

Abstract
Preeclampsia is a pregnancy-induced hypertensive disorder that causes substantial maternal and fetal morbidity and mortality. Despite being a leading cause of maternal death and a major contributor to maternal and perinatal morbidity, the mechanisms responsible for the pathogenesis of preeclampsia are poorly understood. Recent studies indicate that women with preeclampsia have autoantibodies that activate the angiotensin receptor, AT1, and that autoantibody-mediated receptor activation contributes to pathophysiology associated with preeclampsia. The research reviewed here raises the intriguing possibility that preeclampsia may be a pregnancy-induced autoimmune disease.
Preeclampsia12 affects ~3–5% of all pregnancies. It is a multisystem disorder generally appearing after the 20th week of gestation and characterized by hypertension, proteinuria, vascular abnormalities, and often intrauterine growth retardation (1–3). Preeclampsia is a leading cause of maternal death and a major contributor to maternal and perinatal morbidity. The mechanisms responsible for the pathogenesis of preeclampsia are poorly understood. The only effective treatment is delivery of the fetus and placenta, often resulting in serious complications of prematurity for the neonate. Immune mechanisms have long been implicated in the pathogenesis of preeclampsia (4, 5). The results presented here provide additional support for this view by reviewing the evidence that women with preeclampsia have autoantibodies capable of activating the angiotensin AT1 receptor (6, 7). AT1 receptor agonistic Abs, herein termed AT1-AA,3 are rarely seen in normotensive pregnant women. Overall, these results raise the intriguing possibility that preeclampsia may be an autoimmune disease in which pathophysiological symptoms result from autoantibody-induced angiotensin receptor activation. These autoantibodies may represent novel therapeutic targets for preeclampsia.
Initial evidence for angiotensin receptor-activating autoantibodies in women with preeclampsia
The autoantibodies were originally detected by Wallukat et al. based on the ability to activate AT1 angiotensin receptors on cultured neonatal rat cardiac myocytes, resulting in increased cardiomyocyte contraction rates (6). They showed that AT1-AA increase the beating rate of the cultured cardiomyocytes, a feature that was blocked by AT1 receptor antagonists. Using affinity purification and peptide competition experiments, they showed that AT1-AA bind to a seven-amino acid sequence present on the second extracellular loop of the AT1 receptor. The presence of this peptide epitope, AFHYESQ, in the cardiomyocyte contraction assay blocked Ab-induced stimulation of cardiomyocyte contraction. These remarkable findings were the first to show that preeclamptic women possess stimulatory autoantibodies against the AT1 receptor and that these autoantibodies are directed to a common epitope associated with the second extracellular loop.
AT1-AA may contribute to multiple features of preeclampsia
In subsequent studies we showed (7, 8) that these autoantibodies activate AT1 receptors on human trophoblasts, resulting in increased synthesis and secretion of plasminogen activator inhibitor-1 (PAI-1). PAI-1 plays a role in trophoblast invasion by inhibiting the urokinase-type plasminogen activator, resulting in decreased conversion of plasminogen to plasmin, decreased extracellular matrix digestion, and shallow trophoblast invasion. We have also shown that AT1-AA activate AT1 receptors on cultured human mesangial cells resulting in the stimulation of PAI-1 synthesis and secretion, a feature that may contribute to kidney damage leading to proteinuria, a hallmark manifestation of preeclampsia (9). Increased PAI-1 production by trophoblast cells, mesangial cells, and possibly other cell types may contribute to the hypercoagulation sometimes associated with preeclampsia. Dechend et al. provided evidence that AT1-AA stimulate increased production of tissue factor by endothelial cells (10) and NADPH oxidase by vascular smooth muscle cells and trophoblast cells (11), features that may play a role in vascular injury and oxidative stress, respectively. Thus, available evidence indicates that AT1-AA activate AT1 receptors on a variety of cells and provoke biological responses that are relevant to the pathophysiology of preeclampsia (Fig. 1).
FIGURE 1.

AT1-AA may underlie many features of preeclampsia by interacting with AT1 receptors on different cell types. AT1-AA from preeclamptic patients function as Ang II in the activation of AT1 receptors at the surface of many cell types. Autoantibody-induced AT1 receptor activation results in increased contraction rates in cardiac myocytes, increased production of NADPH oxidase by trophoblast and vascular smooth muscle cells, PAI-1, sFlt-1, and NADPH oxidase by trophoblast cells, PAI-1 and IL-6 production by mesangial cells, and tissue factor by endothelial cells. AT1-AA-mediated AT1 receptor activation also results in the mobilization of intracellular calcium and the activation of NFAT-responsive genes. We propose that AT1-AA activate AT1 receptors on other cell types, resulting in the physiological changes associated with preeclampsia. Note that increased NADPH oxidase activity leads to increased production of reactive oxygen species (ROS). SMC, Smooth muscle cell; EC, endothelial cell.
Pregnancy is characterized by significant changes in the abundance of angiogenic factors such as vascular endothelial growth factor and placental growth factor and their antagonist, a soluble form of the vascular endothelial growth factor receptor termed soluble fms-like tyrosine kinase-1 (sFlt-1) (12, 13). The major source of sFlt-1 during pregnancy is the placenta, where angiotensin II (Ang II) stimulates increased synthesis and secretion of sFlt-1 by trophoblast cells late in pregnancy (14). sFlt-1 is significantly elevated in the plasma of woman with preeclampsia in comparison with that of normotensive pregnant women (12, 13, 15–18) and is believed to contribute to elevated blood pressure, proteinuria, and glomerular endotheliosis, classic features of preeclampsia (19). We have recently shown that IgG from women with preeclampsia induces the synthesis and secretion of sFlt-1 by human placental villous explants and human trophoblast cells (22). The secreted sFlt-1 has significant antiangiogenic properties as judged by its impact on in vitro endothelial cell migration and tube formation assays. The introduction of IgG from preeclamptic patients into pregnant mice resulted in increased secretion of sFlt-1 (C. Zhou, Y.J. Zhang, R. Irani, T.J. Mi, R.E. Kellems and Y. Xia, unpublished observations). Autoantibody-induced sFlt-1 secretion resulted from AT1 receptor activation and downstream signaling through the calcineurin-NFAT pathway, leading to Flt-1 transcriptional up-regulation. We also found that Ang II and AT1-AA can function additively to induce sFlt-1 secretion through AT1 receptor activation. Our findings suggest that Ang II is a key regulator of sFlt-1 synthesis and secretion during normal pregnancy (14) and that the excessive accumulation of sFlt-1 observed in women with preeclampsia is due to the additional activation of AT1 receptors mediated by AT1-AA (Fig. 2)
FIGURE 2.

Proposed model of Ang II- and AT1-AA-induced sFlt-1 secretion in normal pregnancy and in preeclampsia. The synthesis and secretion of sFlt-1 is increased late in a normal pregnancy through the action of Ang II (14). We have suggested that the antiangiogenic action of elevated sFlt-1 functions as a brake to inhibit continuing angiogenesis late in pregnancy (14). However, under preeclamptic conditions the additional activation of the AT1 receptor by the maternal circulating AT1-AA results in additional sFlt-1 secretion over that stimulated by Ang II alone. The excessive placenta-derived sFlt-1 has detrimental effects on placental development and maternal vascular and renal function. Our recent results (22) suggest that the excessive production of sFlt-1 can be prevented by the seven-amino acid epitope peptide and that this peptide may have therapeutic potential in the management of preeclampsia. 7-mer, Seven-amino acid epitope peptide.
AT1-AA cross the placenta and enter the fetal circulation
The transfer of Ig from the mother to the fetus through the placenta is a naturally occurring process by which the mother provides passive immunity to the developing fetus. Hence, the opportunity exists for AT1-AA to enter the fetal circulation. Recent findings from Herse et al. provide preliminary evidence to support this expectation (20). Using the neonatal rat cardiomyocyte beating assay, they reported that the offspring of preeclamptic women with AT1-AA also contain the autoantibodies capable of activating the AT1 receptor. Infants born to normotensive pregnant women do not have AT1-AA. In agreement with these findings we have observed AT1-AA in the cord blood of women with preeclampsia (C. Zhou, unpublished observations). AT1-AA was not observed in the cord blood of normotensive pregnant women. Overall, these studies indicate that AT1-AA cross the placenta and enter the fetal circulation. The presence of these biologically active autoantibodies may have detrimental effects on fetal growth and development.
Detection of AT1-AA in pregnant women represents a potentially important presymptomatic risk factor
Important recent work by Walther et al. shows that AT1-AA can be detected before the 20th wk of gestation in women with impaired uterine perfusion by Doppler sonography (21). Using the cardiomyocyte contraction assay they found that AT1-AA were detected between the 18th and 22nd wk of gestation in women with reduced uterine perfusion. When followed to term, these women fell into three groups: those who developed preeclampsia, those characterized by fetuses with intrauterine growth retardation, and those with otherwise normal outcomes. AT1-AA was not observed in second-trimester women with a normal Doppler ultrasound. Thus, AT1-AA tracks women showing reduced uterine perfusion pressure during the second trimester and may serve to identify women at risk for intrauterine growth retardation and/or preeclampsia. The authors of this study suggested, as we had done earlier (7), that AT1-AA may be responsible for reduced trophoblast invasion and impaired placental development. The fact that AT1-AA can be detected many weeks before the symptoms of preeclampsia has significant implications regarding presymptomatic identification of women at risk for preeclampsia.
Therapeutic possibilities based on blocking autoantibody-induced receptor activation
Currently there is no specific treatment for preeclampsia, and severe cases often require premature delivery of the infant. If maternal circulating AT1-AA contribute to the pathophysiology of preeclampsia, as suggested by the data reviewed here, then blocking the action of these autoantibodies may provide significant therapeutic benefit. One approach to this is based on in vitro studies showing that AT1-AA recognize a specific seven-amino acid sequence present on the second extracellular loop of the AT1 receptor (Fig. 1) and that the ability of AT1-AA to activate AT1 receptors on various cell types can be blocked by the presence of this heptapeptide epitope (6, 7). Our recent studies show that this peptide can neutralize AT1-AA in vivo and thereby prevent autoantibody-induced sFlt-1 production in pregnant mice (22). Thus, the use of epitope peptide therapy to block the action of angiotensin receptor-activating autoantibodies has the potential of being a safe and effective treatment of preeclampsia (Fig. 1).
Mechanism of Ab induced receptor activation
G protein-coupled receptors (GPCRs) were long considered to function as monomers. However, a large body of biochemical evidence published in recent years clearly indicates that GPCRs form homodimers, heterodimers, and possibly higher order oligomeric structures (23–25). Agonist-induced dimerization has been shown for a number of GPCRs, a feature that is consistent with recent evidence that GPCR dimers associate with a single heterotrimeric G protein complex. One of the best-studied GPCRs is the angiotensin receptor AT1. AbdAlla et al. have shown that the active form of the AT1 receptor is a homodimer and that homodimer formation is associated with enhanced Ang II responsiveness (26–28). Because AT1-AAs are bivalent, we propose that they exert their agonistic effect by cross-linking and thereby stabilizing AT1 receptor homodimers (Fig. 1). A thorough knowledge of the mechanism of Ab-induced receptor activation may provide insight that is useful in developing therapeutic strategies to block the action of the Abs, thereby reducing the detrimental effects of excessive AT1 receptor activation.
Placenta ischemia and hypoxia result in endovascular damage leading to a maternal inflammatory response; a possible foundation for autoantibody production in preeclampsia?
The etiology of autoimmune disease remains largely unknown. Multiple factors, including genetic predisposition, a maladaptive immune system, and environment challenge have been proposed to be involved in autoantibody production (29–31). Evidence presented below suggests that the generation of AT1-AA may be secondary to the reduced placental perfusion and increased maternal inflammatory response that is associated with preeclampsia.
It is a widely held view that the maternal syndrome of preeclampsia is secondary to placental abnormalities, especially those resulting from placental ischemia (1, 32). In this regard Granger and colleagues have developed a rat model of preeclampsia based on experimentally induced placental ischemia resulting from reduced uterine perfusion pressure (RUPP) (3, 33–35). Such experimentally manipulated pregnant rats developed hypertension, proteinuria, and other features of preeclampsia. Granger and colleagues also investigated the placentas and maternal circulation of rats with experimentally induced RUPP and found that TNF-α expression was elevated several fold (36). More recently, this group found that sera from RUPP-manipulated pregnant rats stimulated increased endothelin synthesis by endothelial cells through AT1 receptor activation (35). Additionally, in collaboration with the Berlin group, Granger and colleagues examined these rat models of preeclampsia for the presence of AT1-AA. Using the cardiomyocyte contraction bioassay, they observed AT1 receptor agonistic autoantibodies in RUPP pregnant rats but not in normal pregnant rats (37). They found that a low dose TNF-α infusion in pregnant rats also resulted in increased blood pressure and the appearance of AT1 receptor agonistic autoantibodies (34). Neither feature was induced by TNF-α infusion into nonpregnant rats, suggesting a role for pregnancy in the production of AT1-AA. These experimentally induced models of preeclampsia in the pregnant rat may provide a valuable model system to determine the immunological origins and pathophysiological role of AT1-AA in preeclampsia (Fig. 3).
FIGURE 3.
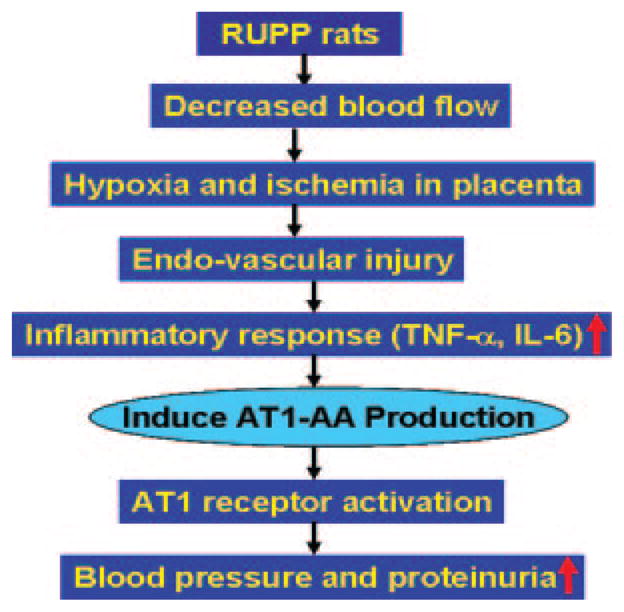
A model to account for autoantibody induction during pregnancy. AT1-AA production is preceded by a maternal inflammatory response to placental ischemia. The decreased blood flow to the placenta in RUPP rats leads to placental ischemia and hypoxia. This may result in endovascular damage, leading to a maternal inflammatory response associated with the increased secretion of inflammatory cytokines (i.e., TNF-α and IL-6). The resulting inflammatory cytokine secretion contributes to AT1-AA production. AT1-AA will directly induce higher blood pressure and proteinuria via AT1 receptor activation. This eventually leads to more hypoxia, endovascular damage, and enhanced inflammatory response, further favoring autoantibody production.
AT1-AA in malignant hypertension and renal allograft rejection
AT1 receptor-activating autoantibodies are not restricted to pregnancy. They have also been associated with other forms of hypertension (38, 39). More recently, AT1-AA were observed in kidney transplant recipients suffering from severe vascular rejection and malignant hypertension (40). Thus, these auto-antibodies have appeared in multiple clinical circumstances associated with hypertension and vascular injury.
Other examples of Ab-mediated receptor activation
Autoantibodies capable of activating GPCRs have also been observed in other human diseases (Table I). One of the best known examples is Graves’ disease, a condition characterized by the presence of autoantibodies capable of activating the thyroid-stimulating hormone receptor on thyroid cells, resulting in the excessive production and secretion of thyroid hormones (41). Agonistic autoantibodies directed against the insulin receptor are associated with some forms of hypoglycemia (42). The autoantibodies directed against the cardiac β1-adrenergic receptor are associated with patients with dilated cardiomyopathy (43). These IgG Abs function as agonists for the β1-adrenergic receptor, resulting in a positive chronotropic effect on cultured cardiomyocytes. Jahns et al. used a rat model to provide direct evidence for a β1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy (44). In a follow-up paper this group showed that in humans these Abs are associated with increased cardiovascular mortality (45). Autoantibodies capable of activating the α1-adrenergic receptor are associated with various forms of hypertension (46, 47). Finally, autoantibodies capable of activating the muscarinic M2 receptor are associated with idiopathic dilated cardiomyopathy (48). Thus, the presence of agonistic autoantibodies directed against GPCRs has been observed in numerous disease states.
Table I.
Examples of autoantibody-mediated receptor activation
| Receptor (Reference) | Disease |
|---|---|
| Thyroid hormone-stimulating receptor (41) | Hyperthyroidism (Grave’s disease) |
| Insulin receptor (42) | Hypoglycemia |
| β1-Adrenergic receptor (43) | Dilated cardiomyopathy |
| α1-Adrenergic receptor (47) | Hypertension |
| Muscarinic M2 receptor (48) | Idiopathic dilated cardiomyopathy |
| Angiotensin AT-1 receptor (6–8) | Preeclampsia |
Concluding remarks
In 1999 Wallukat et al. reported their remarkable findings that sera from women with preeclampsia contain autoantibodies that react with AT1 receptors in a stimulatory fashion (6). Within a few years their important findings were confirmed and extended in numerous ways, showing that these autoantibodies activate AT1 receptors on cardiac myocytes, trophoblast cells, endothelial cells, mesangial cells, vascular smooth cells, and Chinese hamster ovary cells (Fig. 1). Altogether, these studies show that AT1-AA activate AT-1 receptors on a variety of cell types and provoke biological responses that are relevant to the pathophysiology of preeclampsia. We have recently shown that the introduction of these autoantibodies into pregnant mice resulted in the increased secretion of sFlt-1 (22) as well as other key features of preeclampsia, including hypertension and proteinuria (C. Zhou, unpublished observations). Available evidence indicates that the biological properties of these autoantibodies can be blocked by a seven-amino acid peptide that corresponds to a specific epitope associated with the second extracellular loop of the AT1 receptor. This fact has immediate therapeutic implications and also suggests a common immunological origin for these autoantibodies in different individuals. If AT1-AA play a significant role in the etiology and pathophysiology of preeclampsia, as we hypothesize, it may one day be possible to block this Ab response and thus either forestall or prevent the onset of preeclampsia.
Footnotes
This work was supported by National Institutes of Health Grants HL076558 (to Y.X) and HD34130 (to R.E.K) and grants from the March of Dimes Foundation (6-FY06-323) and the Texas Higher Education Coordinating Board (to R.E.K.).
Abbreviations used in this paper: AT1-AA, AT1 receptor agonistic antibody; Ang II, angiotensin II; GPCR, G protein-coupled receptor; PAI-1, plasminogen activator inhibitor-1; sFlt-1, soluble fms-like tyrosine kinase-1; RUPP, reduced uterine perfusion pressure.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Redman CW, I, Sargent L. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 2.Roberts JM. Preeclampsia: what we know and what we do not know. Semin Perinatol. 2000;24:24–28. doi: 10.1016/s0146-0005(00)80050-6. [DOI] [PubMed] [Google Scholar]
- 3.Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension. 2001;38:718–722. doi: 10.1161/01.hyp.38.3.718. [DOI] [PubMed] [Google Scholar]
- 4.Gleicher N. Why much of the pathophysiology of preeclampsia-eclampsia must be of an autoimmune nature. Am J Obstet Gynecol. 2007;196:5.e1–7. doi: 10.1016/j.ajog.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Sargent IL, Borzychowski AM, Redman CW. Immunoregulation in normal pregnancy and preeclampsia: an overview. Reprod Biomed Online. 2006;13:680–686. doi: 10.1016/s1472-6483(10)60659-1. [DOI] [PubMed] [Google Scholar]
- 6.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia Y, Wen HY, Bobst S, Day MC, Kellems RE. Maternal autoantibodies from preeclampsia patients activate angiotensin receptors on human trophoblast cells. J Soc Gyenocol Investig. 2003;10:82–93. doi: 10.1016/s1071-5576(02)00259-9. [DOI] [PubMed] [Google Scholar]
- 8.Xia Y, Wen HY, Kellems RE. Angiotensin II inhibits human trophoblast invasiveness through AT-1 receptor activation. J Biol Chem. 2002;277:24601–24608. doi: 10.1074/jbc.M201369200. [DOI] [PubMed] [Google Scholar]
- 9.Bobst SM, Day MC, Gilstrap LC, III, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. Am J Hypertens. 2005;18:330–336. doi: 10.1016/j.amjhyper.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 11.Dechend R, Viedt C, Muller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107:1632–1639. doi: 10.1161/01.CIR.0000058200.90059.B1. [DOI] [PubMed] [Google Scholar]
- 12.Chaiworapongsa T, Romero R, Espinoza J, Bujold E, Mee Y, Kim L, Goncalves F, Gomez R, Edwin S. Evidence supporting a role for blockade of the vascular endothelial growth factor system in the pathophysiology of preeclampsia. Young Investigator Award. Am J Obstet Gynecol. 2004;190:1541–1547. doi: 10.1016/j.ajog.2004.03.043. discussion 1547–1550. [DOI] [PubMed] [Google Scholar]
- 13.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 14.Zhou CC, Ahmad S, Mi T, Xia L, Abbasi S, Hewett PW, Sun CX, Ahmed A, Kellems RE, Xia Y. Angiotensin II induces soluble fms-like tyrosine kinase-1 (sFlt-1) release via calcineurin signaling pathway in pregnancy. Circ Res. 2007;100:88–95. doi: 10.1161/01.RES.0000254703.11154.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaiworapongsa T, Romero R, Kim YM, Kim GJ, Kim MR, Espinoza J, Bujold E, Goncalves L, Gomez R, Edwin S, Mazor M. Plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated prior to the clinical diagnosis of preeclampsia. J Matern Fetal Neonatal Med. 2005;17:3–18. doi: 10.1080/14767050400028816. [DOI] [PubMed] [Google Scholar]
- 16.Koga K, Osuga Y, Yoshino O, Hirota Y, Ruimeng X, Hirata T, Takeda S, Yano T, Tsutsumi O, Taketani Y. Elevated serum soluble vascular endothelial growth factor receptor 1 (sVEGFR-1) levels in women with preeclampsia. J Clin Endocrinol Metab. 2003;88:2348–2351. doi: 10.1210/jc.2002-021942. [DOI] [PubMed] [Google Scholar]
- 17.Nagamatsu T, Fujii T, Kusumi M, Zou L, Yamashita T, Osuga Y, Momoeda M, Kozuma S, Taketani Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology. 2004;145:4838–4845. doi: 10.1210/en.2004-0533. [DOI] [PubMed] [Google Scholar]
- 18.Tsatsaris V, Goffin F, Munaut C, Brichant JF, Pignon MR, Noel A, Schaaps JP, Cabrol D, Frankenne F, Foidart JM. Overexpression of the soluble vascular endothelial growth factor receptor in preeclamptic patients: pathophysiological consequences. J Clin Endocrinol Metab. 2003;88:5555–5563. doi: 10.1210/jc.2003-030528. [DOI] [PubMed] [Google Scholar]
- 19.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herse F, Dechend R, Harsem NK, Wallukat G, Janke J, Qadri F, Hering L, Muller DN, Luft FC, Staff AC. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension. 2007;49:604–611. doi: 10.1161/01.HYP.0000257797.49289.71. [DOI] [PubMed] [Google Scholar]
- 21.Walther T, Wallukat G, Jank A, Bartel S, Schultheiss HP, Faber R, Stepan H. Angiotensin II type 1 receptor agonistic antibodies reflect fundamental alterations in the uteroplacental vasculature. Hypertension. 2005;46:1275–1279. doi: 10.1161/01.HYP.0000190040.66563.04. [DOI] [PubMed] [Google Scholar]
- 22.Zhou CC, Ahmad A, Xia L, Day MC, Ramin S, Kellems RE, Xia Y. Upregulation of placental soluble fms-like tyrosine kinase 1 (sFlt-) by AT1-receptor agonistic autoantibodies in preeclampsia. Hypertens Pregnancy. 2006;25:38. [Google Scholar]
- 23.Bai M. Dimerization of G-protein-coupled receptors: roles in signal transduction. Cell Signal. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- 24.Breitwieser GE. G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Circ Res. 2004;94:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- 25.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness (see comment) Nat Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 27.AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell. 2004;119:343–354. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 28.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407:94–98. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 29.Pearce SH, Merriman TR. Genetic progress towards the molecular basis of autoimmunity. Trends Mol Med. 2006;12:90–98. doi: 10.1016/j.molmed.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barak Y. The immune system and happiness. Autoimmun Rev. 2006;5:523–527. doi: 10.1016/j.autrev.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Roberts JM, Lain KY. Recent insights into the pathogenesis of preeclampsia. Placenta. 2002;23:359–372. doi: 10.1053/plac.2002.0819. [DOI] [PubMed] [Google Scholar]
- 33.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension. 2006;48:711–716. doi: 10.1161/01.HYP.0000238442.33463.94. [DOI] [PubMed] [Google Scholar]
- 34.Dechend R, Llinas M, Caluwaerts S, Herse F, Lamarca B, Mueller DN, Luft FC, Pijnenborg R, Wallukat G, Granger JP. Agonistic autoantibodies to the AT1 receptor in rat models of preeclampsia: induced by chronic reduction in uterine perfusion pressure (RUPP) and low dose TNF-α infusion. Hypertens Pregnancy. 2006;25:70. Abstr. [Google Scholar]
- 35.Roberts L, LaMarca BB, Fournier L, Bain J, Cockrell K, Granger JP. Enhanced endothelin synthesis by endothelial cells exposed to sera from pregnant rats with decreased uterine perfusion. Hypertension. 2006;47:615–618. doi: 10.1161/01.HYP.0000197950.42301.dd. [DOI] [PubMed] [Google Scholar]
- 36.LaMarca BB, Bennett WA, Alexander BT, Cockrell K, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-α. Hypertension. 2005;46:1022–1025. doi: 10.1161/01.HYP.0000175476.26719.36. [DOI] [PubMed] [Google Scholar]
- 37.Llinas M, Wallukat G, Dechend R, Mueller DN, Luft FC, Alexander BT, Lamarca BB, Granger JP. Agonistic autoantibodies to the AT1 receptor in a rat model of preeclampsia induced by chronic reductions in uterine perfusion pressure (RUPP) Hypertension. 2005;46:883. Abstr. [Google Scholar]
- 38.Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjogren KG, Hjalmarson A, Muller-Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18:945–953. doi: 10.1097/00004872-200018070-00017. [DOI] [PubMed] [Google Scholar]
- 39.Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1-receptor and α1-adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25:641–646. doi: 10.1291/hypres.25.641. [DOI] [PubMed] [Google Scholar]
- 40.Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, Kintscher U, Rudolph B, Hoebeke J, Eckert D, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection (see comment) N Engl J Med. 2005;352:558–569. doi: 10.1056/NEJMoa035717. [DOI] [PubMed] [Google Scholar]
- 41.Chistiakov DA. Thyroid-stimulating hormone receptor and its role in Graves’ disease. Mol Genet Metab. 2003;80:377–388. doi: 10.1016/j.ymgme.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Dozio N, Sarugeri E, Scavini M, Beretta A, Belloni C, Dosio F, Savi A, Fazio F, Sodoyez-Goffaux F, Pozza G. Insulin receptor antibodies inhibit insulin uptake by the liver: in vivo 123I-insulin scintigraphic scanning and in vitro characterization in autoimmune hypoglycemia. J Investig Med. 2001;49:85–92. doi: 10.2310/6650.2001.34094. [DOI] [PubMed] [Google Scholar]
- 43.Wallukat G, Wollenberger A, Morwinski R, Pitschner HF. Anti-β1-adrenoceptor autoantibodies with chronotropic activity from the serum of patients with dilated cardiomyopathy: mapping of epitopes in the first and second extracellular loops.[Published erratum appears in 1995 J. Mol. Cell. Cardiol. 27: 2529.] J Mol Cell Cardiol. 1995;27:397–406. doi: 10.1016/s0022-2828(08)80036-3. [DOI] [PubMed] [Google Scholar]
- 44.Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, Lohse MJ. Direct evidence for a β1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy (see comment) J Clin Invest. 2004;113:1419–1429. doi: 10.1172/JCI20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jahns R, Boivin V, Lohse MJ. β1-Adrenergic receptor-directed auto-immunity as a cause of dilated cardiomyopathy in rats. Int J Cardiol. 2006;112:7–14. doi: 10.1016/j.ijcard.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 46.Fu ML, Herlitz H, Wallukat G, Hilme E, Hedner T, Hoebeke J, Hjalmarson A. Functional autoimmune epitope on α1-adrenergic receptors in patients with malignant hypertension. Lancet. 1994;344:1660–1663. doi: 10.1016/s0140-6736(94)90456-1. [DOI] [PubMed] [Google Scholar]
- 47.Luther HP, Homuth V, Wallukat G. α1-Adrenergic receptor antibodies in patients with primary hypertension. Hypertension. 1997;29:678–682. doi: 10.1161/01.hyp.29.2.678. [DOI] [PubMed] [Google Scholar]
- 48.Wallukat G, Fu HM, Matsui S, Hjalmarson A, Fu ML. Autoantibodies against M2 muscarinic receptors in patients with cardiomyopathy display non-desensitized agonist-like effects. Life Sci. 1999;64:465–469. doi: 10.1016/s0024-3205(98)00589-x. [DOI] [PubMed] [Google Scholar]