

Abstract
New strategies are needed to circumvent increasing outbreaks of resistant strains of pathogens and to expand the dwindling supply of effective antimicrobials. A common impediment to drug development is the lack of an easy approach to determine the in vivo mechanism of action and efficacy of novel drug leads. Towards this end, we describe an unbiased approach to predict in vivo mechanisms of action from NMR metabolomics data. Mycobacterium smegmatis, a non-pathogenic model organism for Mycobacterium tuberculosis, was treated with 12 known drugs and 3 chemical leads identified from a cell-based assay. NMR analysis of drug-induced changes to the M. smegmatis metabolome resulted in distinct clustering patterns correlating with in vivo drug activity. The clustering of novel chemical leads relative to known drugs provides a mean to identify a protein target or predict in vivo activity.
Keywords: NMR, metabolomics, OPLS-DA, perturbation, drug discovery, Mycobacterium smegmatis, Mycobacterium tuberculosis
Emerging and remerging infectious disease outbreaks from numerous gram-negative and grampositive pathogens have increased dramatically over the past decade.1 Further, we are facing the serious likelihood that these pathogens will soon become resistant to all known antibacterial treatments, which may lead to worldwide pandemics.2 Unfortunately, the development and approval of antibiotics have not kept pace with the growing emergence of resistant pathogens.3 Instead, there has been a decline in the approval of new antibiotics.4 Twenty novel classes of marketable antibiotics were produced between 1930 and 1962.5 These classes of antibiotics inhibit a short list of cellular processes: cell wall biosynthesis, DNA supercoiling, transcription, translation and folate biosynthesis. Since 1962, only two new antibiotic classes have received FDA approval: oxazolidinones, which inhibits protein synthesis, and cyclic lipopeptides, which destroys membrane potential. Both compounds are used in the treatment of gram positive bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA).5 However, additional antibiotics are needed to combat the prevalence of other multidrug resistant pathogens, such as Enterococcus faecium, Klebsiella pneumonia, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter species that are infecting the majority of US hospitals.6 Also extreme drug resistant strains of Mycobacterium tuberculosis are a rising threat in the world.
The Infectious Diseases Society of America (IDSA) has proposed an initiative to develop and approve 10 novel antibiotics by the year 2020.7 However, existing drug discovery strategies may not be able to meet these challenges. Drug discovery programs rely heavily on target based high throughput screening (HTS) of large chemical libraries followed by lead optimization.8, 9 Unfortunately, this approach has demonstrated an extremely high rate of failure and erroneous leads. Even when a valid HTS hit is found, it is uncertain if this chemical lead can penetrate into the bacterial cell and demonstrate in vivo activity.
NMR Metabolomics is evolving as a significant component of the drug discovery process and offers an inexpensive route to help overcome the multiple challenges faced by researchers.10 Metabolomics is a relatively new field and is based on the identification and quantification of small molecules found in living cells or biofluids.11 Since small molecules are downstream products of biomolecular processes, the identity and concentration of metabolites provide biochemical signatures for tracking the physiological effects of antibiotic efficacy, selectivity, and toxicity. Characterizing these biochemical signatures relies upon the global determination of numerous endogenous small molecules followed by pattern recognition using multivariate analysis.12 Such comprehensive biochemical information can be readily obtained using 1H NMR spectroscopy with minimal sample handling while providing highly reproducible data in an automated fashion.10 Multivariate statistical analysis, such as orthogonal partial least-squares discriminant analysis (OPLS-DA), is typically employed to extract information from the large and complex NMR data sets.13 Simply, OPLS-DA is used to identify clustering patterns from the major variations between NMR spectra.10
Herein, we describe a new method using 1H NMR and OPLS-DA to profile the in vivo mechanism of action of known antibiotics used to treat M. tuberculosis. More importantly, we aim to use this information to classify compounds with unknown mechanisms of action, but demonstrated anti-tubercular activity. Our approach is predicated on the hypothesis that drugs with similar modes of activity or therapeutic targets will have a similar impact on the metabolome of M. smegmatis and will cluster together in an OPLS-DA scores plot. Thus, the mode of action of a novel chemical lead can be inferred from its clustering in an OPLS-DA scores plot relative to drugs with defined biological targets. Importantly, if the chemical lead is separated from known drugs in the OPLS-DA scores plot, then this result would infer a new mechanism of action and a potentially valuable, new antibiotic.
Our methodology was demonstrated using 12 antibiotics known to inhibit the growth of M. tuberculosis and M. smegmatis (Table 1). The mechanism of action for each antibiotic was identified from the Drug Bank Database,14 and the minimum inhibitory concentrations (MIC) were obtained from the scientific literature.15-23 In addition, three chemical leads were randomly selected from the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) library of compounds (http://www.TAACF.org). The compounds were screened against M. tuberculosis and have comparable MICs to known TB drugs, but the biological target or mechanism of action was not reported by TAACF. The non-pathogenic M. smegmatis was used as a model system for the NMR metabolomics study.
Table 1.
Description of antimicrobial compounds and dosages used in this study.
| Compound | Class | Mechanism of Action | MICa (μg/ml) | Dosageb (μg/ml) |
|---|---|---|---|---|
| Ampicillin | Penicillins | Inhibits transpeptidation and prevents cell wall formation. | 16.0c | 96.0 |
| Chloramphenicol | Amphetamines | Inhibits protein synthesis by binding to the 50S ribosomal subunit. | 6.0 | 6.0 |
| Ciprofloxacin | Fluoroquinolones | Inhibits DNA gyrase and prevents DNA supercoiling. | 0.2 | 2.0 |
| D-cycloserine | Oxazolidinones | Inhibits alanine racemase and alanine ligase and prevents cell wall formation (different from other oxazolidinones that inhibit protein synthesis). | 750 | 750 |
| Ethambutol | Amino Alcohols | Disrupts arabinogalactan formation by inhibiting arabinosyl transferase. | 10.0 | 100.0 |
| Ethionamide | Pyridine Derivatives | Inhibits mycolic acid formation similar to isoniazid. | 20.0 | 160.0 |
| Gentamicin | Aminoglycosides | Inhibits protein synthesis by binding to the 30S ribosomal protein S12 and 16S rRNA. | 2.0 | 8.0 |
| Isoniazid | Pyridine Derivatives | A prodrug that inhibits InhA and prevent mycolic acid synthesis. | 2.0 | 48.0 |
| Kanamycin | Aminoglycosides | Inhibits protein synthesis by binding to the 30S ribosomal protein S12 and 16S rRNA. | 4.0 | 4.0 |
| Rifampicin | Rifampicins | Inhibits RNA polymerase and prevent RNA synthesis. | 30.0 | 60.0 |
| Streptomycin | Aminoglycosides | Inhibits protein synthesis by binding to the 30S ribosomal protein S12 and 16S rRNA. | 0.25 | 1.5 |
| Vancomycin | Glycopeptides | Binds to the D-alanyl-D-alanine dipeptide and prevents cell wall formation. | 50 | 450 |
| Amiodarone | Benzofurans | Unknown | 26.6 | 212.8 |
| Clofazimine | Anilines | Unknown | 0.32 | 3.84 |
| Chlorprothixene | Thioxanthines | Unknown | 36.0 | 216.0 |
Literature values of minimum inhibitory concentrations against M. smegmatis used as a starting point to determine an optimal dosage for the NMR metabolomics study.
Actual dosage used to treat M. smegmatis cells to inhibit growth by ~50% following drug treatment.
Reported for M. smegmatis β-lactamase and ribosomal protein S12 mutants.
In order to analyze changes in the M. smegmatis metabolome, the drug dosage needs to be below lethal levels and only affect cell growth. Typically, a drug concentration that inhibits cell growth by approximately 50% of the growth rate of untreated cells is desirable. While MIC values are available from the literature, these concentrations are based on standardized drug gradients, inoculum sizes, and readout endpoints. Additionally, the reported MICs were obtained with different bacterial strains, at different growth stages or cell densities, and under a variety of experimental conditions that includes either broth or agar methods. Further complicating the situation is the diversity of MICs values reported for a single drug. Thus, the literature MIC values listed in Table 1 were simply used as a starting point to determine an optimal dosage for the NMR metabolomics study under our experimental conditions. Each drug was titrated over a concentration range of 1 to 24 times the literature MIC values. The individual drug concentrations needed to achieve ~50% growth inhibition are reported in Table 1. An average growth inhibition of 43.1 ± 10.5% was observed after the addition of each of the 15 drugs. Preparation of the M. smegmatis cell cultures for metabolomic analysis was then performed using the optimal dosage for each drug.
Due to the inherit variability of biological samples and to provide a robust statistical analysis, 10 cultures inoculated with each antibiotic and 40 cultures of untreated cells were prepared for the NMR metabolomics study. A 1D 1H NMR spectrum was collected for each biological sample, which were normalized using center averaging and analyzed using OPLS-DA. A representative 2D OPLS-DA scores plot displaying a comparison between 6 antibiotics with known mechanisms of action is shown in Figure 1. The OPLS-DA model was cross-validated using a modified leave-one-out method. A quality assessment score (Q2) of 0.82 was obtained, which is an excellent result compared to an ideal score of one. Thus, the cross-validation indicates a highly reliable model. Each point in the 2D OPLS-DA scores plot represents a single 1D 1H NMR spectrum of a specific drug treated or untreated cell culture. The 2D OPLS-DA scores plot consists of 4 separate clustering patterns, which demonstrates that each group has a considerably different impact on the metabolome of M. smegmatis. Importantly, all the drug-treated M. smegmatis cell cultures form distinct and separate clusters from the untreated cell cultures. This is consistent with all the drugs being biologically active and inhibiting M. smegmatis cell growth.
Figure 1.
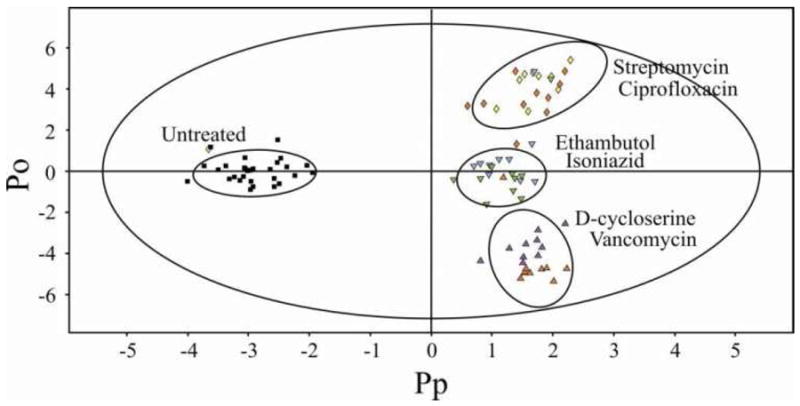
2D OPLS-DA scores plot demonstrating the clustering pattern obtain for six different antibiotics with known and distinct biological targets: untreated M. smegmatis cells (■), ciprofloxacin (
 ), streptomycin (
), streptomycin (
 ), ethambutol (
), ethambutol (
 ), isoniazid (
), isoniazid (
 ), D-cycloserine (
), D-cycloserine (
 ), and vancomycin (
), and vancomycin (
 ) treated M. smegmatis cells. The ellipses correspond to the 95% confidence limits from a normal distribution for each cluster. The untreated M. smegmatis cells (■) was designated the control class and the remainder of the cells were designated as treated. The OPLS-DA used one predictive component and three orthogonal components to yield a R2X of 0.610, R2Y of 0.893 and Q2 of 0.82.
) treated M. smegmatis cells. The ellipses correspond to the 95% confidence limits from a normal distribution for each cluster. The untreated M. smegmatis cells (■) was designated the control class and the remainder of the cells were designated as treated. The OPLS-DA used one predictive component and three orthogonal components to yield a R2X of 0.610, R2Y of 0.893 and Q2 of 0.82.
Antibiotics that share a similar or identical biological target were observed to cluster together in the OPLS-DA scores plot. For example, ethambutol and isoniazid inhibit mycolic acid biosynthesis that prevents the formation of the arabinogalactan-mycolic acid matrix. Streptomycin and ciprofloxacin form the second cluster. Streptomycin prevents protein synthesis and ciprofloxacin inhibits DNA supercoiling that affects replication, transcription, and repair, leading to a similar disruption in protein synthesis. Since these two antibiotics cluster together, it implies that the inhibition of transcription or translation results in a similar impact on the metabolome. Vancomycin and D-cycloserine both affect cell wall formation and form the third cluster. In a principal component analysis (PCA) of the data (see supplemental Figure 1S) there is a more pronounced separation between vancomycin and D-cycloserine along PC2. This reflects a fundamental difference between PCA and OPLS-DA, where PCA is limited to a linear model and does not readily differentiate between within-class and between-class variations.13, 24 Correspondingly, OPLS-DA is preferred as long as cross-validation verifies a reliable model.
The NMR metabolomics analysis was then expanded to include a total of 12 drugs with known biological targets and 3 compounds randomly chosen from the TAACF library. Amiodorone, clofazamine and chlorprothixene are active against TB, but have unknown mechanisms of action according to the TAACF database. Nevertheless, the three compounds are known drugs, where amiodorone is an antiarrhythmic agent that affects potassium efflux, chlorprothixene is an antipsychotic drug that inhibits dopamine receptors, and clofazamine is a 40 year-old leprosy treatment with an ill-defined biological activity. The 2D OPLS-DA scores plot (Figure 2A) identified 4 distinct clusters and yielded a highly reliable cross validation Q2 score of 0.671. As before, the different clusters are correlated with distinct modes of action: inhibition of cell wall formation, inhibition of mycolic acid biosynthesis, and inhibition of transcription, translation or the overall effects of DNA supercoiling.
Figure 2.
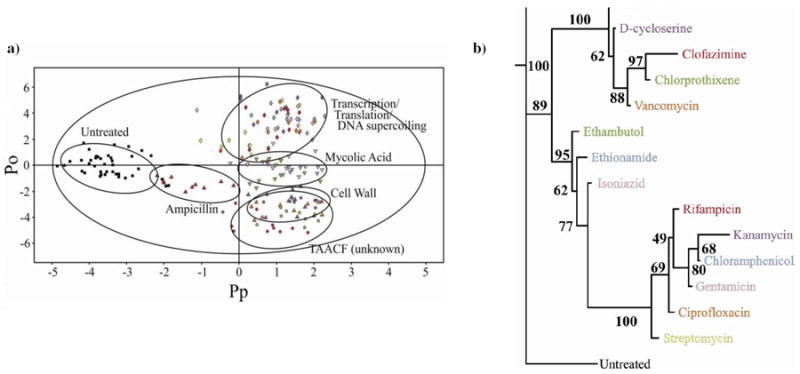
(a) 2D OPLS-DA scores plot demonstrating the clustering pattern for 12 antibiotics with known biological targets and three compounds of unknown in vivo activity: untreated M. smegmatis cells (■), chloramphenicol (
 ), ciprofloxacin (
), gentamicin (
), ciprofloxacin (
), gentamicin (
 ), kanamycin (
), kanamycin (
 ), rifampicin (
), rifampicin (
 ), streptomycin (
), ethambutol (
), ethionamide (
), streptomycin (
), ethambutol (
), ethionamide (
 ), isoniazid (
), ampicillin (
), isoniazid (
), ampicillin (
 ), D-cycloserine (
), vancomycin (
), amiodorone (
), D-cycloserine (
), vancomycin (
), amiodorone (
 ), chlorprothixene (
), chlorprothixene (
 ), and clofazimine (
), and clofazimine (
 )treated M. smegmatis cells. The ellipses correspond to the 95% confidence limits from a normal distribution for each cluster. The untreated M. smegmatis cells (■) was designated the control class and the remainder of the cells were designated as treated. The OPLS-DA used one predictive component and six orthogonal components to yield a R2X of 0.715, R2Y of 0.803 and Q2 of 0.671. (b) Metabolomics tree diagram determined from the OPLS-DA scores plot. The coloring scheme for each compound in the tree diagram correlates with the data point colors in the OPLS-DA scores plot. The bootstrap numbers for each node are indicated on the tree diagram.
)treated M. smegmatis cells. The ellipses correspond to the 95% confidence limits from a normal distribution for each cluster. The untreated M. smegmatis cells (■) was designated the control class and the remainder of the cells were designated as treated. The OPLS-DA used one predictive component and six orthogonal components to yield a R2X of 0.715, R2Y of 0.803 and Q2 of 0.671. (b) Metabolomics tree diagram determined from the OPLS-DA scores plot. The coloring scheme for each compound in the tree diagram correlates with the data point colors in the OPLS-DA scores plot. The bootstrap numbers for each node are indicated on the tree diagram.
The accompanying metabolomics tree diagram25 (Figure 2B) clearly visualizes the relative groupings of the three antibiotic classes. The bootstrap numbers of 89 to 100 indicate a statistically significant separation between the five clusters and the reliability of the general drug and TAACF classifications. The metabolic tree diagram also provides a finer separation between the drugs within each cluster. These separations may reflect actual differences in the specific drug targets. For example, D-cycloserine and vancomycin are on separate branches in the cell wall node potentially because D-cycloserine inhibits alanine racemase and alanine ligase compared to vancomycin binding the D-alanyl-D-alanine dipeptide. Alternatively, the separation may result from differences in the relative activity of the drug. Streptomycin forms a separate branch in the transcription, translation or DNA supercoiling drug cluster despite having a similar target (binding to the 30S ribosomal protein S12 and 16S rRNA) relative to other members within the cluster. But, streptomycin is one of the most active compounds tested, requiring only a dosage of 1.5 μg/ml to inhibit M. smegmatis growth by approximately 50%. Also, over-interpreting these subtle separations may be erroneous since the within cluster differences may simply reflected experimental variability and may not be biologically relevant. For instance, an average growth inhibition of 43.1 ± 10.5% was observed after the addition of each of the 15 drugs. This dosage variability may lead to unintended separations in the 2D OPLS-DA scores plot. Essentially, the reliability of these finer cluster differences is dependent on additional supportive biological data.
Surprisingly, amiodorone, chlorprothixene, and clofazamine were found to cluster together in the 2D OPLS-DA scores plot and metabolic tree diagram. This was an unexpected result given that the three compounds were randomly selected from the large TAACF library and have diverse therapeutic usages. But, it also implies the three compounds share a similar mechanism of action in TB. Importantly, the three TAACF compounds also cluster with the antibiotics that disrupt cell wall formation, ampicillin, D-cycloserine and vancomycin. This infers a similar mode of action between the three TAACF compounds and the antibiotics that are known to interfere with bacterial cell walls. A subsequent literature search indicated that the three TAACF compounds have been previously shown to disrupt bacterial membranes in organisms distinct from TB.26-30 Thus, the literature results are consistent with our NMR metabolomics analysis, which support our general classification of the TAACF compounds as interfering with bacterial cell walls. It is important to note that while ampicillin is a member of this class of antibiotics, it is also skewed toward the untreated cells in the 2D OPLS-DA scores plot. Presumably, this is because of M. smegmatis β-lactamase activity that provides resistance to ampicillin.23, 31 The impact of ampicillin on the metabolome of M. smegmatis is significantly diminished such that ampicillin M. smegmatis is similar to untreated cells. As described previously, there are some differences between the OPLS-DA and PCA scores plot (see supplemental Figure 2S). There is less discrimination between the untreated and drug treated cells in the 2D PCA scores plot. This is not unexpected since PLS is preferred over PCA for discrimination been classes.24 Also, there is a separation between the three TAACF compounds and the cell wall antibiotics in the PCA scores plot, but the TAACF compounds are still closer to the cell wall antibiotics in the associated metabolic tree diagram (see supplemental figure 2S). In fact, the OPLS-DA and PCA metabolomic tree diagrams are quite similar despite these visible differences in the scores plots. Additionally, the quality of the OPLS-DA model is apparent from the fit to the data, R2X > 0.610 and R2Y > 0.803 and the reliability of the model is apparent from the cross-validation Q2 scores > 0.617. Further validation of the OPLS-DA drug and TAACF classifications comes from the analysis of the metabolites identified as the major contributors to the OPLS-DA class separation (see supplemental Figures 3S-6S).
The S-plots and loading plots determined from the OPLS-DA models identify the chemical shifts (and associated metabolites) that contribute to the observed separation between the untreated and treated cells in the 2D OPLS-DA scores plot. The metabolites and corresponding pathways predominately perturbed by the addition of each drug class are listed in supplemental Tables 1S- 3S. While there are some broad similarities in the metabolites and pathways affected by the drugs because the comparisons are all made relative to untreated cells, there are also some distinct differences. For example, proline, cytidine, uridine and inosine (pyrimidine and purine pathways) are all uniquely decreased by drugs that affect transcription, translation or DNA supercoiling. Obviously, nucleotides are essential metabolic precursors to DNA and RNA synthesis. Alternatively, choline phosphate, lysine, spermidine, citruline, ascorbate and dehydroascorbate (glycerophospholipid, lysine biosynthesis, arginine and proline metabolism, and ascorbate metabolism pathways) are decreased by drugs affecting the mycolic acid pathway. Ascorbate metabolism is directly linked to the mycolic acid pathway, where ascorbate leads to arabinose. Arabinose is the primary precursor for the arabinogalactan-mycolic acid complex. Also, the inhibition of spermidine synthesis has been previously observed for drugs targeting the mycolic acid pathway in mycobacteria.32 Importantly, the set of metabolites affected by the TAACF compounds were identical to metabolites perturbed by D-cycloserine and vancomycin. Both show a decrease in oxaloacetate, glutamine, glutamate, methionine and folate and an increase in isoleucine. Clearly, amino acids and their precursors are important components in peptidoglycan, cell wall and cell membrane synthesis. There were some additional metabolites that are increased by the addition of D-cycloserine and vancomycin that were not observed with the TAACF compounds. These include other amino acids (alanine, lysine serine, valine) and other precursors to peptidoglycan synthesis (N-acetyl-D-glucosamine, N-acetylneuraminate). Overall, the identity of the specific metabolites perturbed by each drug class is consistent with the 2D OPLS-DA scores plot clustering pattern and drug classifications.
In conclusion, we have demonstrated that different classes of antibiotics uniquely affect the metabolome of M. smegmatis. These metabolomic changes are directly correlated with broad mechanisms of action that are associated with each TB class of antibiotics, disruption of cell walls or membranes, inhibition of transcription, translation or DNA supercoiling, or the inhibition of mycolic acid biosynthesis. Thus, NMR metabolomics provides an efficient, simple and unbiased approach for providing rapid classification of promising drugs leads that emerge from HTS. This is critical since HTS does not provide any information on mechanisms of action; only relative activity with a high-false positive rate. Instead, the in vivo biological activity of a novel lead can be inferred by its relative clustering to existing drug classes in an OPLS-DA scores plot derived from metabolomics data. Importantly, a chemical lead that forms a distinct cluster from known drugs infers a potential new mechanism of action and a reason to prioritize the chemical lead for a detailed follow-up investigation. Of course, the induced metabolomic changes relative to untreated cells provide further confirmation of in vivo efficacy, which was implied from the HTS results. While the technique was demonstrated with M. smegmatis, it is generally applicable to bacterial pathogens and the effect of therapeutic agents on human cell lines in addition to the analysis of biofluids.
METHODS
Determining Optimal Drug Dosage for NMR Metabolomics Experiments
M. smegmatis mc2155 cells were grown at 37 °C with shaking at 200 rpm in 50 mL of Middlebrook 7H9 media until an average optical density at 600 nm (O.D.600) of 0.6 was achieved. Each drug was titrated over a concentration range of 1 to 24 times the literature MIC values and the cells were grown for an additional 2 hours. The optical density was recorded and the growth rate inhibition was calculated by comparing the optical density of the treated cells to the untreated cells in the 2 hour time period. The desired drug dosage was determined where a drug concentration inhibits cell growth by approximately 50% of the growth rate of untreated cells.
Sample Preparation
A total of 190 M. smegmatis mc2155 cultures were grown in 50 mL of Middlebrook 7H9 at 37 °C with shaking at 200 rpm until an O.D.600 of 0.6 was achieved. A total of 40 untreated cultures were used as a control and 10 cultures were inoculated with each antibiotics at the optimal dosage needed to inhibit cell growth by ~50% as described in Table 1. The cells were then grown for an additional 2 hours. The used media was removed and the cells were washed 3 times and resuspended with 1 mL ice cold double distilled water. The cells were lysed using a FastPrep-24 instrument for 60 seconds at 6 m/s, and the supernatant was extracted and frozen in a dry ice ethanol bath. The samples were lyophilized and then resuspended with 700 μL of 99.8% D2O solution containing 50 mM phosphate buffer (pH 7.2, uncorrected) and 50 μM 3-(trimethylsilyl)propionic acid-2,2,3,3-d4 (TMSP-d4) as an internal standard for chemical shift referencing. The samples were then centrifuged for 5 minutes to remove any insoluble material, and 600 μL of the supernatant was transferred to an NMR tube.
NMR Data Collection and Processing
The NMR spectra were collected on a Bruker 500 MHz Avance spectrometer equipped with a triple resonance and z axis gradient cryoprobe. A BACS-120 sample changer was used for automated data collection. 1D 1H NMR spectra were collected using excitation sculpting to remove the solvent signal and maintain a flat spectral baseline.33 A total of 32K data points with a spectral width of 5482.5 Hz, 16 dummy scans and 128 scans were used to obtain each spectrum. The data was processed automatically using ACD/1D NMR Manager (Advanced Chemistry Development). Intelligent bucketing was used to integrate each spectral region with a bin size of 0.025 ppm. Each NMR spectrum was center averaged to minimize any experimental variations between bacterial cultures as follows:34
| (1) |
where X̄ is the average signal intensity, σ is the standard deviation in the signal intensity, and Xi is the signal intensity within a bin. Noise regions of the spectra were omitted from the PCA analysis by setting the corresponding bins to zero.35
OPLS-DA and PCA was performed using Simca-11.5+ (Umetrics), where each 1H NMR spectra was reduced to a single point in the 2D OPLS-DA and PCA scores plot. The OPLS-DA was calculated with two classes, untreated versus drug treated cell cultures, for the Y matrix with the NMR data incorporated into the X matrix. The OPLS-DA model was cross validated using a modified version of the leave-one-out technique, where 1 out of every 7 samples (spectra) were left out to calculate a model and predict the left out data.36 The procedure was sequentially repeated leaving out a different 1/7th of the data. The predicted data was then compared to the original data, where the quality assessment (Q2) score provides a qualitative measure of the predictability of the model based on the consistency between the predicted and original data. An ideal value for Q2 is one, where a typical value for a biological model is ≥ 0.4.
Metabolomic Tree diagrams with corresponding bootstrap values were created using our PCAtoTree program to interpret the OPLS-DA clustering pattern.25 The metabolomics tree diagram is based on the Euclidean distances between the cluster centers from the 2D OPLS-DA scores plot. Standard bootstrapping techniques are used to generate a set of 100 distance matrices by randomly re-sampling the cluster centers and Euclidean distances. The set of 100 distance matrices are then used by PHYLIP (http://www.phylip.com),37 phylogenetic software package, to generate 100 tree diagrams and a consensus tree diagram. The bootstrap numbers on the consensus tree diagram indicates the number of times each node was present in the set of 100 tree diagrams, where a bootstrap number below 50% indicates a generally insignificant node or insignificant separation between the clusters.
Four additional OPLS-DA models were generated to identify specific metabolites associated with drug activity: (i) inhibition of translation, transcription or DNA supercoiling drug treated cells versus untreated cells, (ii) inhibition of mycolic acid synthesis drug treated cells versus untreated cells, (iii) inhibition of cell wall synthesis drug treated cells versus untreated cells, and (iv) the three TAACF compounds versus untreated cells. S-plots and loading plots were generated from each OPLS-DA model. Bins (chemical shift values) demonstrating a covariance of greater than 0.10 or less than -0.10 were identified as major contributors to the class separation. Metabolites were identified from this list of chemical shifts using the Human Metabolome Database (HMDB, http://www.hmdb.ca/)38 with a chemical shift tolerance of 0.02 ppm. Metabolic network maps were then generated using Cytoscape (http://www.cytoscape.org/)39 with the MetScape40 plugin for the top 100 metabolite predicted by HMDB. Metabolites were excluded if not part of a network or not present in M. smegmatis.
Supplementary Material
Acknowledgments
We acknowledge O. Chacon for helpful and valuable review of this manuscript. S. Halouska was partially supported by O. Chacon’s R21 grant to standardize NMR techniques included in this publication. This work was supported in part by funds from the America Heart Association (0860033Z) and by the National Institute of Allergy and Infectious Diseases (R21 AI087561) to R.P. The research was performed in facilities renovated with support from the National Institutes of Health (NIH, RR015468-01).
Footnotes
Author Contributions S.H carried out experiments to determine drug dosage, growing cell cultures, metabolite extraction, NMR data collection, statistical analysis and contributed to writing the paper. R.J. F. contributed the determination of drug dosage, growing cell cultures and writing the paper. R.G. B. supervised and provided materials to conduct experiments for cell culture growth, and contributed to writing the paper. R.P. designed and supervised all aspects of research, analyzed the data and contributed to writing the paper.
ASSOCIATED CONTENT Supporting Information. Supplemental material includes two figures of PCA scores plots and the associated metabolomic tree, four figures of OPLS-DA S-plots and loading plots, and four tables listing metabolites affected by each drug class. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meya DB, McAdam KPWJ. The TB pandemic: an old problem seeking new solutions. J Intern Med. 2007;261:309–329. doi: 10.1111/j.1365-2796.2007.01795.x. [DOI] [PubMed] [Google Scholar]
- 3.Gwynn MN, Portnoy A, Rittenhouse SF, Payne DJ. Challenges of antibacterial discovery revisited. Ann N Y Acad Sci. 2010;1213:5–19. doi: 10.1111/j.1749-6632.2010.05828.x. [DOI] [PubMed] [Google Scholar]
- 4.Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 5.Coates AR, Halls G, Hu Y. Novel classes of antibiotics or more of the same? Br J Pharmacol. 2011;163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annonymous. The 10 × ’20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis. 2010;50:1081–1083. doi: 10.1086/652237. [DOI] [PubMed] [Google Scholar]
- 8.Brown D. Unfinished business: target-based drug discovery. Drug Discov Today. 2007;12:1007–1012. doi: 10.1016/j.drudis.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Bunnage ME. Getting pharmaceutical R&D back on target. Nat Chem Biol. 2011;7:335–339. doi: 10.1038/nchembio.581. [DOI] [PubMed] [Google Scholar]
- 10.Powers R. NMR metabolomics and drug discovery. Magn Reson Chem. 2009;47(Suppl 1):S2–11. doi: 10.1002/mrc.2461. [DOI] [PubMed] [Google Scholar]
- 11.Baker M. Metabolomics: From small molecules to big ideas. Nat Methods. 2011;8:117–121. [Google Scholar]
- 12.Lindon JC, Holmes E, Nicholson JK. Pattern recognition methods and applications in biomedical magnetic resonance. Prog Nucl Magn Reson Spectrosc. 2001;39:1–40. [Google Scholar]
- 13.Bylesjoe M, Rantalainen M, Cloarec O, Nicholson JK, Holmes E, Trygg J. OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J Chemom. 2007;20:341–351. [Google Scholar]
- 14.Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010;39:D1035–1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung GA, Aktar Z, Jackson S, Duncan K. High-throughput screen for detecting antimycobacterial agents. Antimicrob Agents Chemother. 1995;39:2235–2238. doi: 10.1128/aac.39.10.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephan J, Mailaender C, Etienne G, Daffe M, Niederweis M. Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob Agents Chemother. 2004;48:4163–4170. doi: 10.1128/AAC.48.11.4163-4170.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malik M, Lu T, Zhao X, Singh A, Hattan CM, Domagala J, Kerns R, Drlica K. Lethality of quinolones against Mycobacterium smegmatis in the presence or absence of chloramphenicol. Antimicrob Agents Chemother. 2005;49:2008–2014. doi: 10.1128/AAC.49.5.2008-2014.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu T, Zhao X, Li X, Hansen G, Blondeau J, Drlica K. Effect of chloramphenicol, erythromycin, moxifloxacin, penicillin and tetracycline concentration on the recovery of resistant mutants of Mycobacterium smegmatis and Staphylococcus aureus. J Antimicrob Chemother. 2003;52:61–64. doi: 10.1093/jac/dkg268. [DOI] [PubMed] [Google Scholar]
- 19.Halouska S, Chacon O, Fenton RJ, Zinniel DK, Barletta RG, Powers R. Use of NMR metabolomics to analyze the targets of D-cycloserine in mycobacteria: role of D-alanine racemase. J Proteome Res. 2007;6:4608–4614. doi: 10.1021/pr0704332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burguiere A, Hitchen PG, Dover LG, Dell A, Besra GS. Altered expression profile of mycobacterial surface glycopeptidolipids following treatment with the antifungal azole inhibitors econazole and clotrimazole. Microbiology. 2005;151:2087–2095. doi: 10.1099/mic.0.27938-0. [DOI] [PubMed] [Google Scholar]
- 21.Mick V, Rebollo MJ, Lucia A, Garcia MJ, Martin C, Ainsa JA. Transcriptional analysis of and resistance level conferred by the aminoglycoside acetyltransferase gene aac(2’)-Id from Mycobacterium smegmatis. J Antimicrob Chemother. 2008;61:39–45. doi: 10.1093/jac/dkm440. [DOI] [PubMed] [Google Scholar]
- 22.Chacon O, Feng Z, Harris NB, Caceres NE, Adams LG, Barletta RG. Mycobacterium smegmatis D-Alanine Racemase Mutants Are Not Dependent on D-Alanine for Growth. Antimicrob Agents Chemother. 2002;46:47–54. doi: 10.1128/AAC.46.1.47-54.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flores AR, Parsons LM, Pavelka MS., Jr Characterization of novel Mycobacterium tuberculosis and Mycobacterium smegmatis mutants hypersusceptible to β-lactam antibiotics. J Bacteriol. 2005;187:1892–1900. doi: 10.1128/JB.187.6.1892-1900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barker M, Rayens W. Partial least squares for discrimination. J Chemom. 2003;17:166–173. [Google Scholar]
- 25.Werth MT, Halouska S, Shortridge MD, Zhang B, Powers R. Analysis of metabolomic PCA data using tree diagrams. Anal Biochem. 2009;399:58–63. doi: 10.1016/j.ab.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosa SMLJ, Antunes-Madeira MC, Jurado AS, Madeira VVMC. Amiodarone interactions with membrane lipids and with growth of Bacillus stearothermophilus used as a model. Appl Biochem Biotechnol. 2000;87:165–175. doi: 10.1385/abab:87:3:165. [DOI] [PubMed] [Google Scholar]
- 27.Oliva B, O’Neill AJ, Miller K, Stubbings W, Chopra I. Anti-staphylococcal activity and mode of action of clofazimine. J Antimicrob Chemother. 2004;53:435–440. doi: 10.1093/jac/dkh114. [DOI] [PubMed] [Google Scholar]
- 28.Kristiansen JE, Thomsen VF, Martins A, Viveiros M, Amaral L. Non-antibiotics reverse resistance of bacteria to antibiotics. In Vivo. 2010;24:751–754. [PubMed] [Google Scholar]
- 29.O’Neill AJ, Miller K, Oliva B, Chopra I. Comparison of assays for detection of agents causing membrane damage in Staphylococcus aureus. J Antimicrob Chemother. 2004;54:1127–1129. doi: 10.1093/jac/dkh476. [DOI] [PubMed] [Google Scholar]
- 30.Hurdle JG, O’Neill AJ, Chopra I, Lee RE. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat Rev Microbiol. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon HH, Tomioka H, Saito H. Distribution and characterization of β-lactamases of mycobacteria and related organisms. Tuber Lung Dis. 1995;76:141–148. doi: 10.1016/0962-8479(95)90557-x. [DOI] [PubMed] [Google Scholar]
- 32.Paulin LG, Brander EE, Poso HJ. Specific inhibition of spermidine synthesis in Mycobacteria spp. by the dextro isomer of ethambutol. Antimicrob Agents Chemother. 1985;28:157–159. doi: 10.1128/aac.28.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang T-L, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. J Magn Reson Ser A. 1995;112:275–279. [Google Scholar]
- 34.Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC. Scaling and Normalization Effects in NMR Spectroscopic Metabonomic Data Sets. Anal Chem. 2006;78:2262–2267. doi: 10.1021/ac0519312. [DOI] [PubMed] [Google Scholar]
- 35.Halouska S, Powers R. Negative impact of noise on the principal component analysis of NMR data. J Magn Reson. 2006;178:88–95. doi: 10.1016/j.jmr.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Westeruis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, van VEJJ, van DJPM, van DFA. Assessment of PLSDA cross validation. Metabolomics. 2008;4:81–89. [Google Scholar]
- 37.Retief JD. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 2000;132:243–258. doi: 10.1385/1-59259-192-2:243. [DOI] [PubMed] [Google Scholar]
- 38.Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De SA, Zuniga A, Dawe M, Xiong Y, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledge-base for the human metabolome. Nucleic Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Tarcea VG, Karnovsky A, Mirel BR, Weymouth TE, Beecher CW, Cavalcoli JD, Athey BD, Omenn GS, Burant CF, Jagadish HV. Metscape: a Cytoscape plug-in for visualizing and interpreting metabolomic data in the context of human metabolic networks. Bioinformatics. 2010;26:971–973. doi: 10.1093/bioinformatics/btq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.