

Abstract
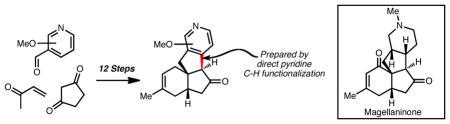
A concise enantioselective approach to the tetracyclic core of the magellanine-type Lycopodium alkaloids is reported. Key to this approach is the use of the Hajos-Parrish reaction to set a challenging quaternary stereocenter, thereby guiding the stereoselectivity for the remainder of the synthesis, as well as the use of a palladium-mediated direct pyridine functionalization reaction to forge the tetracyclic core.
The Lycopodium alkaloid magellanine (1, Figure 1) has served as a forum to highlight creative synthetic strategies and methods for almost two decades. Overman was first to report a highly elegant total synthesis of 1 in 1993, featuring a Prins-pinacol rearrangement.1 Since this initial report, there have been many other reports of inventive syntheses, as well as approaches, toward magellanine.2,3 Magellanine and the related Lycopodium alkaloids4 magellaninone (2) and paniculatine (3) are characterized by a tetracyclic core that possesses up to six contiguous stereocenters, one of which is quaternary. This framework presents a formidable synthetic challenge, which is further heightened by the highly basic tertiary amine group. Not surprisingly, the majority of syntheses of these molecules have focused on introducing the amine group (and therefore the piperidine ring) in the late stages of the synthesis, thus realizing a ‘bottom-up’ strategy. In this report, we present an alternative ‘top-down’ approach (see Figure 1) to the magellanine-type alkaloids, which is enabled by the use of methoxypyridine intermediates.
Figure 1.

Magellanine-type Lycopodium alkaloids
Our laboratory has previously shown the utility of methoxypyridines as surrogates for piperidines and pyridones in complex molecule synthesis.5 This approach offers several advantages, notably the fact that 2-methoxypyridines possess a less basic nitrogen atom than the corresponding unsubstituted pyridine. This is supported by a pKa of 3.06 for 2-methoxypyridinium ion versus a pKa of 5.23 for pyridinium (see Figure 2).6 The mitigated basicity of 2–methoxypyridines can be attributed to inductive electron-withdrawing effects of the alkoxy group.7 Additionally, steric shielding of the nitrogen lone pair by the alkoxy group is pronounced in the favored conformer, B (see Figure 2), which avoids pseudo A1,3 interactions and also minimizes dipoles between the nitrogen and oxygen lone pairs.8
Figure 2.

Basicity considerations of the pyridine nitrogen9
With the appreciation for the mitigated basicity of methoxypyridines in mind, we envisioned a retrosynthetic analysis for magellanine (and congeners) that would bring the natural product(s) back to tetracycle 4 (Scheme 1). Two variants of this late-stage intermediate were planned: a dimethoxypyridine containing tetracycle (4a) as well as a monomethoxypyridine variant (4b). In the forward sense, appropriate functionalization and oxidation level adjustments of rings A and B in 4a/b would set the stage for access to the magellanine-type alkaloids. By analogy to the related precedent of Meyers,3 the methoxypyridine ring could be transformed to the necessary piperidine moiety, bearing the correct stereochemistry at the C/D ring fusion, through a N-methylation/global reduction protocol. Tetracycles 4a/b could arise from vinyl triflate 5a/b by a direct functionalization of the pyridine C4 position using a palladium-mediated C-C bond formation that has its origins in the instructive studies of Fagnou10 and Echavarren.11 This transformation is particulary powerful since it requires no pre-functionalization of the pyridine starting material. Vinyl triflate 5a/b may, in turn, arise from Hajos-Parrish ketone derivative 6a/b, which could be prepared enantioselectively from pyridine aldehyde 7a/b, methyl vinyl ketone (8) and 1,3-cyclopentanedione (9) following the protocol of Brittain and coworkers.12
Scheme 1.

Retrosynthetic analysis of magellanine and congeners
Our synthetic studies to arrive at tetracycle 4a, described herein, are illustrative of the approach to 4b as well, which is presented in its entirety in the Supporting Information. The synthesis of 4a commenced with a Knoevenagel condensation of commercially available aldehyde 7a and 1,3-cyclopentanedione (9) to afford adduct 10 in 83% yield (Scheme 2). Hydrogenation of the double bond proceeded without event to afford C2-functionalized dione/vinylogous acid 11. Acid 11 was subjected to proline-catalyzed Robinson annulation conditions in the presence of methyl vinyl ketone (8) to provide the aldol product, which was smoothly dehydrated in the presence of HCl and MeOH to give enone 6a in 83% yield and 87% ee (absolute configuration was not determined; one enantiomer is arbitrarily depicted). This reaction sets the stereochemistry of the all carbon quaternary center present in the natural product, which we envision using to guide the stereoselectivity for the rest of the synthesis. Of note, an analogous Robinson annulation employing derivatives of 11 which lack the methoxy groups proceeded to give the corresponding bicyclic enones (12 and 13, Figure 3) in drastically lower yield. Furthermore, these reactions could not be scaled above 100 mg without significant accompanying decomposition. The derivative with the methoxy group para to the alkyl substituent (14) was also formed in a very low yield. This outcome likely results from significant basicity of the pyridine nitrogen in 12–14, which curtails the yield of the Robinson annulation reaction. It appears that a methoxy group ortho to the alkyl substituent in the pyridine moeity (as in 6b), has a higher population of conformer B (see Figure 2) to avoid increased A1,3 strain, which more effectively blocks the nitrogen lone pair. This in turn leads to a higher yield of the bicyclic enone products and the reactions can be performed on multigram scale. Additional blocking groups (as in 11) further enhance the yield of the Robinson annulation. Thus, carefully designed methoxypyridines can be used to tune the basicity of the pyridine nitrogen, which faciliatates reactions that would normally be very low yielding.
Scheme 2.

Synthesis of Hajos-Parrish ketone derivative 6a
Figure 3.

Other Hajos Parrish ketone derivatives
As shown in Scheme 3, bicyclic enone 6a was readily advanced to vinyl triflate 5a in 2 steps. This synthetic sequence entailed a tandem ketalization/hydrogenation of enone 6a to afford ketal 15 in 94% yield as a single diastereomer.13 Interestingly, when the two steps to reach 15 were performed separately, the diastereoselectivity was poor and the yield was significantly lower. With ketal 15 in hand, we were able to effect a soft enolization of the carbonyl group to give vinyl triflate 5a.
Scheme 3.

Synthesis of the tetracycles
In the key step to furnish the tetracyclic core (4a/b) of the magellanine-type alkaloids, vinyl triflates 5a/b were subjected to the conditions identified by Echavarren to be optimal for direct palladium-catalyzed arylation of pyridines.11 Gratifyingly, tetracycle 4a was obtained in 62% yield over the 2 steps. This sequence has been conducted on a >3 g scale without a drop in yield. Additionally, vinyl triflate 5b, bearing a mono-methoxy pyridine, afforded a 66% yield of 4b under the same conditions.
With access to tetracycles 4a and 4b, we have begun to explore the functionalization of these late-stage intermediates to afford advanced precursors to the magellanine-type alkaloids. Hydrolysis of the ketal group of 4a followed by addition of methyl magnesium bromide into the resulting carbonyl moiety yields tertiary alcohol 16 as a single diastereomer.14 Hydroboration of the Bring alkene group of 16 was accomplished under standard conditions to provide diol 17 following oxidative work-up. Further oxidation of the secondary hydroxy group using pyridinium chlorochromate gave a keto alcohol that could be dehydrated to afford a single regioisomer of advanced tetracycle 18.
Our efforts thus far toward the magellanine-type alkaloids have yielded several insights. Most importantly, we have found that the regioselectivity of the double bond placement in the A-ring of the tetracycle is highly dependent on the number of sp2-hybridized carbon atoms in the B-ring (see Eqs. 1 and 2). Thus, while 19 yields exclusively one regioisomer of silyl enol ether 20, diol 21 leads to a regioisomeric mixture of silyl enol ether products (22). These observations, were indeed, applied to the synthesis of 18.
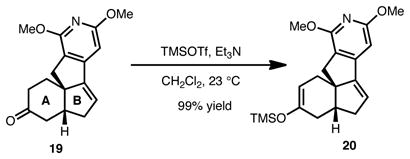 |
(1) |
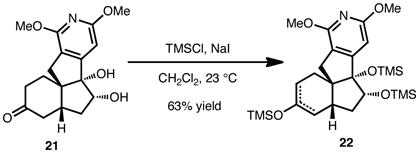 |
(2) |
In conclusion, we have developed an efficient synthetic sequence to the tetracyclic core of the magellanine-type Lycopodium alkaloids. Our strategy features a variant of the enantioselective Robinson annulation, which sets a challenging quaternary center early in the synthesis. This single stereocenter has been shown to guide the stereoselective manipulation of the core. Additionally, a powerful palladium-catalyzed C-C bond forming reaction was utilized to assemble the strained tetracyclic core of the magellanine-type Lycopodium alkaloids without the nescessity of prefunctionalization. Efforts are underway to apply these lessons to the total synthesis of the magellanine-type Lycopodium alkaloids.
Supplementary Material
Scheme 4.

Functionalization of the A/B rings of 4a
Acknowledgments
We are grateful to Ms. Tara Pesce (Columbia University) for optimization studies on the preparation of 11 during her participation in the UC Berkeley Amgen Scholars Program. The NIH (NIGMS RO1 086374) is gratefully acknowledged for financial support of this work.
Footnotes
Supporting Information Available. Experimental details and characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hirst GC, Johnson T, Overman LE. J Am Chem Soc. 1993;115:2992–2993. [Google Scholar]
- 2.For other previous syntheses of magellanine-type alkaloids, see Paquette L, Williams JP, StLaurent D, Friedrich D, Pinard E, Roden B. J Am Chem Soc. 1994;116:4689–4696.Mukai C, Kozaka T, Miyakoshi N. J Org Chem. 2007;72:10146–10154. doi: 10.1021/jo702136b.Liao C. Pure Appl Chem. 2005;77:1221–1234.Takahashi T, Ishizaki M, Niimi Y, Hoshino O, Hara H. Tetrahedron. 2005;61:4053–4065.
- 3.For a particulary pertinent previous approach, see: Meyers AI, Brengel GP. Chem Commun. 1997:1–8.Sandham D, Meyers AI. J Chem Soc, Chem Commun. 1995:2511–2512.
- 4.For a recent review on Lycopodium alkaloids, see: Gang DR, Ma X. Nat Prod Rep. 2004;21:752–772. doi: 10.1039/b409720n.
- 5.For selected examples, see: Sarpong R, Larson KK. J Am Chem Soc. 2009;131:13244–13245. doi: 10.1021/ja9063487.Bisai V, Sarpong R. Org Lett. 2010;12:2551–2553. doi: 10.1021/ol100823t.Bisai A, West SP, Sarpong R. J Am Chem Soc. 2008;130:7222–7223. doi: 10.1021/ja8028069.
- 6.Manallack DT, Gancia E, Pitt WR, Wong MG, Lloyd EJ, Tehan BG. Quant Struct-Act Relat. 2002;21:473–485. [Google Scholar]
- 7.For discussion on inductive effects in substituted pyridines see: Joule JA, Mills K. Heterocyclic Chemistry. 5. Blackwell Science Ltd; Cambridge: 2010. pp. 125–175. (Particularly relevant, pp 125–126.)
- 8.See Ref. 6 and Blonski WJP, Hruska TA, Wildman TA. Org Mag Res. 1984;22:505–509.
- 9.Molecular modeling computations were performed at the B3LYP/6-31G* level of theory using a IEF-PCM simulation of THF as solvent.
- 10.(a) Fagnou K, Campeau L, Parisien M, Leblanc M. J Am Chem Soc. 2004;126:9186–9187. doi: 10.1021/ja049017y. [DOI] [PubMed] [Google Scholar]; (b) Fagnou K, Campeau L, Stuart DR, Leclerc J, Bertrand-Laperle M, Villemure E, Sun H, Lasserre S, Guimond N, Lecavallier M. J Am Chem Soc. 2009;131:3291–3306. doi: 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]
- 11.(a) Echavarren AM, Maseras F, Garcia-Cuadrado D, de Mendoza P, Braga AAC. J Am Chem Soc. 2007;129:6880–6886. doi: 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]; (b) Echavarren AM, Garcia-Cuadrado D, Braga AAC, Maseras F. J Am Chem Soc. 2006;128:1066–1067. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]
- 12.Brittain DE, Kennedy JWJ, Vietrich S, Weinmann H. J Org Chem. 2008;73:5151–5154. doi: 10.1021/jo800638s. [DOI] [PubMed] [Google Scholar]
- 13.Parsons PJ, Hudson P. Synlett. 1992;11:867–868. [Google Scholar]
- 14.The newly formed stereocenter was not rigorously determined.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.