

Abstract
The implicit membrane model IMM1 is extended to include the membrane dipole potential and applied to molecular dynamics simulations of the helical peptides alamethicin, WALP23, influenza hemagglutinin fusion peptide, HIV fusion peptide, magainin, and the pre-sequence of cytochrome c oxidase subunit IV (p25). The results show that the orientation of the peptides in the membrane can be influenced by the dipole potential. The binding affinity of all peptides except for the hemagglutinin fusion peptide decreases upon increase of the dipole potential. The changes in both orientation and binding affinity are explained by the interaction of the dipole potential with the helix backbone dipole and ionic side-chains. In general, peptides that tend to insert the N-terminus in the membrane and/or have positively charged side chains will lose binding affinity upon increase of the dipole potential.
Keywords: Membrane dipole potential, Molecular dynamics simulation, Implicit membrane model, Peptide orientation, Binding affinity
1. Introduction
There are three electrostatic potentials associated with cellular lipid bilayers [1,2]. The transmembrane potential Δψ is caused by the difference in charge concentration in the two bulk phases across the membrane; the surface potential ψs arises from the presence of net charge at the membrane surface; and the dipole potential ψd is thought to arise from the alignment of lipid and water molecule dipoles. The dipole potential ψd was discovered in studies of ion-transport across membranes [3–5] and has attracted great interest since it can significantly influence many important physiological processes such as ion permeability, voltage-gated channels, and function of membrane proteins. The dipole potential is commonly attributed to two factors: a negative contribution from lipid polar groups (the P-N dipole of phosphatidylcholine and the C-O dipole of carbonyl), and a positive contribution from interfacial water molecules [6]. The former is apparently over-compensated by the latter, leading to a positive total potential in the lipid hydrophobic core [7–16]. It should be noted that, despite its name, ψd could contain significant quadrupolar contributions, as was clearly shown for liquid-vapor interfaces [17–19].
Several techniques have been developed to measure ψd in experiments, including ion permeability [20], electron paramagnetic resonance [21], lipid monolayers [22], voltage-sensitive dye fluorescence [23], and cryo-EM [24]. Although it is agreed that the potential is positive in the lipid hydrophobic core, its magnitude varies depending on the technique used [5,7,21,22,24–27]. From potential measurements across a PC monolayer at the air-water interface, values of 390–440 mV were obtained [5,25], whereas from the differential membrane conductance induced by organic cations, values of 197–240 mV were obtained [7,21,25]. The membrane dipole potential could also be influenced by lipid composition. For example, it was found that the potential of a DMPC monolayer is 449.1 mV, while that of an anionic DMPG is 301.8 mV [28], and the potential of a pure DMPC vesicle is 410 mV, while the value for a hybrid DMPC/MMPC vesicle is 268 mV [29].
Another way to modify the membrane dipole potential is to add specific compounds to the solution [30–33]. Among these compounds, 6-ketocholestanol (6-KC) increases ψd, while phloretin or phlorizin decrease it. Cholesterol also raises ψd but less effectively than 6-KC. The reason that ψd is affected by these compounds could be the dipole of these molecules, or that they can change the packing of the headgroups and/or the orientation of interfacial water molecules [34].
The dipole potential might influence the structure and function of membrane-active peptides. In one study it was found that phlorizin modulated alamethicin activity, possibly by affecting its tilt angle or depth of insertion in the membrane [35]. The influence of ψd on peptide orientation could be crucial in physiological processes like membrane fusion, which is facilitated by so called fusion peptides. These peptides are presumed to adopt specific angles with respect to the membrane [36], which may be manipulated by the membrane dipole potential.
Molecular dynamics (MD) simulations with explicit solvent have been extensively used to investigate ψd in lipid membranes [8–16,18,19,37–43]. The dipole potential is calculated as a double integral of charge density along the membrane normal. All simulations give a positive potential in the membrane interior relative to water. As to the magnitude of ψd, however, the simulations are not consistent. With the GROMOS force field and SPC water, Tieleman and Berendsen obtained a value of 250 mV for a DPPC bilayer [11]. The potential was as high as 1000 mV in another simulation with the AMBER force field and TIP4P water [12]. A third study that used the AMBER force field but charges from ab initio calculation, gave a value of 960 mV [13]. With similar parameterization, Smondyrev and Berkowitz found that ψd ranges between 600 mV and 1000 mV for a hybrid DPPC/cholesterol bilayer [16]. Besides the force field, truncation of electrostatic interactions [41] and polarization [19,39] also have profound influence on the calculated potential. It is worth pointing out that the potential obtained from simulations is a theoretical idealization that cannot be experimentally measured, since any experiment would have to use a finite-size probe that would inevitably perturb the membrane [44].
In recent years implicit solvation models have also been applied to MD simulation of peptides in membranes due to their computational efficiency [45–48]. Although the membrane dipole potential has been proven crucial for lipid membranes and their interaction with peptides, it has not yet been incorporated into implicit membrane models. This could cause inaccuracies due to the omission of electrostatic interaction between the potential and the peptide dipole. In this study, we implement the membrane dipole potential into the IMM1 implicit membrane model [46]. With the new model, we investigate the interaction between a zwitterionic bilayer and the helical peptides alamethicin, WALP23, influenza hemagluttin fusion peptide (HAFP), HIV fusion peptide (HIVFP), magainin, and the pre-sequence of cytochrome c oxidase subunit IV (p25). The influence of the membrane dipole potential on peptide orientation and binding energy are determined and discussed.
2. Methods
2.1. Implicit membrane model IMM1
IMM1 [46] is an extension of the EEF1 energy function for proteins in solution [49]. In EEF1 and IMM1 the effective energy (potential of mean force) is the sum of the extended atom CHARMM energy function and a term describing the solvation free energy:
| (1) |
where is the solvation free energy of atom i, rij is the distance between i and j, fi is the solvation free energy density of i (a Gaussian function of rij), Vj is the volume of atom j, and is the solvation free energy of an isolated atom. In addition, the dielectric constant is distance dependent to account for the screening effect of the surrounding solvent and the ionic side-chains are neutralized. The membrane bilayer is represented by a hydrophobic slab centered at the plane z = 0 Å. The thickness of the slab depends on lipid type and typically ranges between 20 Å and 30 Å. In IMM1 the solvation parameters are linear combinations between the values pertaining to water and to cyclohexane, with a continuous transition between water and membrane environments. In addition, the dielectric screening is attenuated in the membrane. The surface potential can be treated using the Gouy-Chapman theory [50].
2.2. The membrane dipole potential model
Explicit MD simulations give a positive potential in the membrane interior relative to water. Its magnitude varies depending on lipid type and simulation force field [8–16,18,19,37–43]. The width of the transition region seems to be around 10 Å, while the position varies slightly depending on lipid composition. In many simulations the potential profile is monotonic [8–13,18,19,34,37–40] but in others a small peak was found on the membrane side of the transition region [11,14–16,41–43]. This could be caused by local changes at the interface due to surface tension, as discussed by Tieleman and Berendsen [11]. In their simulation of a DPPC bilayer, similar peaks were found in the NVT and NγT systems but not in the NPT system. Given the disagreement in simulation results and to keep the model simple, we decided to use a monotonic potential profile.
The following function was chosen to mimic the membrane dipole potential:
| (2) |
where a, b, and c are parameters and z is the distance to the bilayer center. The position and the width of the transition region are controlled by parameters a and b, which are set to 6.23×10−7 and 5.5, respectively. This places the center of the region around 13.5 Å, corresponding to the hydrophobic boundary of the POPC bilayer. This choice of parameters gives an approximate 10 Å for the width of the transition region. While the shape of the potential curve is controlled by a and b, the height is only determined by c. Unlike a and b that are fixed throughout the simulations, c is varied from 0 mV to 400 mV. In this way, the interaction between the membrane bilayer and peptides at various ψd could be investigated. Figure 1 compares the potential profile from our model (Eq. 2) to that from the explicit MD simulation by Zhou and Schulten [10]. It can be seen that there is good correspondence between them. In this implementation the dipole potential profile, like standard IMM1, is symmetric with respect to the bilayer center. It thus describes symmetric bilayers, like those for which the potential profile has been calculated by explicit simulations. More realistic cell membranes with different intracellular and extracellular leaflet compositions could be treated in the future by a straightforward extension of the model.
Figure 1.

Dipole potential profiles across a bilayer from the explicit MD simulation of Zhou and Schulten [10] (dashed line) and from our model (solid line). In our model, the potential is symmetric with respect to the bilayer center, as it should for a symmetric bilayer.
The energy of interaction of a solute with the membrane dipole potential is implemented as an extra term in IMM1:
| (3) |
where qi is the charge of atom i. As was done in the treatment of the surface potential [50], ionic side-chains carry a full charge in the calculation of the dipole potential energy term but are neutral in the calculation of intra- or inter-peptide interactions, as EEF1 and IMM1 require.
2.3. Molecular dynamics simulations
MD simulations were carried out with the CHARMM program [51]. Membrane and water were implicitly represented with IMM1. Six peptides were simulated: alamethicin, WALP23, HAFP, HIVFP, magainin, and p25. Their sequences are shown in Table 1. All peptides were simulated in a zwitterionic (neutral) bilayer except p25, for which an anionic bilayer was used with 30% anionic lipids. This choice was made because it has been reported that little or no helical structure was detected for p25 in membranes containing less than 20% negatively charged lipids [52]. The initial helical structures of the peptides were generated by CHARMM based on their sequences. At the beginning of the simulations, HAFP, HIVFP, magainin, and p25 were placed parallel to the membrane plane with their hydrophobic face toward the membrane. For alamethicin, both interfacial orientation (alamI) and transmembrane orientation (alamT) were simulated, and for WALP23 only the transmembrane orientation was considered. All helices were first simulated for 5 ns without the dipole potential. For interfacial orientations, besides the placement with the hydrophobic face toward the membrane, the helices were also rotated by various angles around their axes and simulated for 25 ns. The simulations gave similar equilibrated structures for all initial orientations, showing that the results are not significantly influenced by the initial conditions. After the simulation without the dipole potential, another 5 ns simulation was run on the final structures at five different ψd values (0 mV, 100 mV, 200 mV, 300 mV, and 400 mV). Each simulation was repeated eight times with different initial velocities, based on which the standard deviation of tilt angle and binding energy were calculated. For comparison, an extended 25 ns simulation was also performed on each peptide at 200 mV, and the energy, the tilt angle, and the RMSD over the trajectory were calculated. The results show that the energy converges rapidly during the simulations. The tilt angle and RMSD also converge within the first 2 ns of the simulations for all the peptides except HIVFP and p25. The tilt angle and RMSD of HIVFP and p25 exhibit larger fluctuations than in the other peptides because, as can be observed during the simulation, the termini of the two peptides tend to unfold. Average structures and energies of the last 2 ns were taken for analysis. The binding energy of the peptides was estimated as the average effective energy difference in membrane and water environments for the conformational ensemble generated by the membrane simulation:
| (4) |
where and are the effective energy in membrane and in water respectively. The index i runs through N = 1000 conformations saved at equal intervals within the last 2 ns of the simulation. Qualitatively similar results were obtained by averaging the effective energy in independent simulations in water and membrane and taking the difference. In that approach the statistical uncertainties were significantly larger.
Table 1.
Simulated peptides and their sequences
| alamethicin | Ac-UPUAUAQUVUGLUPVUUEQF-OH |
| HAFP | Ac-GLFGAIAGFIENGWEGMIDG-CONH2 |
| HIVFP | Ac-AVGIGALFLGFLGAAGSTMGARS-CONH2 |
| magainin | Ac-GIGKFLHSAKKFGKAFVGEIMNS-CONH2 |
| p25 | Ac-MLSLRQSIRFFKPATRTLCSSRYLL-CONH2 |
| WALP23 | Ac-GWWLALALALALALALALALWWA-CONH2 |
U in alamethicin refers to 2-aminoisobutyric acid (Aib). Charged residues are underscored.
3. Results
3.1. Influence of the membrane dipole potential on peptide orientation
The starting configuration of all helices was parallel to the membrane plane except for alamT and WALP23, for which it was perpendicular to the membrane plane. During the course of the simulation they adopted the tilt angles (θ) shown in Table 2. Figures 2–5 show their average orientations at different values of the dipole potential. Magainin remains almost perfectly parallel to the membrane. HAFP adopts a slight tilt. Increasingly larger tilts (smaller tilt angles) are adopted by alamI, HIVFP, and p25. WALP23 (not shown) adopts an almost perpendicular orientation to the membrane and its tilt angle is insensitive to the dipole potential. AlamT is more tilted and its tilt angle decreases with the dipole potential.
Table 2.
Tilt angles of the helices on membrane bilayer
| ψd (mV) | 0 | 100 | 200 | 300 | 400 |
|---|---|---|---|---|---|
| alamI | 69.5 (±0.9) | 71.4 (±1.4) | 84.9 (±0.7) | 86.9 (±0.1) | 88.8 (±0.2) |
| alamT | 28.8 (±1.1) | 26.0 (±1.2) | 22.5 (±0.9) | 18.5 (±0.5) | 16.6 (±0.5) |
| HAFP | 80.0 (±2.1) | 82.6 (±1.1) | 83.0 (±1.6) | 84.3 (±1.6) | 86.0 (±0.7) |
| HIVFP | 33.0 (±6.5) | 31.3 (±8.5) | 36.5 (±1.2) | 36.5 (±2.3) | 37.3 (±2.1) |
| magainin | 86.6 (±0.2) | 88.0 (±0.9) | 89.2 (±1.8) | 90.5 (±1.9) | 95.5 (±5.0) |
| p25 | 61.5 (±4.5) | 60.8 (±3.4) | 62.9 (±7.1) | 66.6 (±3.5) | 65.4 (±5.2) |
| WALP23 | 10.3 (±0.7) | 10.5 (±0.4) | 10.8 (±0.7) | 11.2 (±0.7) | 11.4 (±0.7) |
The tilt angle (in degrees) is calculated as the angle between helix axis and membrane normal. Standard deviation is given in parenthesis. AlamI refers to the interfacial orientation, and alamT refers to the transmembrane orientation.
Figure 2.

Orientations of alamI (top) and alamT (bottom). Orientations at various ψd are labeled as: red, 0 mV; orange, 100 mV; yellow, 200 mV; green, 300 mV; blue, 400 mV. Black lines denote the bilayer boundary. The N-terminus is toward the left for alamI and toward the bottom for alamT.
Figure 5.
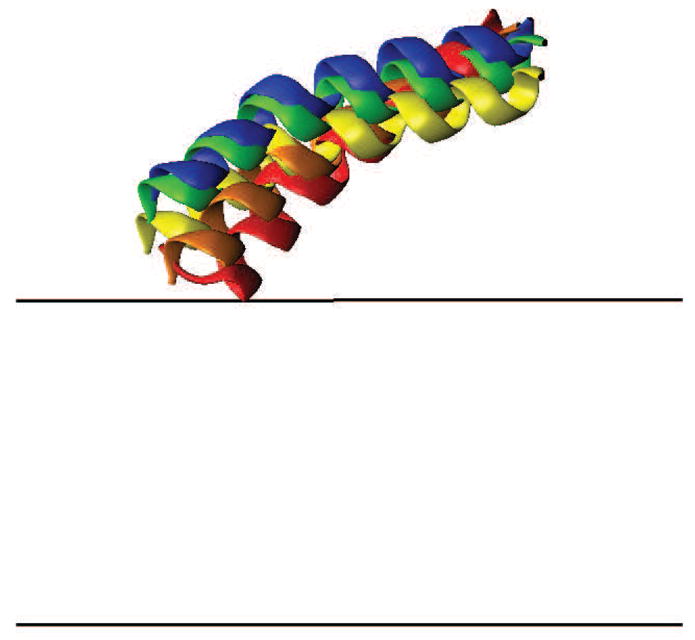
Orientation of p25. The N-terminus is toward the left. The color labeling is the same as in Figure 2.
Figure 2 shows the orientation of alamethicin at various values of ψd. For alamI, it can be seen that when ψd is set to 0 mV, the helix has an average θ = 69.5°. As ψd increases, the N-terminus gradually moves out of the membrane while the C-terminus remains at the same position. This result is consistent with the proposal of Luchian and Mereuta on the effect of phlorizin on alamethicin orientation [35]. Another interesting observation is that the relation between ψd and helix re-orientation is not linear. Re-orientation happens faster at low potential (between 0 mV and 200 mV), and slows down beyond that.
For alamT, the simulation gives an average θ = 28.8° at 0 mV, a little larger than the 17° obtained by Bak et al. in their solid-state NMR experiment [53]. As ψd increases to 400 mV, the tilt angle of alamT decreases to 16.6°. The tilt angle of the other transmembrane helix WALP23 is around 10.0°, smaller than the value around 30° given by previous experimental and explicit simulation studies [54–56]. The reason for this is probably that in those studies thinner bilayers were used, such as DMPC. Unlike alamT, there is no significant change in the angle for WALP23 from 0 mV to 400 mV.
In Figure 3 the orientations of HAFP and magainin are shown. Unlike alamI, the N-termini of HAFP and magainin stay much closer to the membrane surface. The tilt angles of the two helices are close to each other at ψd = 0 mV, and the values of 80.0° and 86.6° are in good agreement with the 77.6° and 80°, respectively, obtained in previous studies [57,58]. The tilt angles of both helices increase slightly with the dipole potential to a final value around 86.0° and 95.5° at 400 mV.
Figure 3.
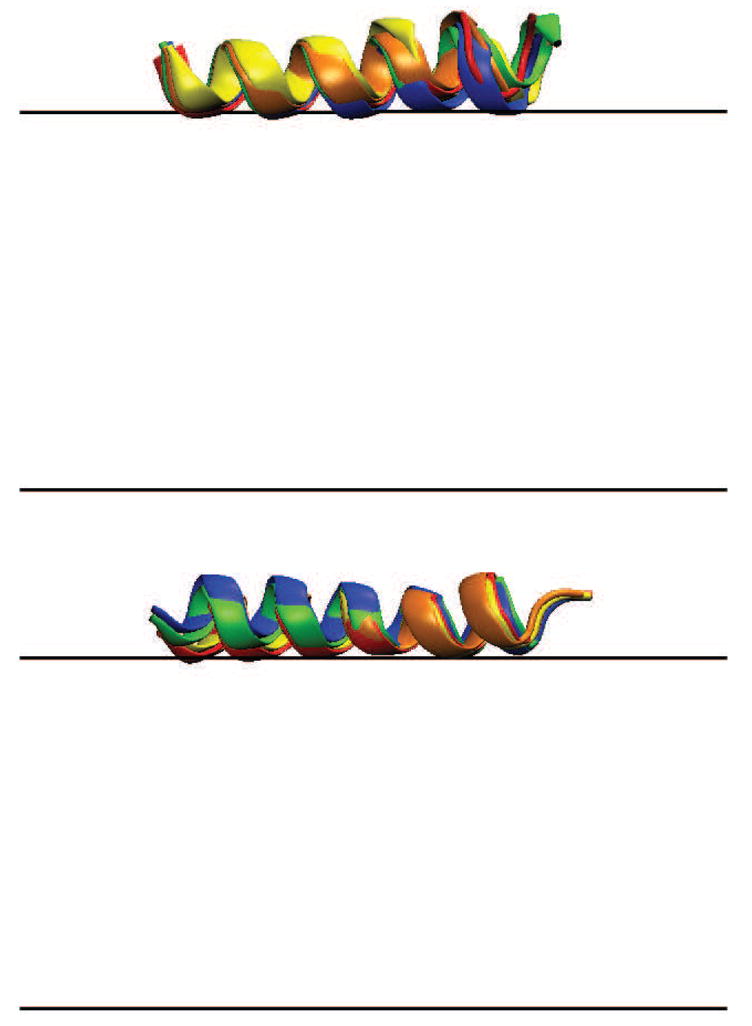
Orientations of HAFP (top) and magainin (bottom). The N-terminus is toward the left for both HAFP and magainin. The color labeling is the same as in Figure 2.
HIVFP (Figure 4) at ψd = 0 mV adopts a large tilt angle, θ = 33.0°, and a very deep insertion of the (acetylated) N-terminus that almost reaches the bilayer center. The tilt angle is in good agreement with the 36.5° in DOPC/Chol/DOPE/DOPG multilayer and the 39.0° in DMPC bilayer obtained spectroscopically [59], and a little smaller than the 44.0° obtained in an explicit MD simulation [60]. The angle increases slightly with dipole potential to 37.3°. Relatively large error bars are observed for this peptide, probably because, as we observed from the simulation trajectory, the helix C-terminus is unstable and easily unfolds.
Figure 4.
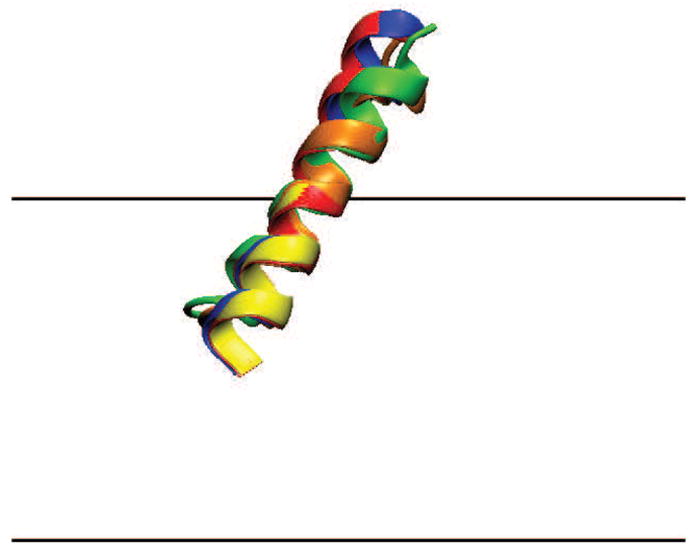
Orientation of HIVFP. The N-terminus is toward the left. The color labeling is the same as in Figure 2.
The behavior of p25 is a little different from that of the other helices (Figure 5). From 0 mV to 400 mV the tilt angle increases slightly. However, it can be seen from the figure that as ψd increases, there is a clear shift along the membrane normal for the whole helix. This is probably due to the five cationic side-chains, which are evenly distributed along the helix axis. As the membrane dipole potential increases, the interaction between the potential and the cationic side-chains pushes both termini away from the bilayer. It was found that the helical structure of p25 is very unstable and easily unfolds, probably because p25 is located in a more aqueous environment than the other helices. This is consistent to what one would expect from simple arguments based on hydrophobicity and electrostatics [61], although some experimental results seem to suggest that the helices are inserted into the lipid membrane parallel to the lipid acyl chains [62].
To explain the re-orientation that takes place for some peptides one has to consider the interaction with the membrane dipole potential of two factors: the backbone dipole and ionic side-chains. It is well known that in a helix the aggregate effect of all the individual dipoles from the CO and NH groups creates an overall dipole moment that points from its N-terminus to C-terminus. When placed in an external electric field, which in this study is the membrane dipole potential induced by lipid headgroups and interfacial water molecules, the dipole of the helix will adjust its orientation to achieve optimal alignment to the field. Since the potential is more positive in the lipid hydrophobic core than in water, the N-terminus tends to be expelled from the membrane interior while the C-terminus is attracted to the membrane. Ionic side-chains could have considerable electrostatic interaction with the dipole potential, although their favorable solvation in water forces them to avoid the membrane interior where the dipole potential is stronger. Still, cationic side-chains should be pushed away from the membrane and anionic side-chains should be attracted to the membrane.
The effect of the backbone dipole is most clearly demonstrated in the case of alamI, which shows a large increase in tilt angle as the dipole potential increases from 0 mV to 400 mV. An increase is also observed for HIVFP but it is much smaller. One possible reason for this difference in behavior between alamI and HIVFP is that on the C-terminal side alamI has an anionic glutamate, while HIVFP has a cationic arginine. These will also interact with the membrane dipole and either act in concert (as in alamI) or oppose (as in magainin) the reorientation induced by the backbone dipole. Another possibility is that the backbone dipole interaction with the membrane dipole is not strong enough in the case of HIVFP to overcome the natural tendency of this peptide for oblique orientations that results from a hydrophobicity gradient along its helical axis [36].
Transmembrane helices such as WALP23 show smaller interactions with the dipole potential because their interactions with each leaflet partly cancel out. AlamT does not completely cross the bilayer and has its N terminus on the opposite leaflet. It can, therefore, interact more strongly with the dipole potential. Its tilt angle decreases and its N-terminus moves towards the opposite interface as the dipole potential is increased. This shift improves favorable interactions of the N-terminus with the dipole potential on the trans side of the bilayer.
3.2. Influence of the membrane dipole potential on binding energy
The membrane binding energy (ΔGo) of the peptides is estimated as the average effective energy change upon moving the peptides from water into the bilayer. The results are summarized in Table 3. For alamI, magainin, and p25, ΔGo increases (binding affinity decreases) by 1.4–3.7 kcal/mol from 0 mV to 400 mV. For transmembrane helices alamT and WALP23, ΔGo increases by smaller amounts, 1.0 kcal/mol and 0.9 kcal/mol from 0 mV to 400 mV. Among all helices, HIVFP has the largest ΔGo increase of 5.1 kcal/mol. HAFP is the only peptide that gains membrane affinity as the dipole potential increases.
Table 3.
Relative binding energy of the peptides to the membrane
| ψd (mV) | 0 | 100 | 200 | 300 | 400 |
|---|---|---|---|---|---|
| alamI | 0.0 (±0.2) | 1.0 (±0.2) | 1.4 (±0.1) | 1.5 (±0.1) | 1.4 (±0.1) |
| alamT | 0.0 (±0.2) | 0.4 (±0.2) | 0.7 (±0.2) | 1.1 (±0.1) | 1.0 (±0.2) |
| HAFP | 0.0 (±0.6) | −0.9 (±0.8) | −0.4 (±0.2) | −1.2 (±0.9) | −3.1 (±0.8) |
| HIVFP | 0.0 (±0.6) | 1.5 (±0.7) | 2.4 (±0.4) | 3.6 (±0.2) | 5.1 (±0.4) |
| magainin | 0.0 (±0.2) | 0.9 (±0.4) | 1.5 (±0.5) | 2.0 (±0.4) | 2.6 (±0.5) |
| p25 | 0.0 (±0.8) | 1.1 (±1.0) | 2.1 (±0.7) | 3.4 (±0.1) | 3.7 (±0.4) |
| WALP23 | 0.0 (±0.5) | 0.1 (±0.5) | 0.7 (±0.5) | 0.7 (±0.5) | 0.9 (±0.5) |
Relative binding energy (kcal/mol) was calculated with free energy at 0 mV as reference (alamI: −8.7 kcal/mol, alamT: −6.7 kcal/mol, HAFP: −7.9 kcal/mol, HIVFP: −8.5 kcal/mol, magainin: −6.9 kcal/mol, p25: −5.1 kcal/mol, WALP23: −23.8 kcal/mol). Standard deviation is given in parentheses. AlamI refers to the interfacial orientation, and alamT refers to the transmembrane orientation. The dipole potential energy term of all peptides at 400 mV was also calculated (alamI: −0.8 kcal/mol, alamT: −0.2 kcal/mol, HAFP: −3.2 kcal/mol, HIVFP: −4.9 kcal/mol, magainin: −0.6 kcal/mol, p25: −1.4 kcal/mol, WALP23: −0.6 kcal/mol)
The loss of membrane binding affinity as the dipole potential increases for alamI and HIVFP can be explained by the interaction between the potential and the helix backbone dipole. Both of these peptides insert their N-termini into the membrane. Since the helix backbone dipole is from N-terminus to C-terminus, and the membrane dipole potential is higher inside the membrane, the electrostatic interaction between the potential and the dipole is unfavorable and becomes more unfavorable as ψd increases.
This explanation, however, does not apply to HAFP, magainin, and p25. HAFP and magainin are almost parallel to the membrane. Therefore, the influence of the membrane dipole potential on the helix backbone dipole should be limited. Here, the change in binding affinity is due to ionic side chains. Magainin carries a total charge of +3 (four lysine and one glutamate residues). Although the dipole potential reaches its largest magnitude in the membrane interior, it also affects to a smaller extent the regions where the ionic side chains are. For example, a positively charged lysine NH3 group in magainin is located around 21 Å from the bilayer center, corresponding to 10 mV in case of a total ψd = 100 mV. This value, although small, is enough to cause a considerable interaction with the ionic side-chains of magainin. For the same reason, p25 with a total charge of +5, loses 3.7 kcal/mol of binding affinity from 0 mV to 400 mV. HAFP has one aspartate residue and two glutamate residues, which gives the helix a total net charge of −3. Unlike magainin and p25, the interaction between the membrane dipole potential and the negatively charged helix should now be favored, which explains the 3.1 kcal/mol energy decrease from 0 mV to 400 mV for HAFP.
The smallest changes in binding energy are observed for the transmembrane peptides, WALP23 and alamT. WALP23 is uncharged and the helix backbone dipole is along the membrane normal. Because of symmetry, the interactions in the upper and lower leaflets essentially cancel out. As a result, the change in binding affinity is within statistical uncertainty. The alamT configuration is not entirely symmetric with respect to the membrane, but its interaction with the membrane dipole potential is also quite small. Unlike the neutral WALP23, alamT carries a net charge of −1 from its glutamate residue. The interaction between the membrane dipole potential and the negative charge provides additional stability to membrane binding, leading to a small favorable change in free energy with increasing dipole potential.
The values in Table 3 are averages from the MD simulations. As such, they are noisy and include contributions not only from the dipole potential-peptide interactions but also from changes in peptide configuration. Sometimes, there could even be secondary structure fluctuations, as was observed in the simulation of HIVFP and p25. In order to see how much the potential itself contributes to the binding energy, the average structure from the MD simulation at 0 mV for each helix was selected and single point energy calculations were performed on it at 0 mV, 100 mV, 200 mV, 300 mV, and 400 mV. Since everything else remains the same except the potential, the differences in energy show the effect of the dipole potential only. The results are summarized in Figure 6 and exhibit the same pattern as that in Table 3. AlamT and WALP23 have the smallest energy change, while HIVFP has the largest one. The values of alamI, magainin, and p25 are in between, changing by around 0.6–1.2 kcal/mol every 100 mV, and the binding of HAFP becomes more stable as ψd increases, consistent with Table 3. In order to check if the extra stability of HAFP is really caused by the interaction between the membrane dipole potential and ionic side-chains, aspartate and glutamate residues were replaced with neutral glycine residues. For the mutant peptide the energy increased by 3.4 kcal/mol from 0 mV to 400 mV, which confirms our explanation.
Figure 6.

Relative energy as a function of ψd for the average structure from the MD simulation at 0 mV. Since the peptide structure remains the same, the energy difference is only related to the membrane dipole potential. The figure shows the same pattern as the binding energy in Table 3.
In experiments, by adding compounds such as phloretin, 6-KC, or cholesterol, or by changing lipid composition, ψd can be altered and the partition coefficient Kp or dissociation constant Kd can be measured [63–68]. The binding free energy ΔGo of the peptides to the membrane can then be deduced from Kp and Kd. Usually, however, the energy changes as a function of ψd is very small, sometimes close to experimental error. In addition, it is not clear that the observed differences are due to the change in the dipole potential or other factors. Despite this limitation, we will now survey the relevant experimental data and compare to our results.
Our simulations show that helices will lose binding affinity unless they carry a considerable amount of negative charge. The results from experiments are not conclusive. In a study of the interaction between p25 peptide and PC liposomes, Cladera and O’Shea found the dissociation constant Kd decreases from 4.0 ± 1.1 μM in the presence of phloretin to 3.1 ± 0.2 μM in the presence of 6-KC, corresponding to a decreasing ΔGo with ψd [64]. In a study of the interaction between HIV fusion peptide and PC/PE LUV, Buson and Cladera also reported a decreasing Kd as more 6-KC is added to the solution [67]. As phloretin decreases and 6-KC increases the dipole potential, these experiments are not in agreement with our simulations. Others, however, are consistent with our findings. In a study of the interaction between the signal sequence of the Escherichia coli LamB protein with egg phosphatidylcholine (EPC), Voglino et al. reported a 0.1 kcal/mol and 2.4 kcal/mol ΔGo increase by adding cholesterol and 6-KC respectively, both of which increase the magnitude of the membrane dipole potential [63]. Similarly, in a study of the interaction of mitochondrial peptide rhodanese (MPR) and melittin with polyethylene glycol (PEG) LUV, Allende et al. reported a 1.0 kcal/mol and 0.2 kcal/mol ΔGo increase for MPR and melittin by adding 6-KC, and a 1.4 kcal/mol and 1.3 kcal/mol by adding cholesterol [66]. Both of the above mentioned peptides are positively charged like p25. However, this loss of membrane affinity was attributed to factors other than the dipole potential in these studies. Voglino et al. pointed out that the ketone group of 6-KC will increase the hydrophilic thickness of the membrane interface by about 4 Å. As a result, fewer of the hydrophobic amino acids in the helix will be exposed to the low-dielectric acyl chain region of the bilayer, and the contribution of the hydrophobic energy to the binding free energy will decrease. Allende et al. proposed that addition of cholesterol increases the compressibility modulus of the bilayer. Therefore, to incorporate a peptide into the bilayer, extra work has to be done to increase the surface area of the bilayer to provide room for the peptide. Experimentally, it is very difficult to dissect these effects. Our results provide a theoretical basis for discussing the contribution of the membrane dipole potential to peptide membrane-bound structure and binding affinity.
4. Conclusions
We proposed a simple approach for implicit modeling of the dipole potential across lipid bilayers. The potential profile features a high value in the lipid hydrophobic core and a low value in water, joined together by a smooth curve around the headgroup region. The center and the width of the transition region were determined based on previous MD simulations with explicit lipids while its height ranges between 0 mV and 400 mV. The energy of interaction of a peptide with the dipole potential is then added as part of the solvation free energy in the implicit membrane model IMM1.
With this updated version of IMM1, we performed MD simulations on the helical peptides alamethicin, WALP23, HAFP, HIVFP, magainin, and p25. The influence of the membrane dipole potential on the helix orientation and binding energy was determined. For all helices, insertion of their N-termini to the cis leaflet of the bilayer leads to an unfavorable alignment between the backbone dipole and the potential (in addition to an also unfavorable interaction due to charge in case the N-terminus is protonated). Therefore, when the potential increases, the helix tilt angles increase as well, as observed for alamI, and to a smaller extent for HAFP, HIVFP, magainin, and p25. However, the interaction of the N-terminus with the trans leaflet is favorable, and leads to deeper membrane insertion and smaller tilt angle of alamT. The only peptide that gains membrane binding affinity with increasing dipole potential is HAFP, due to its net negative charge. All other peptides lose binding affinity with increasing dipole potential due to the interaction of either the backbone dipole or cationic side chains with the membrane dipole potential.
Acknowledgments
This work was supported by the National Science Foundation, grant number MCB- 0615552. Infrastructure support was provided by RCMI grant RR03060 from NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Honig BH, Hubbell WL, Flewelling RF. Electrostatic interactions in membranes and proteins. Annu Rev Biophys Biophys Chem. 1986;15:163–193. doi: 10.1146/annurev.bb.15.060186.001115. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin S. The electrostatic properties of membranes. Annu Rev Biophys Biophys Chem. 1989;18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- 3.Liberman EA, Topaly VP. Selective transport of ions through bimolecular phospholipid membranes. Biochim Biophys Acta. 1968;163:125–136. doi: 10.1016/0005-2736(68)90089-8. [DOI] [PubMed] [Google Scholar]
- 4.Haydon DA, Myers VB. Surface charge, surface dipoles and membrane conductance. Biochim Biophys Acta. 1973;307:429–443. doi: 10.1016/0005-2736(73)90289-7. [DOI] [PubMed] [Google Scholar]
- 5.Hladky SB, Haydon DA. Membrane conductance and surface potential. Biochim Biophys Acta. 1973;318:464–468. [Google Scholar]
- 6.Clarke RJ. The dipole potential of phospholipid membranes and methods for its detection. Adv Colloid Interface Sci. 2001;89:263–281. doi: 10.1016/s0001-8686(00)00061-0. [DOI] [PubMed] [Google Scholar]
- 7.Gawrisch K, Ruston D, Zimmerberg J, Parsegian VA, Rand RP, Fuller N. Membrane dipole potentials, hydration forces, and the ordering of water at membrane surfaces. Biophys J. 1992;61:1213–1223. doi: 10.1016/S0006-3495(92)81931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marrink SJ, Berkowitz M, Berendsen HJC. Molecular-dynamics simulation of a membrane water interface - the ordering of water and its relation to the hydration force. Langmuir. 1993;9:3122–3131. [Google Scholar]
- 9.Chiu SW, Clark M, Balaji V, Subramaniam S, Scott HL, Jakobsson E. Incorporation of surface-tension into molecular-dynamics simulation of an interface - a fluid-phase lipid bilayer-membrane. Biophys J. 1995;69:1230–1245. doi: 10.1016/S0006-3495(95)80005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou F, Schulten K. Molecular-dynamics study of a membrane water interface. J Phys Chem. 1995;99:2194–2207. [Google Scholar]
- 11.Tieleman DP, Berendsen HJC. Molecular dynamics simulations of a fully hydrated dipalmitoyl phosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J Chem Phys. 1996;105:4871–4880. [Google Scholar]
- 12.Shinoda W, Shimizu M, Okazaki S. Molecular dynamics study on electrostatic properties of a lipid bilayer: Polarization, electrostatic potential, and the effects on structure and dynamics of water near the interface. J Phys Chem B. 1998;102:6647–6654. [Google Scholar]
- 13.Essmann U, Berkowitz ML. Dynamical properties of phospholipid bilayers from computer simulation. Biophys J. 1999;76:2081–2089. doi: 10.1016/S0006-3495(99)77364-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu SW, Jakobsson E, Subramaniam S, Scott HL. Combined monte carlo and molecular dynamics simulation of fully hydrated dioleyl and palmitoyl-oleyl phosphatidylcholine lipid bilayers. Biophys J. 1999;77:2462–2469. doi: 10.1016/S0006-3495(99)77082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smondyrev AM, Berkowitz ML. Structure of dipalmitoylphosphatidylcholine/cholesterol bilayer at low and high cholesterol concentrations: Molecular dynamics simulation. Biophys J. 1999;77:2075–2089. doi: 10.1016/S0006-3495(99)77049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson MA, Pohorille A, Pratt LR. Study on the liquid vapor interface of water. 1. Simulation results of thermodynamic properties and orientational structure - comment. J Chem Phys. 1989;90:5211–5213. doi: 10.1063/1.456536. [DOI] [PubMed] [Google Scholar]
- 18.Harder E, MacKerell AD, Jr, Roux B. Many-body polarization effects and the membrane dipole potential. J Am Chem Soc. 2009;131:2760. doi: 10.1021/ja806825g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vorobyov I, Allen TW. The electrostatics of solvent and membrane interfaces and the role of electronic polarizability. J Chem Phys. 2010;132:85101–85101. [Google Scholar]
- 20.Andersen O, Fuchs M. Potential-energy barriers to ion-transport within lipid bilayers. Biophys J. 1975;15:795–830. doi: 10.1016/S0006-3495(75)85856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flewelling RF, Hubbell WL. The membrane dipole potential in a total membrane-potential model - applications to hydrophobic ion interactions with membranes. Biophys J. 1986;49:541–552. doi: 10.1016/S0006-3495(86)83664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brockman H. Dipole potential of lipid-membranes. Chem Phys Lipids. 1994;73:57–79. doi: 10.1016/0009-3084(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 23.Gross E, Bedlack RS, Loew LM. Dual-wavelength ratiometric fluorescence measurement of the membrane dipole potential. Biophys J. 1994;67:208–216. doi: 10.1016/S0006-3495(94)80471-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LG, Bose PS, Sigworth FJ. Using cryo-em to measure the dipole potential of a lipid membrane. Proc Natl Acad Sci USA. 2006;103:18528–18533. doi: 10.1073/pnas.0608714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickar AD, Benz R. Transport of oppositely charged lipophilic probe ions in lipid bilayer membranes having various structures. J Membr Biol. 1978;44:353–376. [Google Scholar]
- 26.Simon SA, McIntosh TJ. Magnitude of the solvation pressure depends on dipole potential. Proc Natl Acad Sci USA. 1989;86:9263–9267. doi: 10.1073/pnas.86.23.9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIntosh TJ, Simon SA, Needham D, Huang CH. Interbilayer interactions between sphingomyelin and sphingomyelin cholesterol bilayers. Biochemistry. 1992;31:2020–2024. doi: 10.1021/bi00122a018. [DOI] [PubMed] [Google Scholar]
- 28.Lairion F, Disalvo EA. Effect of phloretin on the dipole potential of phosphatidylcholine, phosphatidylethanolamine, and phosphatidylglycerol monolayers. Langmuir. 2004;20:9151–9155. doi: 10.1021/la049515k. [DOI] [PubMed] [Google Scholar]
- 29.Starke-Peterkovic T, Clarke RJ. Effect of headgroup on the dipole potential of phospholipid vesicles. Eur Biophys J Biophy. 2009;39:103–110. doi: 10.1007/s00249-008-0392-y. [DOI] [PubMed] [Google Scholar]
- 30.Andersen OS, Finkelstein A, Katz I, Cass A. Effect of phloretin on permeability of thin lipid-membranes. J Gen Physiol. 1976;67:749–771. doi: 10.1085/jgp.67.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melnik E, Latorre R, Hall JE, Tosteson DC. Phloretin-induced changes in ion-transport across lipid bilayer membranes. J Gen Physiol. 1977;69:243–257. doi: 10.1085/jgp.69.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reyes J, Greco F, Motais R, Latorre R. Phloretin and phloretin analogs - mode of action in planar lipid bilayers and monolayers. J Membr Biol. 1983;72:93–103. [Google Scholar]
- 33.Franklin JC, Cafiso DS. Internal electrostatic potentials in bilayers - measuring and controlling dipole potentials in lipid vesicles. Biophys J. 1993;65:289–299. doi: 10.1016/S0006-3495(93)81051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu KC, Klein ML, Tobias DJ. Constant-pressure molecular dynamics investigation of cholesterol effects in a dipalmitoylphosphatidylcholine bilayer. Biophys J. 1998;75:2147–2156. doi: 10.1016/S0006-3495(98)77657-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luchian T, Mereuta L. Phlorizin- and 6-ketocholestanol-mediated antagonistic modulation of alamethicin activity in phospholipid planar membranes. Langmuir. 2006;22:8452–8457. doi: 10.1021/la0613777. [DOI] [PubMed] [Google Scholar]
- 36.Brasseur R, Pillot T, Lins L, Vandekerckhove J, Rosseneu M. Peptides in membranes: Tipping the balance of membrane stability. Trends Biochem Sci. 1997;22:167–171. doi: 10.1016/s0968-0004(97)01047-5. [DOI] [PubMed] [Google Scholar]
- 37.La Rocca P, Shai Y, Sansom MSP. Peptide-bilayer interactions: Simulations of dermaseptin b, an antimicrobial peptide. Biophys Chem. 1999;76:145–159. doi: 10.1016/s0301-4622(98)00232-4. [DOI] [PubMed] [Google Scholar]
- 38.Lin JH, Baker NA, McCammon JA. Bridging implicit and explicit solvent approaches for membrane electrostatics. Biophys J. 2002;83:1374–1379. doi: 10.1016/S0006-3495(02)73908-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis JE, Raharnan O, Patel S. Molecular dynamics simulations of a dmpc bilayer using nonadditive interaction models. Biophys J. 2009;96:385–402. doi: 10.1016/j.bpj.2008.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogberg CJ, Lyubartsev AP. Effect of local anesthetic lidocaine on electrostatic properties of a lipid bilayer. Biophys J. 2008;94:525–531. doi: 10.1529/biophysj.107.104208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patra M, Karttunen M, Hyvonen MT, Falck E, Lindqvist P, Vattulainen I. Molecular dynamics simulations of lipid bilayers: Major artifacts due to truncating electrostatic interactions. Biophys J. 2003;84:3636–3645. doi: 10.1016/S0006-3495(03)75094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurtovenko AA, Patra M, Karttunen M, Vattulainen I. Cationic dmpc/dmtap lipid bilayers: Molecular dynamics study. Biophys J. 2004;86:3461–3472. doi: 10.1529/biophysj.103.038760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mojumdar EH, Lyubartsev AP. Molecular dynamics simulations of local anesthetic articaine in a lipid bilayer. Biophys Chem. 2010;153:27–35. doi: 10.1016/j.bpc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Vorobyov I, Bekker B, Allen TW. Electrostatics of deformable lipid membranes. Biophys J. 2010;98:2904–2913. doi: 10.1016/j.bpj.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spassov VZ, Yan L, Szalma S. Introducing an implicit membrane in generalized born/solvent accessibility continuum solvent models. J Phys Chem B. 2002;106:8726–8738. [Google Scholar]
- 46.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 47.Im W, Feig M, Brooks CL. An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanizaki S, Feig M. A generalized born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes. J Chem Phys. 2005:122. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 49.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 50.Lazaridis T. Implicit solvent simulations of peptide interactions with anionic lipid membranes. Proteins-Struct Funct Bioinform. 2005;58:518–527. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 51.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. Charmm: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamm LK, Bartoldus I. Secondary structure of a mitochondrial signal peptide in lipid bilayer-membranes. FEBS Lett. 1990;272:29–33. doi: 10.1016/0014-5793(90)80441-k. [DOI] [PubMed] [Google Scholar]
- 53.Bak M, Bywater RP, Hohwy M, Thomsen JK, Adelhorst K, Jakobsen HJ, Sorensen OW, Nielsen NC. Conformation of alamethicin in oriented phospholipid bilayers determined by n-15 solid-state nuclear magnetic resonance. Biophys J. 2001;81:1684–1698. doi: 10.1016/S0006-3495(01)75822-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park SH, Opella SJ. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J Mol Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 55.Ozdirekcan S, Etchebest C, Killian JA, Fuchs PFJ. On the orientation of a designed transmembrane peptide: Toward the right tilt angle? J Am Chem Soc. 2007;129:15174–15181. doi: 10.1021/ja073784q. [DOI] [PubMed] [Google Scholar]
- 56.Ramamoorthy A, Kandasamy SK, Lee DK, Kidambi S, Larson RG. Structure, topology, and tilt of cell-signaling peptides containing nuclear localization sequences in membrane bilayers determined by solid-state nmr and molecular dynamics simulation studies. Biochemistry. 2007;46:965–975. doi: 10.1021/bi061895g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sammalkorpi M, Lazaridis T. Configuration of influenza hemagglutinin fusion peptide monomers and oligomers in membranes. BBA-Biomembranes. 2007;1768:30–38. doi: 10.1016/j.bbamem.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 58.Boughton AP, Andricioaei I, Chen Z. Surface orientation of magainin 2: Molecular dynamics simulation and sum frequency generation vibrational spectroscopic studies. Langmuir. 2010;26:16031–16036. doi: 10.1021/la1024394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castano S, Desbat B. Structure and orientation study of fusion peptide fp23 of gp41 from hiv-1 alone or inserted into various lipid membrane models (mono-, bi- and multibi-layers) by ft-ir spectroscopies and brewster angle microscopy. BBA-Biomembranes. 2005;1715:81–95. doi: 10.1016/j.bbamem.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 60.Kamath S, Wong TC. Membrane structure of the human immunodeficiency virus gp41 fusion domain by molecular dynamics simulation. Biophys J. 2002;83:135–143. doi: 10.1016/S0006-3495(02)75155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brasseur R, Deloof H, Ruysschaert JM, Rosseneu M. Conformational-analysis of lipid-associating proteins in a lipid environment. Biochim Biophys Acta. 1988;943:95–102. doi: 10.1016/0005-2736(88)90350-1. [DOI] [PubMed] [Google Scholar]
- 62.Goormaghtigh E, Martin I, Vandenbranden M, Brasseur R, Ruysschaert JM. Secondary structure and orientation of a chemically synthesized mitochondrial signal sequence in phospholipid-bilayers. Biochem Biophys Res Commun. 1989;158:610–616. doi: 10.1016/s0006-291x(89)80093-2. [DOI] [PubMed] [Google Scholar]
- 63.Voglino L, McIntosh TJ, Simon SA. Modulation of the binding of signal peptides to lipid bilayers by dipoles near the hydrocarbon-water interface. Biochemistry. 1998;37:12241–12252. doi: 10.1021/bi9805792. [DOI] [PubMed] [Google Scholar]
- 64.Cladera J, O’Shea P. Intramembrane molecular dipoles affect the membrane insertion and folding of a model amphiphilic peptide. Biophys J. 1998;74:2434–2442. doi: 10.1016/S0006-3495(98)77951-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asawakarn T, Cladera J, O’Shea P. Effects of the membrane dipole potential on the interaction of saquinavir with phospholipid membranes and plasma membrane receptors of caco-2 cells. J Biol Chem. 2001;276:38457–38463. doi: 10.1074/jbc.M103269200. [DOI] [PubMed] [Google Scholar]
- 66.Allende D, Vidal A, Simon SA, McIntosh TJ. Bilayer interfacial properties modulate the binding of amphipathic peptides. Chem Phys Lipids. 2003;122:65–76. doi: 10.1016/s0009-3084(02)00179-2. [DOI] [PubMed] [Google Scholar]
- 67.Buzon V, Cladera J. Effect of cholesterol on the interaction of the hiv gp41 fusion peptide with model membranes. Importance of the membrane dipole potential. Biochemistry. 2006;45:15768–15775. doi: 10.1021/bi060622i. [DOI] [PubMed] [Google Scholar]
- 68.Wessman P, Stromstedt AA, Malmsten M, Edwards K. Melittin-lipid bilayer interactions and the role of cholesterol. Biophys J. 2008;95:4324–4336. doi: 10.1529/biophysj.108.130559. [DOI] [PMC free article] [PubMed] [Google Scholar]