

Abstract
Must higher level biological processes always be derivable from lower level data and mechanisms, as assumed by the idea that an organism is completely defined by its genome? Or are higher level properties necessarily also causes of lower level behaviour, involving actions and interactions both ways? This article uses modelling of the heart, and its experimental basis, to show that downward causation is necessary and that this form of causation can be represented as the influences of initial and boundary conditions on the solutions of the differential equations used to represent the lower level processes. These insights are then generalized. A priori, there is no privileged level of causation. The relations between this form of ‘biological relativity’ and forms of relativity in physics are discussed. Biological relativity can be seen as an extension of the relativity principle by avoiding the assumption that there is a privileged scale at which biological functions are determined.
Keywords: downward causation, biological relativity, cardiac cell model, scale relativity
1. Introduction
Have we reached the limits of applicability of the relativity principle? And could it have relevance to biology?
By ‘relativity principle’ in this context, I mean distancing ourselves in our theories from specific absolute standpoints for which there can be no a priori justification. From Copernicus and Galileo through to Poincaré and Einstein, the reach of this general principle of relativity has been progressively extended by removing various absolute standpoints in turn. People realized that those standpoints represent privileging certain measurements as absolute, for which there is and could be no basis. First, we removed the idea of privileged location (so the Earth is not the centre of the Universe), then that of absolute velocity (since only relative velocities can be observed), then that of acceleration (an accelerating body experiences a force indistinguishable from that of gravity, leading to the idea of curved space–time). Could biology be the next domain for application of the relativity principle? This article will propose that there is, a priori, no privileged level of causality in biological systems. I will present evidence, experimental and theoretical, for the existence of downward causation from larger to smaller scales by showing how mathematical modelling has enabled us to visualize exactly how multi-level ‘both-way’ causation occurs. I will discuss the consequences for attempts to understand organisms as multi-scale systems. Finally, I will assess where some of the extensions of the relativity principle now stand in relation to these goals.
2. The hierarchy of levels: ‘up’ and ‘down’ are metaphors
In biological science, we are used to thinking in terms of a hierarchy of levels, with genes occupying the lowest level and the organism as a whole occupying the highest level of an individual. Protein and metabolic networks, intracellular organelles, cells, tissues, organs and systems are all represented as occupying various intermediate levels. The reductionist causal chain is then represented by upward-pointing arrows (figure 1). In this figure, I have also represented the causation between genes and proteins with a different kind of arrow (dotted) from the rest of the upward causation since it involves a step that is usually described in terms of coding, in which particular triplets of nucleic acids code for specified amino acids so that a complete protein has a complete DNA template (or, more correctly, a complete mRNA template that may be formed from various DNA exons). The standard story is that genes code for proteins, which then go on to form the networks. Coding of this kind does not occur in any of the other parts of the causal chain, although signalling mechanisms at these levels could also be described in terms of coding (a signal can always be described as using a code in this general sense).
Figure 1.
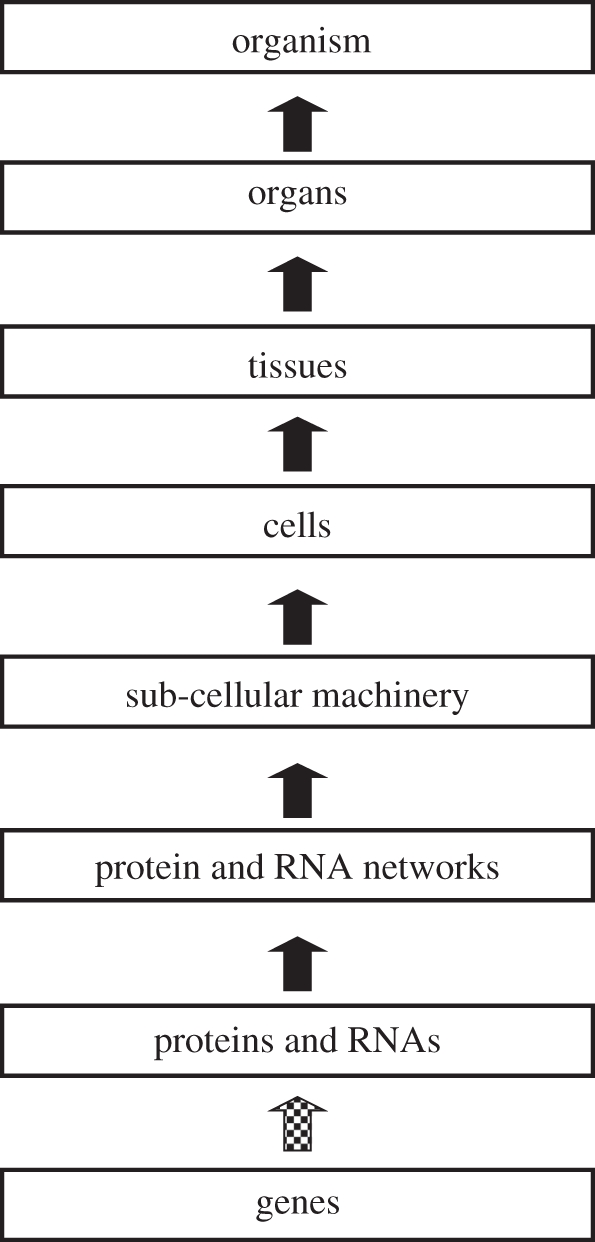
Upward causation: the reductionist causal chain in biology. This is a gross simplification, of course. No one today seriously believes that this diagram represents all causation in biology. Reductive biological discourse, however, privileges this form of causation and regards it as the most important. In particular, the nature and the direction of the lowest arrow (dotted) are fixed and represent the impact of the central dogma of molecular biology. Adapted from Noble [1, fig. 1].
The concepts of level, and of ‘up’ and ‘down’, ‘higher’ and ‘lower’, however, are all metaphors. There is no literal sense in which genes lie ‘below’ cells, for example. Genes are all over the body, so also are cells, and the organism itself, well, that is very much everywhere. This is why I prefer ‘scale’ to ‘level’. The real reason for putting genes, as DNA sequences, at the bottom of the hierarchy is that they exist at the smallest (i.e. molecular) scale in biological systems. The formation of networks, cells, tissues and organs can be seen as the creation of processes at larger and larger scales.
Does the metaphorical nature of the way we represent upward and downward causation matter? The bias introduced by the metaphor is that there is a strong tendency to represent the lower levels as somehow more concrete. Many areas of science have proceeded by unravelling the small elements underlying the larger ones. But notice the bias already creeping in through the word ‘underlying’ in the sentence I have just written. We do not use the word ‘overlying’ with anything like the same causal force. That bias is reinforced by the undeniable fact that, in biology, many of the great advances have been made by inventing more and more powerful microscopical and other techniques that allow us to visualize and measure ever smaller components. I was a graduate student when the first electron microscopes were introduced and I recall the excitement over the ability to visualize individual molecules of, for example, the contractile proteins in muscle cells. This enabled the contractile protein machinery to be understood: and so the sliding filament model of muscle contraction was born [2,3]. Taking a system apart to reveal its bits and then working out how the bits work together to form the machinery is a standard paradigm in science.
That paradigm has been remarkably successful. Breaking the human organism down into 25 000 or so genes and 100 000 or so proteins must be one of the greatest intellectual endeavours of the twentieth century, with completion of the first draft sequencing of the entire human genome occurring appropriately at the turn of the millennium [4,5].
As a scientific approach, therefore, the reductionist agenda has been impressively productive. The question remains though. If ‘up’ and ‘down’ are metaphorical, how can causation in one direction be privileged over that in the reverse direction? Are molecular events somehow causally more important than events that occur at the scales of cells, organs or systems? And are there causally efficacious processes that can only be characterized at higher scales?
3. The central dogma of molecular biology: what does it show?
It is hard to think of an a priori reason why one level in a biological system should be privileged over other levels when it comes to causation. That would run counter to the relativity principle. Moreover, I will outline later in this article how mathematical modelling has enabled us to visualize exactly how multi-level ‘both-way’ causation occurs. If the reductionist view is to be justified, therefore, it must be done a posteriori: we need empirical evidence that information that could be regarded as ‘controlling’ or ‘causing’ the system only passes in one direction, i.e. upwards. In biology, we do not have to look very far for that empirical evidence. The central dogma of molecular biology [6,7] is precisely that. Or is it?
Let us pass over the strange fact that it was called a ‘dogma’, first by Crick and then by very many who followed him. Nothing in science should be a dogma of course. Everything is open to question and to testing by the twin criteria of logic (for mathematical ideas) and experimental findings (for theories with empirical consequences). So, let us look more closely at what is involved. The essence of the central dogma is that ‘coding’ between genes and proteins is one-way. I prefer the word ‘template’ to ‘coding’ since ‘coding’ already implies a program. Another way to express the central point of this article is to say that the concept of a genetic program is part of the problem [1]. I will briefly explain why.
The sequences of DNA triplets form templates for the production of different amino acid sequences in proteins. Amino acid sequences do not form templates for the production of DNA sequences. That, in essence, is what was shown. The template works in only one direction, which makes the gene appear primary. So what does the genome cause? The coding sequences form a list of proteins and RNAs that might be made in a given organism. These parts of the genome form a database of templates. To be sure, as a database, the genome is also extensively formatted, with many regulatory elements, operons, embedded within it. These regulatory elements enable groups of genes to be coordinated [8] in their expression levels. And we now know that the non-coding parts of the genome also play important regulatory functions. But the genome is not a fixed program in the sense in which such a computer program was defined when Jacob and Monod introduced their idea of ‘le programme génétique’ [9–11]. It is rather a ‘read–write’ memory that can be organized in response to cellular and environmental signals [12]. Which proteins and RNAs are made when and where is not fully specified. This is why it is possible for the 200 or so different cell types in an organism such as the human to make those cell types using exactly the same genome. A heart cell is made using precisely the same genome in its nucleus as a bone cell, a liver cell, pancreatic cell, etc. Impressive regulatory circuits have been constructed by those who favour a genetic program view of development [13,14], but these are not independent of the ‘programming’ that the cells, tissues and organs themselves use to epigenetically control the genome and the patterns of gene expression appropriate to each cell and tissue type in multi-cellular organisms. As I will show later, the circuits for major biological functions necessarily include non-genome elements.
That fact already tells us that the genome alone is far from sufficient. It was Barbara McClintock, who received the Nobel Prize for her work on jumping genes, who first described the genome as ‘an organ of the cell’ [15]. And so it is. DNA sequences do absolutely nothing until they are triggered to do so by a variety of transcription factors, which turn genes on and off by binding to their regulatory sites, and various other forms of epigenetic control, including methylation of certain cytosines and interactions with the tails of the histones that form the protein backbone of the chromosomes. All of these, and the cellular, tissue and organ processes that determine when they are produced and used, ‘control’ the genome. For further detail on this issue, the reader is referred to Shapiro's article on re-assessing the central dogma [16] and to his book Evolution: the view from the 21st century [12]. A good example in practice is the way in which neuroscientists are investigating what they call electro-transcription coupling [17], a clear example of downward causation since it involves the transmission of information from the neural synapses to the nuclear DNA.
To think that the genome completely determines the organism is almost as absurd as thinking that the pipes in a large cathedral organ determine what the organist plays. Of course, it was the composer who did that in writing the score, and the organist himself who interprets it. The pipes are his passive instruments until he brings them to life in a pattern that he imposes on them, just as multi-cellular organisms use the same genome to generate all the 200 or so different types of cell in their bodies by activating different expression patterns. This metaphor has its limitations. There is no ‘organist’. The ‘music of life’ plays itself [1], rather as some musical ensembles perform without a conductor. And, of course, the ‘organ’ varies between individuals in a species. But it is quite a good metaphor. The pipes of an organ are also ‘formatted’ to enable subsets to be activated together by the various stops, manuals and couplers. Like the regulatory parts of the genome, these parts of the organ make it easier to control, but both, genome and organ, still do nothing without being activated. The patterns of activation are just as much part of the ‘program’ as the genome itself [18].
So, even at the very lowest level of the reductionist causal chain, we discover a conceptual error. The protein-coding sequences are templates. They determine which set of proteins the organism has to play with, just as a child knows which pieces of Lego or Meccano she has available for construction. Those parts of the genome are best regarded as a database. Even when we add in the regulatory and non-coding regions, there is no program in the genome in the sense that the sequences could be parsed in the way in which we would analyse a computer program to work out what it is specifying. The reason is that crucial parts of the program are missing. To illustrate this, I will use the example of cardiac rhythm to show that the non-genomic parts are essential.
4. Insights from experimental and modelling work on heart cells
Over many years, my research has involved experimental and computational work on heart cells. I was the first to analyse the potassium ion channels in heart muscle [19,20] and to construct a computer model based on the experimental findings [21,22]. Since that time, a whole field of heart modelling has developed [23,24].
How do we construct such models? The trail was blazed by Hodgkin & Huxley [25] in their Nobel prize-winning work on the nerve impulse. The ion channel proteins that sit across the cell membrane control its electrical potential by determining the quantity of charge that flows across the cell membrane to make the cell potential become negative or positive. The gating of these channels is itself in turn controlled by the cell potential. This is a multi-level loop. The potential is a cell-level parameter; the ion channel openings and closings are protein-level parameters. The loop, originally called the Hodgkin cycle, is absolutely essential to the rhythm of the heart. Breaking the feedback (downward causation) between the cell potential and the gating of the ion channels and cellular rhythm are abolished. A simple experiment on one of the cardiac cell models will demonstrate this computationally.
In figure 2 [26], a model of the sinus node (the pacemaker region of the heart) was run for 1300 ms, during which time six oscillations were generated. These correspond to six heartbeats at a frequency similar to that of the heart of a rabbit, the species on which the experimental data were obtained to construct the model. During each beat, all the currents flowing through the protein channels also oscillate in a specific sequence. To simplify the diagram, only three of those protein channels are represented here. At 1300 ms, an experiment was performed on the model. The ‘downward causation’ between the global cell property, the membrane potential and the voltage-dependent gating of the ion channels was interrupted. If there were a sub-cellular ‘program’ forcing the proteins to oscillate, the oscillations would continue. In fact, however, all oscillations cease and the activity of each protein relaxes to a steady value, as also happens experimentally. In this case, therefore, the ‘program’ includes the cell itself and its membrane system. In fact, we do not need the concept of a separate program here. The sequence of events, including the feedback between the cell potential and the activity of the proteins, simply is cardiac rhythm. It is a property of the interactions between all the components of the system. It does not even make sense to talk of cardiac rhythm at the level of proteins and DNA, and it does not make sense to suppose that there is a separate program that ‘runs’ the rhythm.
Figure 2.

Computer model of pacemaker rhythm in the heart [27]. For the first six beats, the model is allowed to run normally and generates rhythm closely similar to a real cell. Then the feedback from cell voltage (a) to protein channels ((b) currents in nanoamps) is interrupted by keeping the voltage constant (voltage clamp). All the protein channel oscillations then cease. They slowly change to steady constant values. Without the downward causation from the cell potential, there is no rhythm. Adapted from Noble [1, fig. 3].
Of course, all the proteins involved in cardiac rhythm are encoded by the genome, but these alone would not generate rhythm. This is the sense (see above) in which I maintain that there is not a program for cardiac rhythm in the genome. The non-genomic structural elements are also essential. Similar arguments apply, for example, to circadian rhythm [1,28] and, indeed, to all functions that require cellular structural inheritance as well as genome inheritance. Indeed, I find it hard to identify functions that do not involve what Cavalier-Smith [29,30] has characterized as the membranome. Much of the logic of life lies in its delicate oily membranes.
5. Generalization of the argument in mathematical terms
We can generalize what is happening here in mathematical terms. The activity of the ion channels is represented by differential equations describing the speed and the direction of the gating processes on each protein. The coefficients in those differential equations are based on experimental data. One might think that, provided all the relevant protein mechanisms have been included in the model and if the experimental data are reliable, and the equations fit the data well, cardiac rhythm would automatically ‘emerge’ from those characteristics. It does not. The reason is very simple and fundamental to any differential equation model. In addition to the differential equations you need the initial and boundary conditions. Those values are just as much a ‘cause’ of the solution (cardiac rhythm) as are the differential equations. In this case, the boundary conditions include the cell structure, particularly those of its membranes and compartments. Without the constraints imposed by the higher level structures, and by other processes that maintain ionic concentrations, the rhythm would not occur. If we were to put all the components in a Petri dish mixed up in a nutrient solution, the interactions essential to the function would not exist. They would lack the spatial organization necessary to do so.
This fact tells us therefore how higher levels in biological systems exert their influence over the lower levels. Each level provides the boundary conditions under which the processes at lower levels operate. Without boundary conditions, biological functions would not exist.
The relationships in such models are illustrated in figure 3. The core of the model is the set of differential equations describing the kinetics of the components of the system (e.g. the channel proteins in figure 2). The initial conditions are represented as being on the same level since they are the state of the system at the time at which the simulation begins. The boundary conditions are represented as being at a higher level since they represent the influence of their environment on the components of the system. So far as the proteins are concerned, the rest of the cell is part of their environment.
Figure 3.

Many models of biological systems consist of differential equations for the kinetics of each component. These equations cannot give a solution (the output) without setting the initial conditions (the state of the components at the time at which the simulation begins) and the boundary conditions. The boundary conditions define what constraints are imposed on the system by its environment and can therefore be considered as a form of downward causation. This diagram is highly simplified to represent what we actually solve mathematically. In reality, boundary conditions are also involved in determining initial conditions and the output parameters can also influence the boundary conditions, while they in turn are also the initial conditions for a further period of integration of the equations. As with the diagrams (see §§2 and 5) of levels in biological systems, the arrows are not really unidirectional. The dotted arrows complete the diagram to show that the output contributes to the boundary conditions (although not uniquely), and determines the initial conditions for the next integration step.
The diagram of figure 1 therefore should look more like figure 4. There are multiple feedbacks from higher levels to lower levels in addition to those from lower to higher levels. In any model of lower level systems, these form the constraints that would need to be incorporated into the boundary and initial conditions. As figure 4 indicates, these include triggers of cell signalling (via hormones and transmitters), control of gene expression (via transcription factors), epigenetic control (via methylation and histone marking), and note also that it is the protein machinery that reads genes—and continually repairs copying errors and so makes the genome reliable. To reverse a popular metaphor, that of the selfish gene [31], it is the ‘lumbering robot’ that is responsible for any ‘immortality’ genes may possess!
Figure 4.

The completion of figure 1 with various forms of downward causation that regulates lower level components in biological systems. In addition to the controls internal to the organism, we also have to take account of the influence of the environment on all the levels (not shown in this diagram). Adapted from Noble [1, fig. 2]. Causation is, therefore, two-way, although this is not best represented by making each arrow two-way. A downward form of causation is not a simple reverse form of upward causation. It is better seen as completing a feedback circuit, as the examples discussed in the text show.
6. Differential and integral views of the relations between genotypes and phenotypes
All of this is fundamental and, even, fairly obvious to integrative physiologists. Physiologists have been familiar with the basic ideas on multi-level control ever since Claude Bernard formulated the concept of control of the internal environment in his book Introduction à l'étude de la médecine expérimentale in 1865 [32] and Walter B. Cannon developed the idea of homeostasis in The wisdom of the Body in 1932 [33]. So, how has mainstream biology tended to ignore it, as has physiology also with some exceptions, for example Guyton's modelling of the circulation [34]? I think the main culprit here has been neo-Darwinism and particularly the popularizations of this theory as a purely gene-centric view [31]. The essential idea of gene-centric theories is what I have called the differential view of the relationships between genes and phenotypes [35–38]. The idea is essential in the sense that it excludes alternative theories by arguing that what matters in evolutionary terms are changes in the genotype that are reflected in changes in phenotype. Selection of the phenotype is therefore, according to this logic, fundamentally equivalent to selection of particular genes (or, more strictly, gene alleles). This view might have been appropriate for a time when genes were regarded as hypothetical entities defined as the cause of each phenotype. It is not appropriate for the current molecular and systems biology-inspired definition of a gene as a particular DNA sequence, replicating and being expressed within cellular and multi-cellular systems. In principle, we can now investigate all the functions that DNA sequence is involved in, though that goal still remains very ambitious in practice. We do not have to be restricted to investigating differences. Anyway, that would be to focus on the tip of the iceberg. Considering just differences at the genetic level is as limiting as it would be for mathematics to limit itself to differential equations without integrating them, as though the integral sign and what it stands for had never been invented [37].
The analogy with the mathematics of differential calculus is strongly revealing. Integration requires knowledge of the initial and boundary conditions in addition to the differential equations themselves (figure 3). One can only ignore those by restricting oneself to the differential equation ‘level’. In a similar way, the neo-Darwinist synthesis tends to ignore downward causation precisely because such causation requires an integral rather than a differential view of genetics for its analysis.
Specifically, when neo-Darwinists refer to the ‘genes’ for any particular phenotype on which selection may act, they are not referring to complete protein-coding sequences of DNA, they are really referring to differences between alleles. The ‘gene’ is, therefore, defined as this inheritable difference in phenotype. It would not even matter whether this difference is a difference in DNA or in some other inheritable factor, such as inherited cytoplasmic changes in Paramecium [39], or the cytoplasmic influences on development observed in cross-species cloning of fish [40].
By contrast, the integral view for which I am arguing does not focus on differences. Instead it asks: what are all the functions to which the particular DNA sequence contributes? Indeed, it would not matter whether those functions are ones that result in a different phenotype. Through the existence of multiple back-up mechanisms, many DNA changes, such as knockouts, do not have a phenotypic effect on their own. As many as 80 per cent of the knockouts in yeast are normally ‘silent’ in this way [41]. Their functionality can be revealed only when the boundary conditions, such as the nutrient environment, are changed. The analogy that I am drawing with differential and integral calculus draws its strength precisely through this dependence on the boundary conditions. A differential equation, on its own, has an infinite set of solutions until those are narrowed down by the boundary conditions. Similarly, a difference in DNA sequence may have a wide variety of possible phenotypic effects, including no effect at all, until the boundary conditions are set, including the actions of many other genes, the metabolic and other states of the cell or organism, and the environment in which the organism exists.
7. A (biological) theory of relativity
I and my colleagues have expressed many of the ideas briefly outlined here in the form of some principles of systems biology [1,42–44]. One of those principles is that, a priori, there is no privileged level of causation in biological systems. Determining the level at which a function is integrated is an empirical question. Cardiac rhythm is clearly integrated at the level of the pacemaker sinus node cell, and does not even exist below that level. The principle can be restated in a more precise way by saying that the level at which each function is integrated is at least partly a matter of experimental discovery. There should be no dogmas when it comes to causation in biological systems.
8. Connecting levels
One way to connect levels in biological simulation can be derived immediately from figure 3. Since the boundary conditions for integration are set by the higher level, determining those conditions at that level either by measurement or by computation can enable them to be inserted into the equations at the lower level. This is the way, for example, in which the structural organization of the whole heart is used to constrain the ordinary and partial differential equations describing the protein channels and the flow of ionic current through the structure—conduction is faster along a fibre axis, for example, than across and between fibres. These kinds of constraints turn out to be very important in studying cardiac arrhythmias, where the sequence of events from ordered rhythm to tachycardia and then to fibrillation is dependent on the high-level structure [45–52].
A similar approach could be used to simulate other biological processes such as development. If we had a sufficiently detailed knowledge of the fertilized egg cell structure and networks, including particularly the concentrations and locations of transcription factors and the relevant epigenetic influences, we could imagine solving equations for development involving gene expression patterns determined by both the genome and its non-DNA regulators. In this case, the various levels ‘above’ the cell (better viewed as ‘around’ the cell) would actually develop with the process itself, as it moves through the various stages, so creating the more global constraints in interaction with the environment of the organism. We cannot do that kind of ambitious computation at the present time, and the reason is not that we do not know the genome that has been sequenced. The problem lies at a higher level. We cannot yet characterize all the relevant concentrations of transcription factors and epigenetic influences. It is ignorance of all those forms of downward causation that is impeding progress. Even defining which parts of the DNA sequence are transcribed (and so to identify ‘genes’ at the DNA level—and here I would include sequences that form templates for RNAs as ‘genes’) requires higher level knowledge. This approach would naturally take into account the role of cell and tissue signalling in the generation of organizing principles involved in embryonic induction, originally identified in the pioneering work of Spemann & Mangold [53–55]. The existence of such induction is itself an example of dependence on boundary conditions. The induction mechanisms emerge as the embryo interacts with its environment. Morphogenesis is not entirely hard-wired into the genome.
9. Emergence and boundary conditions
Reference to emergence leads me to a fundamental point about the limits of reductionism. An important motivation towards reductionism is that of reducing complexity. The idea is that if a phenomenon is too complex to understand at level X then go down to level Y and see, first, whether the interactions at level Y are easier to understand and theorize about, then, second, see whether from that understanding one can automatically understand level X. If indeed all that is important at level X were to be entirely derivable from a theory at level Y, then we would have a case of what I would call ‘weak emergence’, meaning that descriptions at level X can then be seen to be a kind of shorthand for a more detailed explanatory analysis at level Y. ‘Strong emergence’ could then be defined as cases where this does not work, as we found with the heart rhythm model described above. They would be precisely those cases where what would be merely contingent at level Y is systematic at level X. I am arguing that, if level Y is the genome, then we already know that ‘weak emergence’ does not work. There is ‘strong emergence’ because contingency beyond what is in the genome, i.e. in its environment, also determines what happens.
This kind of limit to reductionism is not restricted to biology. Spontaneous symmetry breaking in particle physics is a comparable case. An infinitesimal change can determine which way symmetry is broken [56]. How that happens in particular cases is not derivable from particle theory itself. Biological reductionists whose motivation is that of reducing biology to physics need to be aware that physics itself also displays the kind of limits I am describing here. Nor are these limits restricted to particle theory.
Connecting levels through setting initial and boundary conditions derived from multi-level work has served biological computation very well so far. The successes of the Physiome Project attest the same [23,57]. But there are two reasons why I think it may not be enough.
10. Computability
The first is the problem of computability.
Consider the heart again. Since the very first supercomputer simulations [58,59] in which cell models were incorporated into anatomical structures representing heart tissue and the whole organ [23,60,61], we have continually pushed up against the limits of computer speed and memory. Even today, we are only beginning to be within reach of whole organ simulations of electrical activity running in real time, i.e. that it should take only 1 s of computer time to calculate a second of heart time. Yet, such models represent only a few per cent of the total number of proteins involved in cardiac function, although, of course, we hope we have included the most important ones for the functions we are representing. And the equations for each component are the simplest that can capture the relevant kinetics of ion channel function. Expanding the models to include most, rather than a very few, gene products, extending the modelling of each protein to greater detail, and extending the time scale beyond a few heartbeats would require orders of magnitude increases in computing power.
In fact, it is relatively easy to show that complete bottom-up reconstructions from the level of molecules to the level of whole organs would require much more computing power than we are ever likely to have available, as I have argued in a previous article [37]. In that article, I began by asking two questions. First, ‘are organisms encoded as molecular descriptions in their genes?’ And, second, ‘by analysing the genome, could we solve the forward problem of computing the behaviour of the system from this information, as was implied by the original idea of the “genetic program” and the more modern representation of the genome as the “book of life”?’ (for a recent statement of these ideas see [62]). The answer to both questions was ‘no’. The first would have required that the central dogma of molecular biology should be correct in excluding control of the genome by its environment, while the second runs into the problem of combinatorial explosion. The number of possible interactions between 25 000 genes exceeds the total number of elementary particles in the whole-known Universe [63], even when we severely restrict the numbers of gene products that can interact with each other (see also [64]). Conceivably, we might gain some speed-up from incorporating analogue computation to go beyond the Turing limits [65], but it is still implausible to expect that increased computer power will provide all we need or that it is the best way forward [66].
11. Scale relativity
The second reason why connecting levels via boundary conditions may not be enough is that it assumes that the differential equations themselves remain unchanged when they form part of a hierarchy of levels. This is what we would expect in a classical analysis. But is this necessarily correct?
One of the reasons I introduced this article with some remarks on the general principle of relativity and its history of distancing us from unwarranted assumptions concerning privileged standpoints is that we can ask the same question about levels and scales. If there is no privileged level of causation, then why should there be a privileged scale? This is the question raised by Laurent Nottale's theory of scale relativity [67,68]. As Nottale et al. [69] shows in his recent book, the consequences of applying the relativity principle to scales are widespread and profound, ranging from understanding the quantum–classical transition in physics to potential applications in systems biology [70,71].
I will conclude this article, therefore, by describing what that theory entails, how it relates to the general theory of biological relativity I have outlined here and what is the status of such theories now?
The central feature from the viewpoint of biological modelling can be appreciated by noting that the equations for structure and for the way in which elements move and interact in that structure in biology necessarily depend on the resolution at which it is represented. Unless we represent everything at the molecular level which, as argued above, is impossible (and fortunately unnecessary as well), the differential equations should be scale-dependent. As an example, at the level of cells, the equations may represent detailed compartmentalization and non-uniformity of concentrations, and hence include intracellular diffusion equations, or other ways of representing non-uniformity [72–74]. At the level of tissues and organs, we often assume complete mixing (i.e. uniformity) of cellular concentrations. At that level, we also usually lump whole groups of cells into grid points where the equations represent the lumped behaviour at that point.
These are practical reasons why the equations we use are scale-dependent. The formal theory of scale relativity goes much further since it proposes that it is theoretically necessary that the differential equations should be scale-dependent. It does this by assuming that space–time itself is continuous but generally non-differentiable, therefore fractal, not uniform. The distance between two points, therefore, depends on the scale at which one is operating and that, in the limit, as dx or dt tend to zero, the differential is most often not defined. This does not mean that differential equations cannot be used, simply that terms corresponding to scale should be included as an extension of the usual differential equations as explicit influences of scale on the system. The derivation of these extension terms can be found in Auffray & Nottale [70, pp. 93–97] and in Nottale [69, pp. 73–141].
The idea of fractal space–time may seem strange. I see it as an extension of the general relativity principle that space–time is not independent of the objects themselves found within it, i.e. space–time is not uniform. We are now used to this idea in relation to the structure of the Universe and the way in which, according to Einstein's general relativity, space–time is distorted by mass and energy to create phenomena such as gravitational lensing [75,76]. But, it is usually assumed that, on smaller scales, the classical representations of space–time are sufficient. It is an open question whether that is so and whether scale should be incorporated in explicit terms in the equations we use in multi-scale models. Remember also that the utility of a mathematical concept does not depend on how easily we can visualize the entities involved. We find it difficult to imagine a number like √−1, but it has great utility in mathematical analysis of the real world. We may need to think the unimaginable in order fully to understand the multi-scale nature of biology. The concept of scale is, after all, deeply connected to our conception of space–time.
12. Conclusions
While I think we can be certain that multi-level causation with feedbacks between all the levels is an important feature of biological organisms, the tools we have to deal with such causation need further development. The question is not whether downward causation of the kind discussed in this article exists, it is rather how best to incorporate it into biological theory and experimentation, and what kind of mathematics needs to be developed for this work.
Acknowledgements
This article is based on a presentation of a meeting on Downward Causation held at the Royal Society in September 2010. I should like to acknowledge valuable discussion with many of the participants of that meeting. I also thank Charles Auffray, Jonathan Bard, Peter Kohl and Laurent Nottale for suggesting improvements to the manuscript, and the journal referees for valuable criticism. I acknowledge support from an EU FP7 grant for the VPH-PreDiCT project.
Following acceptance of this article, my attention was drawn to the article on downward causation by Michel Bitbol [77]. He approaches the issue of downward causation from Kantian and quantum mechanical viewpoints, but I would like to acknowledge that many of his insights are similar to and compatible with the views expressed here, particularly on the role of boundary conditions and the relativistic stance.
References
- 1.Noble D. 2006. The music of life. Oxford, UK: Oxford University Press [Google Scholar]
- 2.Huxley A. F. 1957. Muscle structure and theories of contraction. Prog. Biophys. Mol. Biol. 7, 255–318 [PubMed] [Google Scholar]
- 3.Huxley H. 2004. Fifty years of muscle and the sliding filament hypothesis. Eur. J. Biochem. 271, 1403–1415 10.1111/j.1432-1033.2004.04044.x (doi:10.1111/j.1432-1033.2004.04044.x) [DOI] [PubMed] [Google Scholar]
- 4.International Human Genome Mapping Consortium 2001. A physical map of the human genome. Nature 409, 934–941 10.1038/35057157 (doi:10.1038/35057157) [DOI] [PubMed] [Google Scholar]
- 5.Venter C., et al. 2001. The sequence of the human genome. Science 291, 1304–1351 10.1126/science.1058040 (doi:10.1126/science.1058040) [DOI] [PubMed] [Google Scholar]
- 6.Crick F. H. C. 1958. On protein synthesis. Symp. Soc. Exp. Biol. 12, 138–163 [PubMed] [Google Scholar]
- 7.Crick F. H. C. 1970. Central dogma of molecular biology. Nature 227, 561–563 10.1038/227561a0 (doi:10.1038/227561a0) [DOI] [PubMed] [Google Scholar]
- 8.Jacob F., Perrin D., Sanchez C., Monod J., Edelstein S. 1960. The operon: a group of genes with expression coordinated by an operator. C. R. Acad. Sci. Paris 250, 1727–172914406329 [Google Scholar]
- 9.Jacob F. 1970. La Logique du vivant, une histoire de l'hérédité. Paris, France: Gallimard [Google Scholar]
- 10.Jacob F. 1982. The possible and the actual. New York, NY: Pantheon Books [Google Scholar]
- 11.Monod J., Jacob F. 1961. Teleonomic mechanisms in cellular metabolism, growth and differentiation. Cold Spring Harbor Symp. Quant. Biol. 26, 389–401 [DOI] [PubMed] [Google Scholar]
- 12.Shapiro J. A. 2011. Evolution: a view from the 21st century. Upper Saddle River, NJ: Pearson Education Inc [Google Scholar]
- 13.Davidson E. H. 2006. The regulatory genome: gene regulatory networks in development and evolution. New York, NY: Academic Press [Google Scholar]
- 14.Davidson E. H., et al. 2002. A provisional regulatory gene network for specification of endomesoderm in the sea urchin embryo. Dev. Biol. 246, 2–13 10.1006/dbio.2002.0635 (doi:10.1006/dbio.2002.0635) [DOI] [PubMed] [Google Scholar]
- 15.McClintock B. 1984. The significance of responses of the genome to challenge. Science 226, 792–801 10.1126/science.15739260 (doi:10.1126/science.15739260) [DOI] [PubMed] [Google Scholar]
- 16.Shapiro J. A. 2009. Revisiting the central dogma in the 21st century. Ann. N. Y. Acad. Sci. 1178, 6–28 10.1111/j.1749-6632.2009.04990.x (doi:10.1111/j.1749-6632.2009.04990.x) [DOI] [PubMed] [Google Scholar]
- 17.Deisseroth K., Mermelstein P. G., Xia H., Tsien R. W. 2003. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol. 13, 354–365 10.1016/S0959-4388(03)00076-X (doi:10.1016/S0959-4388(03)00076-X) [DOI] [PubMed] [Google Scholar]
- 18.Coen E. 1999. The art of genes. Oxford, UK: Oxford University Press [Google Scholar]
- 19.Hutter O. F., Noble D. 1960. Rectifying properties of heart muscle. Nature 188, 495. 10.1038/188495a0 (doi:10.1038/188495a0) [DOI] [PubMed] [Google Scholar]
- 20.Noble D. 1965. Electrical properties of cardiac muscle attributable to inward-going (anomalous) rectification. J. Cell. Comp. Physiol. 66(Suppl. 2), 127–136 10.1002/jcp.1030660520 (doi:10.1002/jcp.1030660520) [DOI] [Google Scholar]
- 21.Noble D. 1960. Cardiac action and pacemaker potentials based on the Hodgkin–Huxley equations. Nature 188, 495–497 10.1038/188495b0 (doi:10.1038/188495b0) [DOI] [PubMed] [Google Scholar]
- 22.Noble D. 1962. A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pacemaker potentials. J. Physiol. 160, 317–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bassingthwaighte J. B., Hunter P. J., Noble D. 2009. The cardiac physiome: perspectives for the future. Exp. Physiol. 94, 597–605 10.1113/expphysiol.2008.044099 (doi:10.1113/expphysiol.2008.044099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noble D. 2007. From the Hodgkin–Huxley axon to the virtual heart. J. Physiol. 580, 15–22 10.1113/jphysiol.2006.119370 (doi:10.1113/jphysiol.2006.119370) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodgkin A. L., Huxley A. F. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noble D., Denyer J. C., Brown H. F., DiFrancesco D. 1992. Reciprocal role of the inward currents ib,Na and if in controlling and stabilizing pacemaker frequency of rabbit sino-atrial node cells. Proc. R. Soc. Lond. B 250, 199–207 10.1098/rspb.1992.0150 (doi:10.1098/rspb.1992.0150) [DOI] [PubMed] [Google Scholar]
- 27.Noble D., Noble S. J. 1984. A model of sino-atrial node electrical activity based on a modification of the DiFrancesco–Noble (1984) equations. Proc. R. Soc. Lond. B 222, 295–304 10.1098/rspb.1984.0065 (doi:10.1098/rspb.1984.0065) [DOI] [PubMed] [Google Scholar]
- 28.Foster R., Kreitzman L. 2004. Rhythms of life. London, UK: Profile Books [Google Scholar]
- 29.Cavalier-Smith T. 2000. Membrane heredity and early choroplast evolution. Trends Plant Sci. 5, 174–182 10.1016/S1360-1385(00)01598-3 (doi:10.1016/S1360-1385(00)01598-3) [DOI] [PubMed] [Google Scholar]
- 30.Cavalier-Smith T. 2004. The membranome and membrane heredity in development and evolution. In Organelles, genomes and eukaryite phylogeny: an evolutionary synthesis in the age of genomics (eds Hirt R. P., Horner D. S.), pp. 335–351 Boca Baton, FL: CRC Press [Google Scholar]
- 31.Dawkins R. 1976, 2006. The selfish gene. Oxford, UK: Oxford University Press [Google Scholar]
- 32.Bernard C. Introduction à l'étude de la médecine expérimentale. Paris, France: Bailliere: 1865. (Reprinted by Flammarion 1984) [Google Scholar]
- 33.Cannon W. B. 1932. The wisdom of the body. Norton, MA: Boston [Google Scholar]
- 34.Guyton A. C., Coleman T. G., Granger H. J. 1972. Circulation: overall regulation. Annu. Rev. Physiol. 34, 13–46 10.1146/annurev.ph.34.030172.000305 (doi:10.1146/annurev.ph.34.030172.000305) [DOI] [PubMed] [Google Scholar]
- 35.Noble D. 2008. Genes and causation. Phil. Trans. R. Soc. A 366, 3001–3015 10.1098/rsta.2008.0086 (doi:10.1098/rsta.2008.0086) [DOI] [PubMed] [Google Scholar]
- 36.Noble D. 2010. Biophysics and systems biology. Phil. Trans. R. Soc. A 368, 1125–1139 10.1098/rsta.2009.0245 (doi:10.1098/rsta.2009.0245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noble D. 2011. Differential and integral views of genetics in computational systems biology. J. R. Soc. Interface Focus 1, 7–15 10.1098/rsfs.2010.0444 (doi:10.1098/rsfs.2010.0444) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noble D. 2011. Neo-Darwinism, the modern synthesis, and selfish genes: are they of use in physiology? J. Physiol. 589, 1007–1015 10.1113/jphysiol.2010.201384 (doi:10.1113/jphysiol.2010.201384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonneborn T. M. 1970. Gene action on development. Proc. R. Soc. Lond. B 176, 347–366 10.1098/rspb.1970.0054 (doi:10.1098/rspb.1970.0054) [DOI] [PubMed] [Google Scholar]
- 40.Sun Y. H., Chen S. P., Wang Y. P., Hu W., Zhu Z. Y. 2005. Cytoplasmic impact on cross-genus cloned fish derived from transgenic common carp (Cyprinus carpio) nuclei and goldfish (Carassius auratus) enucleated eggs. Biol. Reprod. 72, 510–515 10.1095/biolreprod.104.031302 (doi:10.1095/biolreprod.104.031302) [DOI] [PubMed] [Google Scholar]
- 41.Hillenmeyer M. E., et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 10.1126/science.1150021 (doi:10.1126/science.1150021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohl P., Crampin E., Quinn T. A., Noble D. 2010. Systems biology: an approach. Clin. Pharmacol. Ther. 88, 25–33 10.1038/clpt.2010.92 (doi:10.1038/clpt.2010.92) [DOI] [PubMed] [Google Scholar]
- 43.Kohl P., Noble D. 2009. Systems biology and the virtual physiological human. Mol. Syst. Biol. 5, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noble D. 2008. Claude Bernard, the first systems biologist, and the future of physiology. Exp. Physiol. 93, 16–26 10.1113/expphysiol.2007.038695 (doi:10.1113/expphysiol.2007.038695) [DOI] [PubMed] [Google Scholar]
- 45.Niederer S. A., Ter Keurs H. E., Smith N. P. 2009. Modelling and measuring electromechanical coupling in the rat heart. Exp. Physiol. 94, 529–540 10.1113/expphysiol.2008.045880 (doi:10.1113/expphysiol.2008.045880) [DOI] [PubMed] [Google Scholar]
- 46.Panfilov A., Holden A. V. 1993. Computer simulation of re-entry sources in myocardium in two and three dimensions. J. Theor. Biol. 161, 271–285 10.1006/jtbi.1993.1055 (doi:10.1006/jtbi.1993.1055) [DOI] [PubMed] [Google Scholar]
- 47.Panfilov A., Keener J. 1993. Re-entry generation in anisotropic twisted myocardium. J. Cardiovasc. Electrophysiol. 4, 412–421 10.1111/j.1540-8167.1993.tb01280.x (doi:10.1111/j.1540-8167.1993.tb01280.x) [DOI] [PubMed] [Google Scholar]
- 48.Panfilov A., Kerkhof P. 2004. Quantifying ventricular fibrillation: in silico research and clinical implications. IEEE Trans. Biomed. Eng. 51, 195–196 10.1109/TBME.2003.820608 (doi:10.1109/TBME.2003.820608) [DOI] [PubMed] [Google Scholar]
- 49.Plank G., et al. 2009. Generation of histo-anatomically representative models of the individual heart: tools and application. Phil. Trans. R. Soc. A 367, 2257–2292 10.1098/rsta.2009.0056 (doi:10.1098/rsta.2009.0056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trayanova N., Eason J. 2002. Shock-induced arrhythmogenesis in the myocardium. Chaos 12, 962–972 10.1063/1.1483955 (doi:10.1063/1.1483955) [DOI] [PubMed] [Google Scholar]
- 51.Trayanova N., Eason J., Aguel F. 2002. Computer simulations of cardiac defibrillation: a look inside the heart. Comput. Vis. Sci. 4, 259–270 10.1007/s00791-002-0082-8 (doi:10.1007/s00791-002-0082-8) [DOI] [Google Scholar]
- 52.Whiteley J. P., Bishop M. J., Gavaghan D. J. 2007. Soft tissue modelling of cardiac fibres for use in coupled mechano-electric simulations. Bull. Math. Biol. 69, 2199–2225 10.1007/s11538-007-9213-1 (doi:10.1007/s11538-007-9213-1) [DOI] [PubMed] [Google Scholar]
- 53.De Robertis E. M. 2006. Spemann's organizer and self-regulation in amphibian embryos. Nat. Rev. Mol. Cell Biol. 7, 296–302 10.1038/nrm1855 (doi:10.1038/nrm1855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sander K., Faessler P. E. 2001. Introducing the Spemann-Mangold organizer: experiments and insights that generated a key concept in developmental biology. Int. J. Dev. Biol. 45, 1–11 [PubMed] [Google Scholar]
- 55.Spemann H., Mangold H. 1924. Über induktion von Embryonalagen durch Implantation Artfremder Organisatoren. Wilhelm Roux's Arch. Dev. Biol. 100, 599–638 [Google Scholar]
- 56.Anderson P. W. 1972. More is different. Science 177, 393–396 10.1126/science.177.4047.393 (doi:10.1126/science.177.4047.393) [DOI] [PubMed] [Google Scholar]
- 57.Hunter P., Smaill B. H., Smith N. P., Young A., Nash M., Nielsen P. F., Vaughan-Jones R. D., Omholt S., Paterson D. J. In press The Heart physiome project. WIREs Syst. Biol. Med. [Google Scholar]
- 58.Winslow R., Kimball A., Varghese A., Noble D. 1993. Simulating cardiac sinus and atrial network dynamics on the connection machine. Physica D Non-linear Phenom. 64, 281–298 10.1016/0167-2789(93)90260-8 (doi:10.1016/0167-2789(93)90260-8) [DOI] [Google Scholar]
- 59.Winslow R., Varghese A., Noble D., Adlakha C., Hoythya A. 1993. Generation and propagation of triggered activity induced by spatially localised Na-K pump inhibition in atrial network models. Proc. R. Soc. Lond. B 254, 55–61 10.1098/rspb.1993.0126 (doi:10.1098/rspb.1993.0126) [DOI] [PubMed] [Google Scholar]
- 60.Nash M. P., Hunter P. J. 2001. Computational mechanics of the heart. J. Elast. 61, 113–141 10.1023/A:1011084330767 (doi:10.1023/A:1011084330767) [DOI] [Google Scholar]
- 61.Smith N. P., Pullan A. J., Hunter P. J. 2001. An anatomically based model of transient coronary blood flow in the heart. SIAM J. Appl. Math. 62, 990–1018 10.1137/S0036139999359860 (doi:10.1137/S0036139999359860) [DOI] [Google Scholar]
- 62.Brenner S. 2010. Sequences and consequences. Phil. Trans. R. Soc. B 365, 207–212 10.1098/rstb.2009.0221 (doi:10.1098/rstb.2009.0221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feytmans E., Noble D., Peitsch M. 2005. Genome size and numbers of biological functions. Trans. Comput. Syst. Biol. 1, 44–49 10.1007/978-3-540-32126-2_4 (doi:10.1007/978-3-540-32126-2_4) [DOI] [Google Scholar]
- 64.Lewontin R. C. 1974. The genetic basis of evolutionary change. New York, NY: Columbia University Press [Google Scholar]
- 65.Siegelmann H. T. 1995. Computation beyond the Turing limit. Science 268, 545–548 10.1126/science.268.5210.545 (doi:10.1126/science.268.5210.545) [DOI] [PubMed] [Google Scholar]
- 66.Garny A., Noble D., Kohl P. 2005. Dimensionality in cardiac modelling. Progr. Biophys. Mol. Biol. 87, 47–66 10.1016/j.pbiomolbio.2004.06.006 (doi:10.1016/j.pbiomolbio.2004.06.006) [DOI] [PubMed] [Google Scholar]
- 67.Nottale L. 1993. Fractal space-time and microphysics: towards a theory of scale relativity. Singapore: World Scientific [Google Scholar]
- 68.Nottale L. 2000. La relativité dans tous ses états. Du mouvements aux changements d'échelle. Paris, France: Hachette [Google Scholar]
- 69.Nottale L. 2011. Scale relativity and fractal space-time: a new approach to unifying relativity and quantum mechanics. London, UK: Imperial College Press [Google Scholar]
- 70.Auffray C., Nottale L. 2008. Scale relativity theory and integrative systems biology. I. Founding principles and scale laws. Progr. Biophys. Mol. Biol. 97, 79–114 10.1016/j.pbiomolbio.2007.09.002 (doi:10.1016/j.pbiomolbio.2007.09.002) [DOI] [PubMed] [Google Scholar]
- 71.Nottale L., Auffray C. 2008. Scale relativity and integrative systems biology. II. Macroscopic quantum-type mechanics. Progr. Biophys. Mol. Biol. 97, 115–157 10.1016/j.pbiomolbio.2007.09.001 (doi:10.1016/j.pbiomolbio.2007.09.001) [DOI] [PubMed] [Google Scholar]
- 72.Hinch R., Greenstein J. L., Tanskanen A. J., Xu L. 2004. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 87, 3723–3736 10.1529/biophysj.104.049973 (doi:10.1529/biophysj.104.049973) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hinch R., Greenstein J. L., Winslow R. L. 2006. Multi-scale modelling of local control of calcium induced calcium release. Progr. Biophys. Mol. Biol. 90, 136–150 10.1016/j.pbiomolbio.2005.05.014 (doi:10.1016/j.pbiomolbio.2005.05.014) [DOI] [PubMed] [Google Scholar]
- 74.Tanskanen A. J., Greenstein J. L., Chen A., Sun X., Winslow R. L. 2007. Protein geometry and placement in the cardiac dyad influence macroscopic properties of calcium-induced calcium release. Biophys. J. 92, 3379–3396 10.1529/biophysj.106.089425 (doi:10.1529/biophysj.106.089425) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Einstein A. 1936. Lens-like action of a star by the deviation of light in the gravitational field. Science 84, 506–507 10.1126/science.84.2188.506 (doi:10.1126/science.84.2188.506) [DOI] [PubMed] [Google Scholar]
- 76.Petters A. O., Levine H., Wambsganss J. 2001. Singularity theory and gravitational lensing. Boston, MA: Birkhäuser [Google Scholar]
- 77.Bitbol M. In press Downward causation without foundations. Synthese. 10.1007/s11229-010-9723-5 (doi:10.1007/s11229-010-9723-5) [DOI] [Google Scholar]