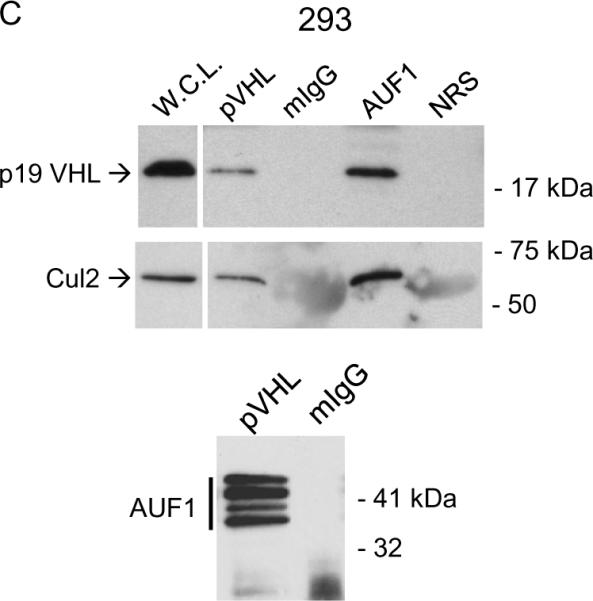
Co-immunoprecipitation of pVHL and AUF1. A, pVHL-positive 786-O G7F RCC cells; B, WT7 RCC cells; or C, 293T cells were extracted with Igepal lysis buffer, and immunoprecipitations were performed using 500 μg cellular protein and antibodies directed against AUF1 or pVHL (Ig32 for 293T cells or Flag for 786-O G7F RCC cells) or irrelevant mouse or rabbit IgG (mIgG or rIgG). Cul2 blotting in was performed to confirm co-immunoprecipitation of AUF1 with the pVHL CRL complex. L.C., immunoglobulin light chain.
pVHL targets p45AUF1 and p42AUF1 for ubiquitylation
Since the pVHL CRL complex associates with AUF1 and AUF1 protein levels exhibited pVHL- and oxygen-dependence, we asked whether pVHL targets AUF1 for ubiquitylation. UOK121 RCC cells, which do not express VHL mRNA or protein (34), were co-transfected with plasmids expressing Myc-tagged pVHL and HA-tagged ubiquitin and cultured in the presence of proteasome inhibitor MG132. pVHL co-immunoprecipitated with high molecular weight, polyubiquitylated AUF1 species (Figure 3A, lane 2), and the levels of polyubiquitylated AUF1 species were enhanced when cells also overexpressed HA-tagged ubiquitin (Figure 3A, lanes 1 and 6). We additionally performed in vitro ubiquitylation assays and found that p45AUF1 was targeted for polyubiquitylation when incubated with p25VHL (Figure 3B, compare lanes 2 and 4 with lane 6). p45AUF1 polyubiquitylation was reduced in the absence of added E2 (Figure 3B, compare lanes 2 and 4), which may be due to low-level E2 activity contained within the reticulocyte extracts that were used to synthesize these proteins. Under our experimental conditions pVHL appeared to target p42AUF1 for polyubiquitylation less efficiently than p45AUF1, since the high molecular weight species were decreased relative to p45AUF1 (Figure 3B, compare lanes 2 and 3). In addition, anti-ubiquitin western blotting showed that the major p42AUF1 ubiquitylated species migrated with relative molecular weights of ~50 to 100 kDa (Figure 3B, lane 3), suggesting multiubiquitylation rather than polyubiquitylation. Ubiquitylation of p42AUF1 was dependent on addition of both p25VHL and E2 ubiquitin-conjugating enzyme (Figure 3B, lanes 1 and 5 versus lane 3). In similar experiments we failed to detect polyubiquitylation of p37AUF1 or p40AUF1 (Figure 3C). It is possible that pVHL may not target these isoforms for ubiquitylation, or that our experimental conditions (e.g., the use of a single E2 enzyme, UbcH5b) were not optimal for these in vitro reactions.
Figure 3.

pVHL-dependent ubiquitylation of AUF1. A, UOK121 RCC cells were transfected with plasmids expressing Myc-tagged pVHL, HA-tagged ubiquitin, or both plasmids. MG132 (10 μM) was added 24 h post-transfection, and after an additional 8 h, cells were extracted with Igepal lysis buffer, and immunoprecipitations were performed with anti-myc antibody. Washed immunoprecipitates were eluted by boiling in Laemmli SDS sample buffer, loaded equally into each of 2 lanes, and resolved by 12% SDS-PAGE. Western blotting was performed with AUF1 and HA antibodies. Blots were stripped and re-probed for pVHL (bottom). H.C. and L.C., immunoglobulin heavy chain and light chain, respectively. B, p25VHL, p42AUF1, and p45AUF1 were synthesized in vitro in separate reactions. Five μl of each programmed reticulocyte lysate was mixed and incubated with recombinant E1 ubiquitin-activating enzyme and UbcH5b E2 ubiquitin-conjugating enzyme, ubiquitin, ubiquitin aldehyde, and an ATP regeneration system at 37°C for 4 hours. Samples were resolved on 8% SDS-polyacrylamide gels and western immunoblots blots were probed first with anti-AUF1 (top) and then stripped and reprobed with anti-ubiquitin (bottom). C, p25VHL, p37AUF1, and p40AUF1 were translated in vitro and ubiquitylation reactions were performed as described. Reactions in the experiment shown contained all in vitro ubiquitylation components and were incubated in the presence or absence of p25VHL. Samples were resolved by 8% SDS-PAGE and western blots were probed with anti-AUF1 and then stripped and re-probed with anti-ubiquitin.
Mapping pVHL-AUF1 binding domains in vitro
To characterize pVHL-AUF1 interactions, we performed GST-fusion protein pull-down studies. We found that all four AUF1 isoforms bound to GST-VHL1–213 (i.e., full length p25VHL) but not to GST alone (Figure 4A). Since GST-pVHL associated with each AUF1 isoform in vitro, we further determined binding of the largest AUF1 isoform, p45, to a series of progressive GST-pVHL C-teminal deletions (Figure 4B). We found that both GST-pVHL1–173 and GST-pVHL1–114 were able to bind p45AUF1, indicating that deletion of the Elongin C-binding pVHL α domain did not affect AUF1 binding (Figure 4B). We also tested GST-pVHL β domain mutations that correspond to truncation at each of the first 4 β sheets as indicated in Figure 4B (3, 4). Deletion of β sheet 4 (construct 1–101) and β sheet 3 (construct 1–89) did not disrupt p45 AUF1 binding, while p45 AUF1 binding to the GST-pVHL1–78 construct was diminished (Figure 4B), suggesting that AUF1 requires either β sheet 2, the loop between β sheet 1 and 2, or both for binding to pVHL. Therefore, pVHL appears to bind AUF1 through its target protein recognition domain.
Figure 4.


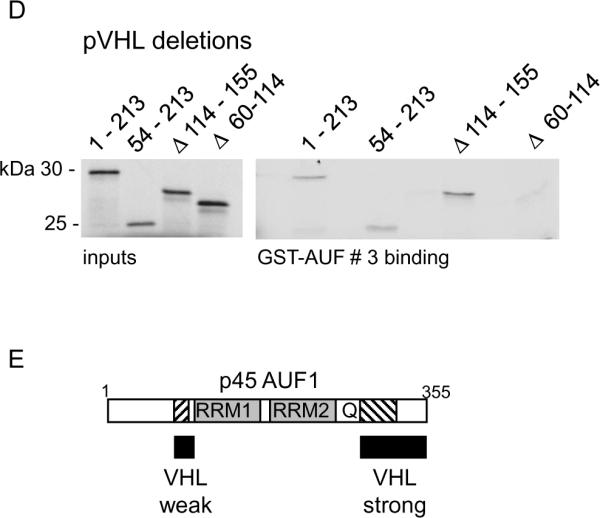
In vitro interaction domain mapping of pVHL and AUF1. A, Each AUF1 isoform (as indicated) was translated in vitro in the presence of 35S-methionine and tested for binding to GST or GST-VHL proteins that had been pre-bound to glutathione-Sepharose. GST, binding to GST alone; GST-VHL, binding to the fusion protein. B, Schematic diagram of pVHL indicating the portions of the protein forming the α and β domains and the first four of the β sheets (arrows 1 – 4). p45AUF1 was translated in vitro in the presence of 35S-methionine and tested for binding to GST or GST-VHL (full length or progressive C-terminal pVHL deletions). The amino acid at the pVHL C-terminal deletion endpoint is indicated above each lane. Inputs represent 10% of the amount used in each binding assay. C, Schematic diagram of p45AUF1 indicating two RNA recognition motifs (RRM1, RRM2) and a glutamine-rich sequence (Q, [QYQQQQQ]). The cross-hatched areas represent sequences encoded by the alternatively spliced exons 2 and 7. GST-AUF1 constructs used in these studies are indicated, with the amino acid numbering system corresponding to p45AUF1. Full-length p25VHL was translated in vitro in the presence of 35S-methionine and tested for binding to GST or the indicated GST-AUF1 constructs that were pre-bound to glutathione-Sepharose beads. D, The indicated pVHL deletion mutations were translated in vitro in the presence of 35S-methionine and tested for binding to GST AUF1 construct # 3. 1–213, full-length pVHL; 54–213, deletion of amino acids 1–53; Δ114–155, deletion of amino acids 114–155; Δ60–114, deletion of amino acids 60–114. Inputs represent 10% of the amount used in each binding assay. E, Schematic diagram of p45AUF1 indicating pVHL binding domains.
To determine the region of AUF1 that binds to pVHL we tested a series of GST-AUF1 deletion constructs (Figure 4C). pVHL bound most strongly to GST-AUF1 constructs containing C-terminal sequences, and in particular those GST-AUF1 constructs that contained amino acids 285–334, which are encoded by the AUF1 alternatively spliced exon 7 and the C-terminus (compare pVHL binding to GST-AUF1 constructs 3 or 5 with weak binding GST-AUF1 construct 2; Figure 4C). Our data suggest that the RNA recognition motifs (RRM1 and RRM2) or the glutamine-rich domain (Q) are not necessary for pVHL binding to AUF1 (compare pVHL binding to GST-AUF1 constructs 3, 4, and 5; Figure 4C). However, weak pVHL binding was also detected with AUF1 amino acids 1–112 (Figure 4C), and sequences encoded by the AUF1 alternatively spliced exon 2 (amino acids 78–98) appeared to be required for this interaction (compare pVHL binding to GST-AUF1 constructs 6 and 7; Figure 4C). It is likely that pVHL binding to GST-AUF1 construct 1 is also mediated through AUF1 amino acids 78–98 (Figure 4C). Our in vitro interaction studies suggest that pVHL may associate with two separate AUF1 domains, which are focused in or around amino acids encoded by the alternatively spliced AUF1 exons 2 and 7 (see Figure 4E). While there does not appear to be significant sequence similarity between these regions of AUF1, it is possible that there may be some structural similarity. Alternatively, different regions of the pVHL β domain may interact with these regions of AUF1. In addition, the contribution of AUF1 exon 2 encoded sequences for binding to pVHL may account for the weaker ubiquitylation activity that pVHL exhibited against p42AUF1 (Figure 3B), which lacks these sequences.
We also tested the ability of internal pVHL deletions to bind to GST-AUF1 construct # 3, containing AUF1 amino acids 174–355. Full length p25 VHL (1–213) and p19 VHL (54–213) both bound GST-AUF1174–355 equally well (Figure 4D), indicating that the pVHL acidic N-terminus is not required for AUF1 binding. Deletion of pVHL amino acids 115 –155 (encoded by alternatively spliced exon 2 and corresponding to β sheets 5, 6 and 7 of the pVHL β domain) also did not affect VHL binding to GST-AUF1174–355 (Figure 4D). However, deletion of pVHL amino acids 60–114 (deleting β sheets 1 – 4) abrogated VHL binding to GST-AUF1174–355 (Figure 4D). Taken together these results further suggest that the N-terminal portion of the pVHL β domain is required for AUF1 binding.
AUF1 and HuR bind to VEGFA ARE RNA
The relative roles of AUF1 and HuR in regulating VEGFA mRNA expression were determined through expression silencing experiments. Suppression of AUF1 expression in 293T cells resulted in a 1.7-fold increase in VEGFA mRNA levels (Figure 5A), suggesting that AUF1 expression is associated with decreased VEGFA mRNA levels. In contrast, suppression of HuR expression resulted in a 30% decrease in VEGFA mRNA levels (Figure 5A), which suggests that HuR binding to VEGFA mRNA is associated with mRNA stability and is in accord with earlier studies (23, 35). Suppression of AUF1 or HuR expression was verified by RT-PCR analyses (Figure 5A) as well as by western blotting (Supplemental Figure 2). These experiments were performed using 293T cells that were cultured under normoxic conditions. When similar silencing experiments were performed with cells that were cultured in hypoxia we did not detect efficient suppression of AUF1 or HuR mRNA or protein levels (data not shown) relative to control cells. The reason for this is unclear but suggests that the silencing apparatus may be sensitive to cellular oxygen levels.
Figure 5.

AUF1 binds to VEGFA ARE RNA. A, 293T cells were transfected with a vector expressing AUF1 shRNA (32), an HuR siRNA pool, or appropriate controls. Total RNA was extracted 48 h post-transfection and quantitative real-time RT-PCR analyses were performed using VEGF-, AUF1, or HuR-specific PCR primers. Expression levels of the target mRNAs were determined in triplicate and averages are expressed relative to TBP mRNA levels, which were unaffected in these experiments. B, Recombinant 6×His-tagged AUF1 isoforms were expressed in bacteria and purified by metal affinity chromatography. For EMSA 100 ng recombinant protein was incubated with radiolabeled VEGF 3'UTR ARE1 probe, treated with RNase A + T1, and resolved on non-denaturing 8% polyacrylamide gels followed by autoradiography. For UV-crosslinking (UV XL) complexes were exposed to UV light (300 mjoules), treated with RNase A+T1, resolved by 12% SDS-PAGE, and autoradiographed. C, Radiolabeled VEGF 3' UTR ARE1 probe was incubated in the presence of 100 μg protein cytosolic extracts that were prepared from 786-O RCC cells. Protein-RNA complexes were incubated with anti-AUF1, irrelevant rabbit anti-serum, or 1 mg/ml BSA. Samples were resolved on non-denaturing 8% polyacrylamide gels followed by autoradiography. AUF1-specific supershift complexes are indicated.
The results presented above suggest that AUF1 may play a role in VEGF mRNA instability in normoxia. Each of the AUF1 isoforms in purified, recombinant form bound to VEGFA ARE1-RNA (see Supplemental Figure 3A) as determined by RNA EMSA or UV crosslinking (Figure 5B). In addition, AUF1 antibody supershift assays resulted in retarded migration of protein-ARE complexes when 786-O RCC cytosolic extracts were incubated with VEGFA ARE1 RNA (Figure 5C). While supershift complexes were gained only in the presence of AUF1 antibody, we did not detect an associated loss of protein-ARE RNA complexes. We feel this is due to the likelihood that other ARE RNA binding proteins in addition to AUF1 are present in the 786-O RCC cytosolic extract (e.g., HuR) and that these complexes will persist even after the addition of the AUF1 antibody. The intense protein-RNA complexes seen in the presence of the AUF1 antibody may represent protein-RNA complexes that are stabilized by the antibody. AUF1 bound not only to VEGFA ARE1 but to all VEGFA ARE-RNA riboprobes that were tested (Supplementary Figure 3B and 3C), suggesting further that AUF1 may play a role in VEGFA regulation.
We next determined the functional effects of pVHL-AUF1 interactions by examining the role of AUF1 in regulating VEGFA mRNA stability. VEGFA mRNA levels were low in 786-O G7F RCC cells that were cultured in normoxia but were up-regulated nearly 15-fold when these cells were cultured under hypoxic conditions (Figure 6A). pVHL-negative 786-O 157Δ RCC cells expressed similar VEGFA mRNA levels when cultured either in normoxia or hypoxia (Figure 6A). Since AUF1 and HuR regulate ARE-containing mRNA stability and appear to have opposing effects on VEGFA mRNA levels (Figure 5A), we asked whether these proteins associate with VEGFA mRNA in RCC cells. Both AUF1 and HuR were found to be associated with VEGFA mRNA in 786-O 157Δ RCC cell extracts (Figure 6B). Actin mRNA was not immunoprecipitated by AUF1 or HuR antibodies from these cells under our experimental conditions, and VEGFA mRNA was not amplified from irrelevant IgG nor from anti-pVHL immunoprecipitations (Figure 6B). These results suggest that the RNP-immunoprecipitations did not co-purify mRNAs non-specifically, and that AUF1 and HuR may play a role in controlling VEGFA mRNA levels in RCC cells.
Figure 6.

AUF1 and HuR associate with VEGFA mRNA in vivo and regulate VEGFA mRNA levels.(A) pVHL-positive RCC cells (786-O G7F) and pVHL-negative RCC cells (786-O 157Δ) were cultured either under normoxic conditions (room air) or hypoxic conditions (1% O2) for 18 hours. Total cellular RNA was prepared and quantitative real time RT-PCR reactions were performed with VEGFA-specific PCR primers. VEGFA expression levels were determined in triplicate and averages are expressed relative to 18S rRNA levels. B, RNP-immunoprecipitations were performed from pVHL-negative 786-O 157Δ RCC cells with antibodies directed against AUF1, HuR, or pVHL, or an irrelevant rabbit IgG (rIgG). Immunoprecipitates were washed in lysis buffer, RNA was extracted, and RT-PCR was performed using VEGF-specific or actin-specific PCR primers. Products of PCR reactions were resolved on 2% low molecular weight agarose gels containing ethidium bromide and photographed under UV light. Reverse photographic images are shown. The VEGFA PCR primers amplify products of 567, 495, 363, and 267 base pairs due to expression of alternatively spliced forms of the VEGF mRNA, and the β-actin primers amplify a 600 base pair product (30).
pVHL associates with RNP-ARE RNA complexes under normoxic but not hypoxic conditions
As discussed above, VEGFA mRNA levels are regulated both transcriptionally and post-transcriptionally. In order to focus on post-transcriptional regulation, a heterologous luciferase reporter construct was tested in which VEGFA ARE1 was subcloned downstream of the luciferase open reading frame (Figure 7A). This region contains a consensus AUUUA ARE site as well as (UA)x repeats, which form stem loop structures (25). Luciferase activity was detected in all of the cells that were transiently transfected with the luciferase-ARE1 construct, and when cells that express wild type pVHL (293T cells and WT7 RCC cells) were cultured in hypoxia, luciferase activity was increased, 1.5-fold and 4-fold, respectively (Figure 7B). Luciferase activity was comparable in the pVHL-negative ARZ2 cells when cultured under normoxic or hypoxic conditions (Figure 7B). Luciferase activity of a construct containing a control fragment from the VEGFA 3' UTR lacking ARE sequences (Figure 7A) did not exhibit regulation by pVHL nor by hypoxia (Figure 7B). Luciferase mRNA levels were also determined by RT-PCR. In WT7 RCC cell transfectants luciferase-ARE1 mRNA levels were induced 3- to 5-fold in hypoxia, while luciferase-control mRNA levels or actin mRNA levels were unaffected by hypoxia in those cells (Figure 7C). Therefore, hypoxic induction of luciferase enzymatic activity reflected differences in luciferase mRNA levels.
Figure 7.



Oxygen-dependent and pVHL-dependent expression of luciferase-VEGFA ARE1 constructs. A, A schematic diagram of the proximal portion of the VEGF 3' UTR showing the regions that were independently subcloned into the 3' UTR of the luciferase reporter. Nucleotide numbering is relative to the VEGFA translation termination codon. B, Cell lines 293T, WT7 (pVHL-positive), and ARZ2 (pVHL-negative) were transiently transfected with plasmids containing either VEGF ARE1 (Luc-ARE1) or control sequences (Luc-control). After 24 h transfected cultures were trypsinized and divided evenly into two dishes, and after an additional 6 h of culture, one dish was transferred to a hypoxia (1% O2) incubator, and one dish was maintained in an incubator in room air (normoxia). Cell lysates were harvested after 12 h and luciferase assays were performed. A CMV-β-gal plasmid was co-transfected and β-galactosidase activity was measured and used to normalize for transfection efficiency. *, p=0.00071; **, p=0.0093 (Student's two-tailed t test). C, 293T cells were transiently transfected with pGL3-Promoter luciferase reporter plasmid containing either VEGFA ARE1 (Luc-ARE1) or VEGF control sequences (Luc-control), and cells were cultured under normoxic (room air) or hypoxic (1% O2) conditions as described above. Total RNA was extracted and RT-PCR using luciferase-specific PCR primers was performed. D, 293T cells were transfected and cultured under normoxic (room air) or hypoxic (1% O2) conditions as described above. Actinomycin D (10 μg/ml) was added and total RNA was isolated at the indicated time points. RT-PCR analyses for luciferase mRNA expression were performed and resolved on 2% low molecular weight agarose gels containing ethidium bromide. Band intensities were quantitated using the Quantity One software package (Bio-Rad) and luciferase mRNA levels were plotted relative to the untreated “0” time point. Non-linear regression analyses showed that the luciferase-VEGF ARE1 mRNA had a half-life of 0.8 h in normoxic cells and 3.8 h in hypoxic cells. E, 293T cells were transfected as described above, and RNP-immunoprecipitations were performed with antibodies directed against pVHL, AUF1, HuR, or irrelevant rabbit IgG (rIgG). Immunoprecipitates were washed in RNP- immunoprecipitation buffer, RNA was extracted, and RT-PCR was performed using luciferase-specific PCR primers. Products of PCR reactions were resolved on 2% low molecular weight agarose gels containing ethidium bromide and photographed under UV light. Reverse photographic images are shown. F, 293T cell extracts were prepared using RNP-immunoprecipitation buffer and incubated in the presence of biotinylated TNFα ARE RNA or ARE-mutated RNA. Protein-RNA complexes were isolated on streptavidin paramagnetic beads, washed in lysis buffer, and resolved by 12% SDS-PAGE. Western blots were probed with AUF1 or HuR antibodies. G, Radiolabeled TNFα ARE RNA probe was incubated with cytosolic extracts that were prepared from 293T cells that had been cultured under normoxic (room air) or hypoxic (1% O2) conditions. The protein-RNA complexes were then subjected to UV-crosslinking followed by RNase A+T1 digestion, immunoprecipitation with the indicated antibodies, and electrophoresis on 12% SDS-polyacrylamide gels. Phosphor images were obtained from dried gels. H, A model for pVHL regulation of VEGFA mRNA stability in normoxia through an indirect association of pVHL with AUF1 and HuR. In hypoxia pVHL levels are suppressed and AUF1 and/or HuR binding with VEGFA mRNA is associated with mRNA stability.
Luciferase-ARE1 activity was consistently lower in pVHL-positive RCC cell transfectants as compared to pVHL-negative RCC cells. The SP1 transcription factor binds to GC-boxes contained within the SV40 promoter of this vector and stimulates constitutive luciferase transcription. Since pVHL binds to SP1 and suppresses its activity (16, 36), it is possible that we are observing both transcriptional and post-transcriptional effects in these studies. In order to determine whether pVHL played a role in regulating luciferase-ARE1 mRNA stability in RCC cells, WT7 RCC cells were transfected with the luciferase-ARE1 construct and then cultured in normoxia or hypoxia in the presence of the transcription inhibitor, actinomycin D. RT-PCR and nonlinear regression analyses of the data determined that the half-life of luciferase-ARE1 mRNA was 0.8 h in normoxic WT7 RCC cells and 3.8 h in hypoxic WT7 RCC cells (Figure 7D). Our results suggest that the VEGFA ARE1 sequence functions as a destabilizing element when expressed in the luciferase mRNA 3' UTR, and that this mRNA is stabilized in hypoxic pVHL-expressing RCC cells.
Since the luciferase-ARE1 mRNA exhibited both pVHL- and hypoxia-dependent post-transcriptional regulation, we determined whether AUF1 and HuR associate with the luciferase-ARE1 mRNA in transiently transfected 293T cells. Both AUF1 and HuR antibodies were able to co-immunoprecipitate luciferase mRNA from luciferase-ARE1 transfectants that had been cultured either in normoxia or in hypoxia (Figure 7E). Neither AUF1 nor HuR antibodies immunoprecipitated the luciferase-control mRNA (Figure 7E). We also performed parallel RNP-immunoprecipitations with pVHL antibody and found that pVHL co-immunoprecipitated with luciferase-ARE1 mRNA in 293T cell transfectants that were cultured in normoxia but not when cultured in hypoxia when pVHL levels are suppressed (Figure 7E). Therefore, pVHL is found in a complex with ARE mRNA, but we cannot determine from these studies whether pVHL binds ARE RNA directly or indirectly.
Association of AUF1 or HuR complexes with an independent ARE from the TNFα mRNA was also determined. Both AUF1 and HuR exhibited specific binding to the TNFα ARE RNA but little to no binding to the ARE-mutated RNA that was tested (Figure 7F). We next incubated extracts from normoxic and hypoxic 293T cells with radiolabeled TNFα ARE RNA, subjected the protein-RNA complexes to UV-crosslinking, and performed immunoprecipitations. We found that AUF1, HuR, and pVHL antibodies each co-immunoprecipitated TNFα ARE RNA from from normoxic 293T cell extracts (Figure 7G). In hypoxia, when pVHL levels are suppressed, anti-pVHL did not immunoprecipitate TNFα ARE RNA (Figure 7G). The RNP complexes that were identified in anti-pVHL immunoprecipitations migrated in SDS-polyacrylamide gels between 30–50 kDa and were similar to the RNP complexes seen in AUF1 or HuR immunoprecipitations. These results suggest that pVHL did not bind directly to the TNFα ARE RNA, since we did not detect a signal migrating at ~19 kDa in the anti-pVHL immunoprecipitations. Since pVHL associates with AUF1 (this study) and with HuR (37), our results suggest an indirect association of pVHL with ARE RNA through AUF1 and/or HuR complexes.
Discussion
Our focus in this report was on interactions between pVHL and the ARE RNA binding protein, AUF1. AUF1 protein levels were elevated in the pVHL-negative RCC cells as compared to the isogenic pVHL-positive RCC cells that were tested. In addition, in pVHL-positive cells AUF1 protein levels were increased in hypoxia or with culture in the presence of proteasome inhibitors. pVHL binds to AUF1 through its target protein recognition domain and targeted p45AUF1 and p42AUF1 for ubiquitylation in vitro. In hypoxia, when pVHL expression is suppressed, AUF1 protein levels were elevated. Taken together our results are consistent with a model in which pVHL controls at least some AUF1 isoforms through ubiquitin-mediated proteasomal degradation and that AUF1 proteins may be commonly overexpressed in clear cell RCC with inactivated pVHL.
We also found that AUF1 bound to ARE RNA under both normoxic and hypoxic culture conditions. The association of AUF1 with VEGFA ARE RNA in normoxia is consistent with a destabilizing activity of AUF1. However, since pVHL levels are suppressed in hypoxia (7) and AUF1 levels are increased in hypoxia (Figure 1), our results also indicate that AUF1 binding to ARE RNA may be associated with VEGFA mRNA stability in hypoxia. These results suggest that the presence or absence of pVHL in the ARE-binding complex constitutes at least one determining factor for VEGFA ARE-mediated decay or stability, respectively (Figure 7H). Whether pVHL ubiquitin-ligase activity is required for the targeting of ARE RNA for degradation remains to be determined. This is a difficult question to address experimentally, since uncoupling pVHL from the CRL complex renders pVHL unstable (38).
The relationship between proteasome activity and turnover of ARE-containing mRNA has been explored previously. Proteasome inhibition resulted in suppressed ARE-mediated RNA decay, suggesting the importance of the ubiquitin-proteasome system for mRNA degradation (39). p37AUF1 and p40AUF1 were shown to be targeted for ubiquitylation in response to heat shock (40), and p37AUF1 and p40AUF1, but not p42AUF1 or p45AUF1, were found to be ubiquitylated in vitro in studies that used HeLa cell extracts as a source of E1, E2, and E3 enzymes (41). Although we did not detect pVHL-dependent ubiquitylation of p37AUF1 and p40AUF1 under our experimental conditions (Figure 3), it is clear that the ubiquitin-proteasome system plays a role in ARE-mediated RNA decay.
AUF1 was found to bind ARE RNA in both normoxia and hypoxia. Suppression of AUF1 expression resulted in elevated VEGFA mRNA levels in normoxia, suggesting that AUF1 destabilizes VEGFA mRNA (Figure 5). However, we could not determine the effects of AUF1 suppression on VEGFA mRNA in hypoxia. Since VEGFA mRNA is stabilized in hypoxia (Figures 6 and 7), this would suggest that AUF1 may stabilize this mRNA in hypoxia, or alternatively, AUF1 may play some other regulatory role. For example, the association of AUF1 with VEGFA ARE RNA in hypoxia could provide a mechanism to regulate mRNA translation. AUF1 complexes with translation initiation factor 4G (eIF4G) and poly(A)-binding protein (PABP) (40), suggesting that AUF1 may play a role in translation initiation. AUF1 may stimulate VEGFA translation in a manner similar to its enhancing effect on c-Myc translation by competing for ARE binding with a translation inhibitor, TIAR (42). Since AUF1 protein levels were found to increase in hypoxia (Figure 1), this would provide an advantage for AUF1 binding in competition with other ARE-binding factors. On the other hand, several reports demonstrated that ARE-mediated RNA degradation was dependent on translation (43–45). Association of AUF1 with VEGFA mRNA in hypoxia may limit the amount of VEGFA protein produced or provide a mechanism to rapidly degrade VEGFA mRNA when hypoxic stress is alleviated.
We characterized here pVHL interactions with AUF1. Earlier studies showed that pVHL binds to other ARE-binding proteins: hnRNP A2 (46), HuR (37), and Tis11b (26). The interaction between hnRNP A2 and pVHL required formation of the pVHL CRL complex, and hnRNP A2 protein levels were downregulated by pVHL in RCC cells. These results are similar to those shown in this report for AUF1. Datta et al. (37) found that pVHL modulated binding of HuR to ARE RNA. They concluded that pVHL expression leads to VEGFA mRNA decay by blocking the ARE RNA stabilizing activity of HuR. We did not detect modulation of HuR protein levels in the cell lines tested in our studies (Figure 1), and we detected HuR association with ARE sequences in both pVHL-positive and –negative RCC cells. However, our RNP-immunoprecipitation studies were not quantitative assays, and therefore, we cannot determine whether total levels of HuR or AUF1 that bound to the ARE RNAs tested were modulated by pVHL or hypoxia.
AUF1 and HuR have been shown to bind independently to some mRNAs and concurrently to other mRNAs (32), which was not addressed in our studies. Since AUF1 binds ARE RNA as hexamers (47) and active HuR binds as a homodimer (48), it is possible that higher order complexes containing pVHL, AUF1, and HuR exist. Other proteins may also be involved. For example AUF1 associates with eIF4G and PABP (discussed above), as well as with Hsc70-Hsp70 and Hsp27. This complex has been referred to as an AUF1- and signal transduction-regulated complex (ASTRC), which recruits RNases to degrade ARE-containing RNA (49). Thus, the stoichiometry of protein-protein interactions and protein-ARE RNA interactions may ultimately determine mRNA stability or decay. Stoichiometry will be affected by the relative expression levels of the various proteins, and we predict that events such as pVHL loss in RCC or hypoxic induction of AUF1 would lead to stabilization of ARE-containing mRNAs. In addition, various post-translational modifications, including phosphorylation, ubiquitylation, SUMOylation, and/or methylation, of pVHL, AUF1, HuR, or other interacting partners are likely to play a role in determining ARE-mediated RNA stability or decay.
The relationship between ARE-mediated mRNA stability or decay and miRNAs is not yet clear. Protein-ARE RNA interactions may recruit miRNA-containing RNA-induced silencing complex (RISC) or they may directly block miRNA interactions with target sequences. Protein binding may also influence RNA secondary structure as was recently shown in the VEGFA 3' UTR (25), which could in turn affect miRNA interactions. While our data suggest that VEGFA mRNA stability was mediated through protein-ARE RNA interactions, the relationship between AUF1 and/or HuR association with ARE RNA and miRNAs remains to be explored.
Supplementary Material
Acknowledgements
We thank Dr. Arthur Haas for support and discussions, and Drs. W. Marston Linehan and William G Kaelin for providing RCC cell lines.
Grant Support This work was supported by grants from the National Institutes of Health, CA78335 and CA125931 (J.G.) and CA052443 (G.B.), and the Louisiana Cancer Research Consortium. H.X. and C.G. received support from the Cancer Association of Greater New Orleans and the Stanley S. Scott Cancer Center. This project used the UPCI Genomics Facility and was supported in part by award P30CA047904.
Footnotes
H. Xin, J.A. Brown, and C. Gong contributed equally to this work.
The authors have no conflicts of interest to disclose.
Reference List
- 1.Maher ER, Neumann HPH, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iliopoulos O, Ohh M, Kaelin WG., Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A. 1998;95:11661–6. doi: 10.1073/pnas.95.20.11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stebbins CE, Kaelin WG, Jr., Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–61. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 4.Leonardi E, Murgia A, Tosatto SCE. Adding structural information to the von Hippel-Lindau (VHL) tumor suppressor interaction network. FEBS Letters. 2009;583:3704–10. doi: 10.1016/j.febslet.2009.10.070. [DOI] [PubMed] [Google Scholar]
- 5.Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12:605–20. doi: 10.1038/nrm3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–28. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 7.Liu W, Xin H, Eckert DT, Brown JA, Gnarra JR. Hypoxia and cell cycle regulation of the von Hippel-Lindau tumor suppressor. Oncogene. 2011;30:21–31. doi: 10.1038/onc.2010.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–85. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Molecular Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Roretz C, Marco SD, Mazroui R, Gallouzi I-E. Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wiley Interdisciplinary Reviews: RNA. 2010:n/a–n/a. doi: 10.1002/wrna.55. [DOI] [PubMed] [Google Scholar]
- 11.Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics. 1998;48:195–202. doi: 10.1006/geno.1997.5142. [DOI] [PubMed] [Google Scholar]
- 12.DeMaria CT, Sun Y, Long L, Wagner BJ, Brewer G. Structural determinants in AUF1 required for high affinity binding to A + U-rich elements. J Biol Chem. 1997;272:27635–43. doi: 10.1074/jbc.272.44.27635. [DOI] [PubMed] [Google Scholar]
- 13.Zucconi BE, Wilson GM. Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Front Biosci. 2011;17:2307–25. doi: 10.2741/3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danilin S, Sourbier C, Thomas L, Lindner V, Rothhut S, Dormoy V, et al. Role of the RNA-binding protein HuR in human renal cell carcinoma. Carcinogenesis. doi: 10.1093/carcin/bgq052. bgq052. [DOI] [PubMed] [Google Scholar]
- 15.Rini BI. New Strategies in Kidney Cancer: Therapeutic Advances through Understanding the Molecular Basis of Response and Resistance. Clin Cancer Res. 2010;16:1348–54. doi: 10.1158/1078-0432.CCR-09-2273. [DOI] [PubMed] [Google Scholar]
- 16.Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S, Sukhatme VP. The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol Cell Biol. 1997;17:5629–39. doi: 10.1128/mcb.17.9.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pal S, Claffey KP, Cohen HT, Mukhopadhyay D. Activation of Sp1-mediated vascular permeability factor/vascular endothelial growth factor transcription requires specific interaction with protein kinase C zeta. J Biol Chem. 1998;273:26277–80. doi: 10.1074/jbc.273.41.26277. [DOI] [PubMed] [Google Scholar]
- 18.Stein I, Neeman M, Shweiki D, Itin A, Keshet E. Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Molecular & Cellular Biology. 1995;15:5363–8. doi: 10.1128/mcb.15.10.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy AP, Levy NS, Goldberg MA. Hypoxia-inducible protein binding to vascular endothelial growth factor mRNA and its modulation by the von Hippel-Lindau protein. J Biol Chem. 1996;271:25492–7. doi: 10.1074/jbc.271.41.25492. [DOI] [PubMed] [Google Scholar]
- 20.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–53. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- 21.Levy AP. Hypoxic Regulation of VEGF mRNA Stability by RNA-binding Proteins. Trends in Cardiovascular Medicine. 1998;8:246–50. doi: 10.1016/s1050-1738(98)00020-6. [DOI] [PubMed] [Google Scholar]
- 22.Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114:2015–9. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- 23.Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273:6417–23. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 24.Shih SC, Claffey KP. Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J Biol Chem. 1999;274:1359–65. doi: 10.1074/jbc.274.3.1359. [DOI] [PubMed] [Google Scholar]
- 25.Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457:915–9. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciais D, Cherradi N, Bailly S, Grenier E, Berra E, Pouyssegur J, et al. Destabilization of vascular endothelial growth factor mRNA by the zinc-finger protein TIS11b. Oncogene. 2004;23:8673–80. doi: 10.1038/sj.onc.1207939. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, et al. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol. 1993;13:7652–65. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy NS, Goldberg MA, Levy AP. Sequencing of the human vascular endothelial growth factor (VEGF) 3' untranslated region (UTR): conservation of five hypoxia-inducible RNA-protein binding sites. Biochim Biophys Acta. 1997;1352:167–73. doi: 10.1016/s0167-4781(97)00052-3. [DOI] [PubMed] [Google Scholar]
- 29.Mukherjee D, Gao M, O'Connor JP, Raijmakers R, Pruijn G, Lutz CS, et al. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. Embo J. 2002;21:165–74. doi: 10.1093/emboj/21.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gnarra JR, Zhou S, Merrill MJ, Wagner JR, Krumm A, Papavassiliou E, et al. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93:10589–94. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy AP, Levy NS, Loscalzo J, Calderone A, Takahashi N, Yeo KT, et al. Regulation of vascular endothelial growth factor in cardiac myocytes. Circ Res. 1995;76:758–66. doi: 10.1161/01.res.76.5.758. [DOI] [PubMed] [Google Scholar]
- 32.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. Embo J. 2004;23:3092–102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 34.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91:9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg-Cohen I, Furneauxb H, Levy AP. A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J Biol Chem. 2002;277:13635–40. doi: 10.1074/jbc.M108703200. [DOI] [PubMed] [Google Scholar]
- 36.Cohen HT, Zhou M, Welsh AM, Zarghamee S, Scholz H, Mukhopadhyay D, et al. An important von Hippel-Lindau tumor suppressor domain mediates Sp1-binding and self-association. Biochem Biophys Res Commun. 1999;266:43–50. doi: 10.1006/bbrc.1999.1767. [DOI] [PubMed] [Google Scholar]
- 37.Datta K, Mondal S, Sinha S, Li J, Wang E, Knebelmann B, et al. Role of elongin-binding domain of von Hippel Lindau gene product on HuR-mediated VPF/VEGF mRNA stability in renal cell carcinoma. Oncogene. 2005;24:7850–8. doi: 10.1038/sj.onc.1208912. [DOI] [PubMed] [Google Scholar]
- 38.Kamura T, Brower CS, Conaway RC, Conaway JW. A molecular basis for stabilization of the von Hippel-Lindau (VHL) tumor suppressor protein by components of the VHL ubiquitin ligase. J Biol Chem. 2002;277:30388–93. doi: 10.1074/jbc.M203344200. [DOI] [PubMed] [Google Scholar]
- 39.Laroia G, Sarkar B, Schneider RJ. Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1842–6. doi: 10.1073/pnas.042575699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laroia G, Cuesta R, Brewer G, Schneider RJ. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science. 1999;284:499–502. doi: 10.1126/science.284.5413.499. [DOI] [PubMed] [Google Scholar]
- 41.Laroia G, Schneider RJ. Alternate exon insertion controls selective ubiquitination and degradation of different AUF1 protein isoforms. Nucleic Acids Research. 2002;30:3052–8. doi: 10.1093/nar/gkf444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao B, Hu Y, Brewer G. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat Struct Mol Biol. 2007;14:511–8. doi: 10.1038/nsmb1249. [DOI] [PubMed] [Google Scholar]
- 43.Savant-Bhonsale S, Cleveland DW. Evidence for instability of mRNAs containing AUUUA motifs mediated through translation-dependent assembly of a > 20S degradation complex. Genes Dev. 1992;6:1927–39. doi: 10.1101/gad.6.10.1927. [DOI] [PubMed] [Google Scholar]
- 44.Winstall E, Gamache M, Raymond V. Rapid mRNA degradation mediated by the c-fos 3' AU-rich element and that mediated by the granulocyte-macrophage colony-stimulating factor 3' AU-rich element occur through similar polysome-associated mechanisms. Mol Cell Biol. 1995;15:3796–804. doi: 10.1128/mcb.15.7.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curatola AM, Nadal MS, Schneider RJ. Rapid degradation of AU-rich element (ARE) mRNAs is activated by ribosome transit and blocked by secondary structure at any position 5' to the ARE. Mol Cell Biol. 1995;15:6331–40. doi: 10.1128/mcb.15.11.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pioli PA, Rigby WF. The von Hippel-Lindau protein interacts with heteronuclear ribonucleoprotein a2 and regulates its expression. J Biol Chem. 2001;276:40346–52. doi: 10.1074/jbc.M105391200. [DOI] [PubMed] [Google Scholar]
- 47.DeMaria CT, Sun Y, Wagner BJ, Long L, Brewer GA. Structural determination in AUF1 required for high affinity binding to A + U-rich elements. Nucleic Acids Symp Ser. 1997:12–4. [PubMed] [Google Scholar]
- 48.Meisner NC, Hintersteiner M, Mueller K, Bauer R, Seifert JM, Naegeli HU, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol. 2007;3:508–15. doi: 10.1038/nchembio.2007.14. [DOI] [PubMed] [Google Scholar]
- 49.Sinsimer KS, Gratacos FM, Knapinska AM, Lu J, Krause CD, Wierzbowski AV, et al. Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol Cell Biol. 2008;28:5223–37. doi: 10.1128/MCB.00431-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270:13333–40. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.