

Abstract
The decomposition of H2O2 on iron minerals can generate •OH, a strong oxidant that can transform a wide range of contaminants. This reaction is critical to In Situ Chemical Oxidation (ISCO) processes used for soil and groundwater remediation, as well as advanced oxidation processes employed in waste treatment systems. The presence of dissolved silica at concentrations comparable to those encountered in natural waters decreases the reactivity of iron minerals toward H2O2, because silica adsorbs onto the surface of iron minerals and alters catalytic sites. At circumneutral pH values, goethite, amorphous iron oxide, hematite, iron-coated sand and montmorillonite that were pre-equilibrated with 0.05 – 1.5 mM SiO2 were significantly less reactive toward H2O2 decomposition than their original counterparts, with the H2O2 loss rates inversely proportional to the SiO2 concentration. In the goethite/H2O2 system, the overall •OH yield, defined as the percentage of decomposed H2O2 producing •OH, was almost halved in the presence of 1.5 mM SiO2. Dissolved SiO2 also slows the H2O2 decomposition on manganese(IV) oxide. The presence of dissolved SiO2 results in greater persistence of H2O2 in groundwater, lower H2O2 utilization efficiency and should be considered in the design of H2O2-based treatment systems.
Introduction
The injection of oxidants into the subsurface is a widely used approach for remediating soils and groundwater contaminated with organic compounds. This method, known as In Situ Chemical Oxidation (ISCO), is attractive because it requires less infrastructure investment and has lower maintenance and operation cost than pump-and-treat remediation.1 Furthermore, the relatively fast production of oxidants expedites completion of site remediation.
Among various oxidants employed in ISCO (i.e., permanganate, hydrogen peroxide, ozone and persulfate)1, hydrogen peroxide (H2O2) is probably the most widely used, because it is relatively inexpensive, easy to transport and introduce into the subsurface, and the byproducts of H2O2 decomposition, namely O2 and H2O, are benign. H2O2-based ISCO technologies rely on the conversion of H2O2 into hydroxyl radical (•OH), either by reactions with subsurface materials (e.g., iron-containing clays and minerals) or by reactions with dissolved ferrous ions that are sometimes co-injected with H2O2.1,2 However, the rapid loss of H2O2 upon injection is often problematic because H2O2 may decompose before it reaches contaminated zones.1,3 Consequently, a large excess of H2O2 is often used and injection wells have to be constructed immediately proximate to contaminated areas.
The rate at which H2O2 decomposes and the fraction of the H2O2 converted into •OH depends upon the composition of the aquifer materials and groundwater. Iron oxides (e.g., ferrihydrite or goethite) convert H2O2 into •OH through a surface-initiated chain reaction analogous to the Haber-Weiss mechanism.2,4–6 Iron oxides also can convert H2O2 directly into O2 and H2O via two-electron transfer mechanisms.5,6 In contrast, manganese oxides do not generate •OH when they decompose H2O2.5,7 In the subsurface, H2O2 can also be decomposed by (e.g., catalases and peroxidases) via pathways that also do not produce •OH.5 Conversely, the presence of phosphate8 or metal complexing ligands, such as citrate and phytate9, enhance the stability of H2O2 because they can bind metals and decrease their reactivity. The efficacy of H2O2-based ISCO, therefore, depends on the H2O2 persistence as well as the pathways through which it is decomposed, because only those that produce •OH will be beneficial for oxidative contaminant removal. A thorough understanding of how different subsurface components affect the decomposition of H2O2 will, therefore, help to predict its fate and could lead to an ability to improve the performance of H2O2-based ISCO.
The aim of this research was to investigate the effect of dissolved SiO2 on the rate of H2O2 decomposition catalyzed by different types of iron- and manganese- containing materials. Silica is particularly important to ISCO, because depending on local geology, groundwater can contain dissolved SiO2 at concentration ranging from 5 mg/L to 85 mg/L (i.e., 0.08 – 1.42mM SiO2).10 Although dissolved SiO2 adsorbs on the surface of iron oxides11,12 and SiO2 is known to act as corrosion inhibitor, its effect on H2O2 decomposition in the subsurface, to the best of our knowledge, has not been investigated previously. Therefore, the rate of H2O2 decomposition on goethite, hematite, amorphous iron oxyhydroxide, iron-coated sand, montmorillonite and pyrolusite was studied in solutions containing various amount of dissolved SiO2 (i.e., 0 – 1.5 mM SiO2). To gain insight into the impact of SiO2 on the performance of ISCO, the effect of dissolved SiO2 on the overall •OH yield, defined as the percentage of decomposed H2O2 producing •OH, was also investigated.
Materials and Methods
Chemicals
Amorphous iron oxyhydroxide (i.e., FeOOH) was obtained from Aldrich, while pyrolusite (β-MnO2) was obtained from Fisher. Wyoming montmorillonite (Swy-2, 31.8 m2/g, 2.59 wt% Fe) was obtained from the Source Clays Repository (The Clay Minerals Society). All other chemicals were reagent grade from Fisher Scientific and were used without further purification.
Goethite and hematite were synthesized following procedures reported in the literature13 and their identity was verified by X-ray diffraction. Briefly, goethite was synthesized by aging freshly made ferrihydrite in a strong alkaline solution (NaOH) at 70°C for 60 hours. Hematite was synthesized using the same method except that the aging was conducted at pH 8 – 8.5 in the presence of NaHCO3 at 90°C for 48 hrs. Amorphous FeOOH 50 – 80 mesh was ground using a mortar and pestle prior to sieving through a 150 micron sieve.
The surface area of these solids, determined using the 5 point BET (Brunauer–Emmett–Teller) nitrogen physisorption method, was 21.8 m2/g for hematite, 19 m2/g for goethite, 165.8 m2/g for FeOOH, and less than 1 m2/g for MnO2. Iron-coated sand (1 wt% Fe, 4.8 m2/g) was kindly provided by Peter Nico (Lawrence Berkeley National Laboratory). The synthesis and characterization of iron-coated sand have been reported elsewhere.14
A stock solution of 15 mM silica was prepared daily from Na2SiO3.9H2O. For simplicity, all species of dissolved silica (e.g., H4SiO4, H3SiO4− and polymeric silica) are denoted as SiO2. All solutions were prepared using 18 MΩ Milli-Q water from a Millipore system.
Experimental setup
All experiments were carried out at 25 ± 1°C in the dark in a 50-mL polypropylene flask open to the atmosphere. The temperature was controlled with a water bath. The pH of solutions was buffered with 1 mM piperazine-N,N'-bis(ethanesulfonic acid) (PIPES) for pH 7 or 4 mM borate for pH 8 – 9. The ionic strength of the solutions was maintained with 0.1 M NaNO3. The pH was measured throughout each experiment and was adjusted when it deviated from the initial value by more than 0.1 unit. Experiments were conducted at least in triplicate and average values along with one standard deviation are presented.
Adsorption of dissolved SiO2 by the solids
Silica was added from a 15 mM stock solution to the buffered solutions and the pH was adjusted with 1 M NaOH or 0.5 M H2SO4. To minimize the polymerization and avoid SiO2(s) precipitation, SiO2 concentrations never exceeded 1.5 mM.15 Next, a solid (i.e., iron oxide, iron coated sand, Swy-2 or pyrolusite) was added to the solution and the pH again was adjusted if necessary. Samples were withdrawn at pre-determined time intervals. Within 5 minutes, the solid was separated by centrifugation, then the supernatant was filtered immediately through a 0.2-μm nylon filter and analyzed for dissolved SiO2.
H2O2 decomposition and phenol oxidation
The decomposition of H2O2 catalyzed by the solids was investigated in the absence and presence of dissolved SiO2. Prior to the addition of H2O2, suspensions were mixed for 24 hrs to equilibrate the solids with SiO2. All experiments with iron-containing minerals were performed in pH 7 ± 0.1 solutions. Experiments with β-MnO2 were conducted at pH 8.4 because the 1 mM PIPES buffer was ineffective at pH 7.0. At pH 8.4, the pH never changed by more than 0.1 units during the experiments.
To investigate the effect of dissolved SiO2 on •OH production, the transformation of 0.2 mM phenol in the goethite/H2O2 system was studied. Phenol was chosen as a model target contaminant because it is not significantly adsorbed by any of the solids and reacts with •OH at a near-diffusion controlled rate. Samples were withdrawn at predetermined time intervals and divided into two parts. In the first aliquot, the solids were separated by centrifugation followed by filtration and the solution was analyzed for H2O2. Acetonitrile was added to the second aliquot (acetonitrile:sample = 1:1) and the mixture was agitated vigorously for 2 minutes with a vortex mixer to extract any adsorbed phenol from the solids. The solids were then separated by centrifugation and filtration and the solution was analyzed for phenol. Phenol recovery by acetonitrile extraction was always above 98% in H2O2-free controls. The stoichiometric efficiency, defined as the amount of phenol transformed per mole of hydrogen peroxide decomposed6, was used to evaluate the effect of dissolved SiO2 on •OH production.
Analytical methods
Phenol was analyzed using HPLC as described previously.6 H2O2 was analyzed spectrophotometrically by the titanium sulfate method.16 An inductively coupled plasma optical emission spectrometer (ICP-OES) was used to measure dissolved SiO2; all results are reported in molar based on the SiO2 formula. Total dissolved iron was quantified using the 1,10-phenanthroline method17 after adding hydroxylamine hydrochloride to the filtered samples. The concentration of dissolved iron was always below the detection limit (i.e., 5 μM).
Goethite surfaces, pre-equilibrated with SiO2 solutions, were examined with a Philips CM200/FEG transmission electron microscope coupled with an energy dispersive X-ray unit (EDX). The instrument was operated in scanning mode (STEM/EDX) with a probe size 1.4 nm. Samples for STEM/EDX analysis were prepared as follows: after the adsorption experiment, the solid was collected by centrifugation and then resuspended in 2 mL fresh Milli-Q water. An aliquot of this suspension was spread on the copper grid, the excess water was gently removed with a Kimwipe tissue and the grid was dried under air at room temperature.
Results
Silica adsorption
The rate of silica adsorption onto goethite, hematite and Swy-2 in pH 7 solutions with different [SiO2]initial was investigated. In all cases, SiO2 adsorption approached equilibrium within 24 hrs (inset of Figure 1 and Figure S1 in Supporting Information). Therefore, SiO2 adsorption as a function of [SiO2]initial was measured after a 24 hr equilibration period. This equilibration period was also employed in the study of H2O2 decomposition and phenol transformation.
Figure 1.

Adsorption isotherm (24 hour equilibration) of dissolved SiO2 on goethite. [goethite] = 4 g/L, [PIPES] = 1 mM, [NaNO3] = 0.1 M, pH = 7. [SiO2]initial = 0 – 1.5 mM (inset: adsorption kinetics).
Higher initial SiO2 concentrations resulted in more SiO2 adsorption onto goethite (Figure 1). Except for the last data point in Figure 1 ([SiO2]equilibrium = 1.14 mM), the adsorption isotherm followed a Langmuir-type isotherm, with a maximum adsorption density (ΓSiO2) of approximately 0.062 mmol SiO2/g goethite (Figure 1). At [SiO2]equilibrium = 1.14 mM, the amount SiO2 sorbed was significantly higher (0.09 mmol SiO2/g goethite).
STEM/EDX analysis indicated that SiO2 was not uniformly adsorbed on the goethite surface. For example, EDX spectra of a goethite sample that was pre-equilibrated with 0.5 mM SiO2 for 24 hrs showed that the surface elemental composition varied among locations (Figure 2), with Si peaks not observed in some locations (Figure 2a), co-occurring with iron in others (Figure 2b) and existing in the absence of an iron peak in others (Figure 2c). The fraction of sites that were fully coated with Si (i.e., sites having EDX spectra similar to that of Figure 2c) increased as the [SiO2]initial increased.
Figure 2.

EDX spectra from three different locations on a goethite surface that was pre-equilibrated with 0.5 mM dissolved silica solution for 24 hrs. Carbon peaks come from the grid support.
H2O2 decomposition and phenol transformation
In the SiO2-free system, the half-life of 5 mM H2O2 in the presence of 4 g/L goethite was 7.77 ± 0.34 hr (Table 1). Addition of dissolved SiO2 slowed the rate of H2O2 decomposition, increasing the H2O2 half-life to 21.7 ± 1.2 and 28.2 ± 1.8 at an [SiO2]initial of 0.5 mM and 1.5 mM, respectively (Table 1 and Figure 3a). The half-life of H2O2 in the presence of [SiO2]initial = 1.5 mM was comparable to that observed in a solution containing 2 mM phosphate (t1/2 = 31.6 ± 1.4, Table 1). Under the experimental conditions employed in this study, the H2O2 decomposition rate was limited by the intrinsic chemical reactivity of the solids and not by diffusion of H2O2 to the surface or the number of sites available for H2O2 adsorption (see Supporting Information for detailed discussion). The observed-first order rate constant of H2O2 decomposition (kobs) was inversely proportional to the amount of SiO2 in the solution (Figure 3b). At adsorption densities below 0.04 mmol/g goethite, kobs decreased linearly with the SiO2 adsorption density, with a slope of −0.303 hr−1.mmol−1.gram. At adsorption densities above 0.04 mmol/g goethite, kobs was much less sensitive to increasing adsorption density (the slope of the regression line was −0.036 hr−1.mmol−1.gram). The presence of dissolved SiO2 also diminished the rate of H2O2 loss catalyzed by other iron-containing minerals (Table 1). In the presence of 0.5 mM [SiO2]initial, the half-life of H2O2 increased by at least a factor of two compared with the SiO2-free system in all cases.
Table 1.
Observed-first order rate constants (kobs) for H2O2 decomposition catalyzed by iron-containing minerals under various conditions.
| Experiment condition | H2O2kobs(h−1) | H2O2 half-life (h) | |
|---|---|---|---|
| 1 | 4g/L goethite, 0 mM SiO2 | 0.089 ± 0.003 | 7.77 ± 0.34 |
| 2 | 4g/L goethite, 0.5 mM SiO2 | 0.032 ± 0.002 | 21.7 ± 1.2 |
| 3 | 4g/L goethite, 1.5 mM SiO2 | 0.025 ± 0.002 | 28.2 ± 1.8 |
| 4 | 4g/L goethite, 2 mM phosphate | 0.022 ± 0.001 | 31.6 ± 1.4 |
| 5 | 4g/L goethite, 0.5 mM SiO2 and 2 mM phosphate | 0.021 ± 0.001 | 33.5 ± 1.3 |
| 6 | 4g/L hematite, 0 mM SiO2 | 0.018 ± 0.002 | 39.4 ± 3.5 |
| 6 | 4g/L hematite, 0.5 mM SiO2 | 0.009 ± 0.001 | 77.7 ± 8.7 |
| 7 | 1 g/L FeOOH, 0 mM SiO2 | 0.562 ± 0.005 | 1.23 ± 0.01 |
| 8 | 1 g/L FeOOH, 0.5 mM SiO2 | 0.165 ± 0.015 | 4.22 ± 0.37 |
| 9 | 1 g/L FeOOH, 100 mM H2O2 | 0.539 ± 0.014 | 1.29 ± 0.03 |
| 10 | 1 g/L FeOOH, 100 mM H2O2, 0.5 mM SiO2 | 0.15 ± 0.02 | 4.81 ± 0.69 |
| 11 | 5 g/L iron coated sand, 0 mM SiO2 | 0.134 ± 0.012 | 5.21 ± 0.45 |
| 12 | 5 g/L iron coated sand, 0.5 mM SiO2 | 0.036 ± 0.010 | 20.3 ± 6.5 |
| 13 | 4 g/L montmorillonite, 0 mM SiO2, [H2O2]initial = 50 mM. | 0.0094 ± 0.0008 | 74.4 ± 6.6 |
| 14 | 4 g/L montmorillonite, 0.5 mM SiO2, [H2O2]initial = 50 mM. | 0.00283 ± 0.00005 | 244.7 ± 4.9 |
Unless otherwise noted, [H2O2]initial = 5 mM, pH = 7, [NaNO3] = 0.1 M. The rate constants were obtained by fitting the experimental data to the first order decay reaction rate law. The r2 values of the fittings were always r2 > 0.99.
Figure 3.

Effect of dissolved SiO2 on H2O2 decomposition by goethite. [goethite] = 4 g/L, [H2O2]initial = 5.1 ± 0.1 mM, pH = 6.9 ± 0.1, [PIPES] = 1 mM, [NaNO3] = 0.1 M. Solid lines are first-order fit of H2O2 decomposition (a) and linear fits of first order rate constant kobs vs. SiO2 sorbed (b).
To understand the effect of dissolved SiO2 on the efficiency of the conversion of H2O2 into •OH by iron-containing minerals, the oxidation of phenol in the goethite/H2O2 system was investigated. Typical phenol transformation data are presented in Figure 4a, which shows that silica slowed the rate of both H2O2 decomposition and phenol transformation. A control experiment indicated no phenol loss in the absence of H2O2. Addition of 100 mM tert-butanol, a •OH scavenger, completely eliminated phenol degradation (data not shown), confirming that the loss of phenol observed was due to reaction with •OH. In the absence of dissolved SiO2, the stoichiometric efficiency throughout the course of the experiment ranged from 0.25 to 0.3%. The stoichiometric efficiency was slightly lower in the presence of [SiO2]initial = 0.5 mM, while at [SiO2]initial = 1.5 mM the efficiency ranged from 0.14 to 0.2% (Figure 4b).
Figure 4.

(a): H2O2 decomposition (left axis) and phenol transformation (right axis) catalyzed by goethite. Solid line: first order fit to the data. (b): stoichiometric efficiency in the presence of dissolved silica. Experiments were conducted at least triplicate and, instead of present the average value and standard deviation, all results were presented. [goethite] = 4 g/L, pH = 7, [PIPES] = 1 mM, [NaNO3] = 0.1 M. Except for the control experiment (inversed triangles), the H2O2 initial concentration in all experiments was [H2O2]0 = 20 mM.
Pyrolusite
The adsorption of dissolved silica onto β-MnO2 and its effect on the catalytic activity of β-MnO2 toward H2O2 decomposition were also investigated (Figure 5). In the SiO2-free system, 50 mM H2O2 was decomposed within 2 hours. As with iron-containing materials, addition of dissolved SiO2 slowed the rate of H2O2 decomposition in proportion to the concentration of added SiO2. The half-life of H2O2 was approximately 0.15 hr for the SiO2-free system and 0.5 hr for the experiment with 1.5 mM SiO2. Unlike the case with iron oxides, the adsorption of SiO2 was not measurable even at a β-MnO2 concentration of 20 g/L (inset of Figure 5).
Figure 5.
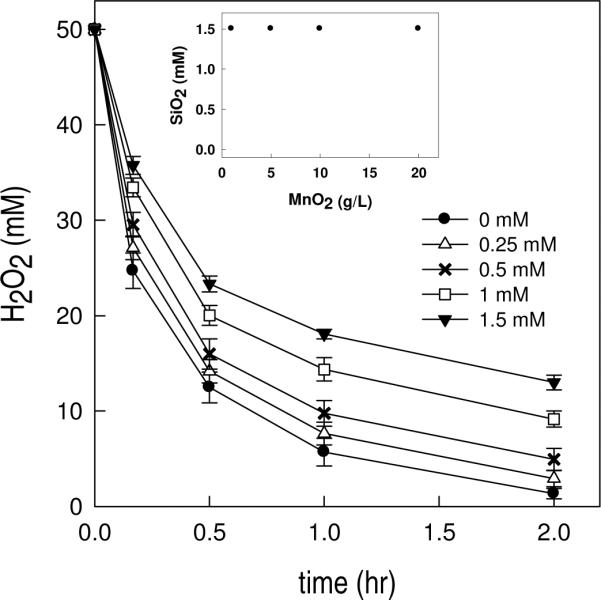
Decomposition of H2O2 catalyzed by pyrolusite (β-MnO2) in the presence of various concentrations of dissolved SiO2. [β-MnO2] = 1 g/L, pH = 8.4, [NaNO3] = 0.1 [borate] = 4 mM. Inset: [SiO2] remaining in the solution after 24 hr equilibration with various amount of MnO2. [SiO2]initial = 1.5 mM, [MnO2] = 1 – 20 g/L, other conditions were similar to those above.
Discussion
Adsorption of SiO2 affects the surface properties and reactivity of metal oxides and clays in natural and engineered processes. For example, the presence of dissolved SiO2 inhibits the nucleation and growth of iron precipitates18 as well as the transformation of amorphous iron (hydr)oxide into more stable phases (e.g., goethite).19 Dissolved SiO2 also appears to stabilize iron oxide colloids, enhancing their mobility in natural waters and decreasing the efficiency of iron-based coagulation processes employed in drinking and wastewater treatment operation.20–22 Adsorption of SiO2 also alters the surface area, charge and surface complexation sites on iron oxides, thereby affecting the adsorption of various solutes.19,23–25 The presence of SiO2 in water also affects the corrosion of iron, with sorbed SiO2 forming a protective layer that inhibits corrosion26,27 or an impurity that destabilizes protective iron oxide scale layers.27
In our experimental system, silica slowed the rate of H2O2 decomposition on iron and manganese mineral surfaces (Figure 3 and 5). To understand how SiO2 affects the reactivity of minerals in this process, it is necessary to understand how SiO2 and H2O2 interact with metal-containing surfaces. The adsorption behavior of SiO2 on iron minerals is presented below, followed by a discussion of H2O2 decomposition mechanisms and the possible effects of SiO2. As the mechanism through which H2O2 is decomposed on MnO2 differs from that of iron minerals, the MnO2 system is discussed separately.
Iron minerals
SiO2 adsorption often exhibits a fast and a slow stage, with more than 90% of adsorption taking place within the first few hours and the remaining 10% of adsorption occurring over several weeks (Figure 1 and S1).12,28 Solution conditions such as pH and SiO2-to-iron oxide ratio strongly affect this process.12,28 In addition to the bulk solution measurements, spectroscopic techniques such as ATR-IR29 and XAFS18, as well as surface modeling tools, have been used to infer the bonding of sorbed SiO2 and the mechanism through which adsorption occurs. The adsorption process generally has been described as a complexation reaction between surface hydroxyl groups and SiO2. However, no consensus has been reached on the exact nature of this interaction at a molecular level. Some investigators argued that the process involves the reaction between a monomeric SiO2 species and one hydroxyl group (reaction (1) and (2) in Table 2).11,30 However, Davis et al. invoked SiO2 adsorption by both monomeric and dimeric species (reaction (3) and (4)) to explain adsorption data along with the zeta potential data.12 It also has been suggested that adsorption can involve a bidentate complex between an SiO2 monomer and 2 hydroxyl groups (reaction (5))18,28and that the siloxane linkages could form between two adjacent sorbed SiO2 monomers or/and between sorbed monomers and dissolved SiO2 (reaction (6)).29,31 The latter scenario might lead to the formation of oligomeric silica species (e.g., a linear trimer29, or cyclic tetramer32) on the surface. The difference in the behavior of surface-adsorbed SiO2 could be attributable to differences in solution conditions that were employed in these studies. Polymeric silica species tend to be important at high pH and high SiO2 concentrations12 while a high SiO2-to- iron oxide ratio could result in a high density of sorbed SiO2 (ΓSi), leading to the formation of oligomeric species.
Table 2.
Possible surface complexation reactions between iron oxides and dissolved SiO2
| Reaction | Reference | |
|---|---|---|
| ≡FeOH + Si(OH)4 → ≡FeSiO(OH)3 + H2O | (1) | 11, 12, 30 |
| ≡FeOH + Si(OH)4 → ≡FeSiO2(OH)2− + H2O + H+ | (2) | 11, 12, 30 |
| ≡FeOH + Si2O2(OH)5− + H+ → ≡FeSi2O2(OH)5 + H2O | (3) | 12 |
| ≡FeOH + Si2O2(OH)5− → ≡FeSi2O3(OH)4− + H2O | (4) | 12 |
| 2 ≡FeOH + Si(OH)4 → ≡Fe2O2Si(OH)2 + 2 H2O | (5) | 18, 28 |
| 2 ≡FeOH + 3 Si(OH)4 → ≡Fe2H6-nSi3O10n− + n H+ + 4 H2O | (6) | 29 |
In the present study, we used STEM/EDX to investigate the distribution of Si on the surface of goethite that had been pre-equilibrated with solutions containing varying amounts of dissolved SiO2. With a nano-sized probe (1.4 nm in this study), STEM/EDX is capable of providing a high resolution elemental distribution map that cannot be obtained by other techniques (e.g., scanning electron microscopy/EDX or X-ray photoelectron spectroscopy). A nano-sized probe technique is also needed to evaluate heterogeneity of the small goethite particles employed in this study (less than 100 nm, Figure S4). The relatively heterogeneous distribution of Si on the goethite surface and the presence of regions that were fully coated with Si (Figure 2) suggest that the adsorption did not take place in a “layer-by-layer” mode, presumably because adsorption is more favorable on some crystallographic faces than on others. The presence of more regions that were fully coated with Si at higher [SiO2]initial supports the hypothesis that a high SiO2-to-iron oxide ratio leads to the formation of oligomeric species.
Hydrogen peroxide decomposition
Iron oxides and iron-containing (e.g., ferrihydrite, goethite, iron-containing clays and iron-coated sand) can catalyze the decomposition of H2O2. This process can generate hydroxyl radical (•OH), presumably through a Haber – Weiss mechanism analogous to that observed in the homogeneous Fenton system:2,4,5,33
| (7) |
| (8) |
| (9) |
Some investigators have postulated that, as in the homogeneous Fenton system, reaction (7) actually consists of a series of reactions, beginning with the formation of a complex between the surface and H2O2:33,34
| (10) |
| (11) |
| (12) |
Assuming that reaction (10) is the first step in H2O2 decomposition, sorbed SiO2 may alter the reactivity of iron minerals by occupying iron surface hydroxyl groups, thereby preventing the formation of ≡Fe-OH3+(H2O2)(s). As mentioned above, the SiO2 adsorption mechanism is not totally understood. Therefore, we did not try to estimate the number of hydroxyl groups that were occupied by SiO2 and consequently, no correlation between the number of available hydroxyl groups with kobs has been made. However, the ΓSi – kobs profile (Figure 3b) supports the hypothesis that the slower decomposition of H2O2 was due to occupation of surface sites by SiO2. At a ΓSi < 0.04 mmol/g goethite, where regions that were fully covered with Si were negligible, kobs drastically decreased as ΓSi increased. Due to the formation of oligomeric species at higher ΓSi, the number of iron sites that were occupied by SiO2 only increased slightly, resulting in a much slower kobs decrease in this range. The higher ΓSi observed at [SiO2]equilibrium = 1.14 mM (Figure 1) also could be attributable to the formation of oligomeric species. Finally, the presence of sites that were not occupied with Si (Figure 2a) could help to explain why H2O2 decomposition in all experiments was still observed at high SiO2 concentration.
It was previously observed that under circumneutral pH conditions, H2O2 decomposes mainly through pathways that do not produce •OH.5,6 Consequently, understanding the branching between different pathways is important because only those that produce •OH will be beneficial for contaminant oxidation. Our data (Figure 4) indicate that dissolved SiO2 has a detrimental effect on the overall stoichiometric efficiency. A possible explanation might be that surface sites have different reactivity toward •OH production and the preferential adsorption of SiO2 on “more •OH productive” sites would lower the stoichiometric efficiency. Although assigning surface sites with different affinities is used widely in describing adsorption on iron oxides35, the above hypothesis is speculative and further research is needed to address this issue.
Pyrolusite
Manganese oxides (such as birnessite and pyrolusite) are very reactive in catalyzing H2O2 decomposition. Although the mechanism of this process remains unclear, our data (Figure S5) and those of other investigators1,5,7 indicate that this process does not produce •OH. Consequently, the presence of MnO2 in aquifer materials is detrimental for H2O2-based ISCO.
H2O2 decomposition by β-MnO2 also slowed in the presence of dissolved SiO2, although the mechanism through which the loss of H2O2 was inhibited is unclear. In a batch experiment with β-MnO2, SiO2 adsorption was not measurable (inset of Figure 5). We were also unable to find any reports of adsorption of SiO2 onto MnO2, suggesting that SiO2 does not adsorb on MnO2 to an appreciable extent. However, it would be difficult to measure minor adsorption in the batch tests, even with the highest solids density tested. A column experiment was used to assess the potential for SiO2 adsorption at higher β-MnO2 concentrations (refer to the Supporting Information for experimental setup). The results show that there was, indeed, a modest degree of silica uptake onto the β-MnO2 from the solution during the first few minutes of the test, the uptake being proportional to the amount of MnO2 in the column (Figure S6). This modest uptake of silica is likely to be responsible for the lower reactivity of MnO2 observed experimentally. However, further research is needed to address this issue.
Environmental implications
The results of this study suggest that in H2O2-based ISCO systems, H2O2 should last longer if groundwater contains a significant amount of dissolved SiO2. In systems where the subsurface is deficient in SiO2, dissolved silica could be injected together with H2O2 to increase the persistence of H2O2 to assure remediation of areas further from the injection well. Dissolved SiO2 has a potential to be a better H2O2-stabilizing agent than phosphate because SiO2 is inexpensive and does not stimulate bacterial growth. Although SiO2 decreased the stoichiometric yield of •OH from iron minerals, this effect was relatively modest, and would be outweighed in in situ applications by the greater longevity of H2O2 in the presence of SiO2. The effect of SiO2, however, will vary among aquifers with different mineral compositions (e.g., iron and manganese content and crystalinity, soil organic matter content) and groundwater chemistry. These factors should be considered in the design and operation of H2O2- based ISCO. Additionally, because silica adsorption is a reversible process (Figure S7), it would appear advisable to inject dissolved SiO2 and H2O2 simultaneously to assure H2O2 lifetime enhancement.
Our study also indicates that many bench-scale studies performed in the absence of dissolved SiO2 may have underestimated the lifetime of H2O2 or overestimated the effect of stabilizing agents. For example, in the SiO2-free system (H2O2 half-life of 7.77 hr, Table 1), 2 mM phosphate increased the H2O2 half-life by approximately a factor of four (t1/2 = 31.6 hr). In the presence of 0.5 mM SiO2, however, phosphate provided much less of an effect, increasing the half-life of H2O2 by about only 50% (t1/2 = 21.7 hr and 33.5 hr in the absence and presence of phosphate, respectively).
SiO2 also suppressed H2O2 decomposition by MnO2. Depending on the relative amount of iron- and manganese-containing solids, SiO2 could enhance the overall efficiency of the remediation process, especially in soils with high Mn content. Additional research is needed to assess the contribution of different iron- and manganese-containing solids to H2O2 loss in soils and aquifer materials.
Finally, our data also suggest that dissolved silica can affect the reactivity of iron-containing catalysts used in H2O2-based advanced oxidation processes. Although the SiO2 concentrations in surface waters and industrial wastes are often lower than those observed in groundwater, a gradual loss in catalyst activity due to SiO2 adsorption will likely occur during long-term catalyst use.
Acknowledgments
This research was funded by the U.S. National Institute for Environmental Health Sciences (NIEHS) Superfund Basic Research Program (Grant P42 ES004705). The authors acknowledge support of the National Center for Electron Microscopy, Lawrence Berkeley National Laboratory, which is supported by the U.S. Department of Energy under Contract # DE-AC02-05CH11231. A.L.P. was supported in part by Vietnam Education Foundation (VEF).
Footnotes
Supporting Information Figure S1–S7, Column experiments with β-MnO2 and FeOOH, role of H2O2 diffusion and H2O2 adsorption on the rate of H2O2 decomposition by goethite. This information is availablefree of charge via the Internet at http://pubs.acs.org/
References
- 1.Huling SG, Pivetz BE. In-Situ Chemical Oxidation. U.S. Environmental Protection Agency Engineering Issue. 2006 http://www.epa.gov/ada/topics/oxidation_pubs.html.
- 2.Pignatello JJ, Oliveros E, MacKay A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Critical Reviews in Environmental Science and Technology. 2006;36:1. [Google Scholar]
- 3.Watts RJ, Teel AL. Chemistry of modified Fenton's reagent (catalyzed H2O2 propagations-CHP) for in situ soil and groundwater remediation. Journal of Environmental Engineering-ASCE. 2005;131:612–622. [Google Scholar]
- 4.Kwan WP, Voelker BM. Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environmental Science & Technology. 2002;36:1467–1476. doi: 10.1021/es011109p. [DOI] [PubMed] [Google Scholar]
- 5.Petigara BR, Blough NV, Mignerey AC. Mechanisms of hydrogen peroxide decomposition in soils. Environmental Science & Technology. 2002;36:639–645. doi: 10.1021/es001726y. [DOI] [PubMed] [Google Scholar]
- 6.Pham AL-T, Lee C, Doyle FM, Sedlak DL. A silica-supported iron oxide catalyst capable of activating hydrogen peroxide at neutral pH values. Environmental Science & Technology. 2009;43:8930–8935. doi: 10.1021/es902296k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furman O, Laine DF, Blumenfeld A, Teel AL, Shimizu K, Cheng IF, Watts RJ. Enhanced reactivity of superoxide in water–solid matrices. Environmental Science & Technology. 2009;43:1528–1533. doi: 10.1021/es802505s. [DOI] [PubMed] [Google Scholar]
- 8.Kakarla PKC, Watts RJ. Depth of Fenton-like oxidation in remediation of surface soil. Journal of Environmental Engineering-ASCE. 1997;123:11–17. [Google Scholar]
- 9.Watts RJ, Finn DD, Cutler LM, Schmidt JT, Teel AL. Enhanced stability of hydrogen peroxide in the presence of subsurface solids. Journal of Contaminant Hydrology. 2007;91:312–326. doi: 10.1016/j.jconhyd.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Langmuir D. Aqueous Environmental Geochemistry. Prentice-Hall, Inc.; 1997. [Google Scholar]
- 11.Sigg L, Stumm W. The interaction of anions and weak acids with the hydrous goethite (α-FeOOH) surface. Colloids and Surfaces. 1981;2:101–117. [Google Scholar]
- 12.Davis CC, Chen H-W, Edwards M. Modeling silica sorption to iron hydroxide. Environmental Science & Technology. 2002;36:582–587. doi: 10.1021/es010996t. [DOI] [PubMed] [Google Scholar]
- 13.Schwertmann U, Cornell RM. Iron Oxides in the Laboratory: Preparation and Characterization. 2000 [Google Scholar]
- 14.Nico PS, Stewart BD, Fendorf S. Incorporation of oxidized uranium into Fe (hydr)oxides during Fe(II) catalyzed remineralization. Environmental Science & Technology. 2009;43:7391–7396. doi: 10.1021/es900515q. [DOI] [PubMed] [Google Scholar]
- 15.Kohn T, Kane SR, Fairbrother DH, Roberts AL. Investigation of the inhibitory effect of silica on the degradation of 1,1,1-Trichloroethane by granular iron. Environmental Science & Technology. 2003;37:5806–5812. doi: 10.1021/es034495e. [DOI] [PubMed] [Google Scholar]
- 16.Eisenberg G. Colorimetric determination of hydrogen peroxide. Industrial & Engineering Chemistry Analytical Edition. 1943;15:327. [Google Scholar]
- 17.Tamura H, Goto K, Yotsuyanagi T, Nagayama M. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III) Talanta. 1974;21:314. doi: 10.1016/0039-9140(74)80012-3. [DOI] [PubMed] [Google Scholar]
- 18.Pokrovski GS, Schott J, Farges F, Hazemann J-L. Iron (III)-silica interactions in aqueous solution: insights from X-ray absorption fine structure spectroscopy. Geochimica et Cosmochimica Acta. 2003;67:3559–3573. [Google Scholar]
- 19.Anderson PR, Benjamin MM. Effect of silicon on the crystallization and adsorption properties of ferric oxides. Environmental Science & Technology. 1985;19:1048–1053. doi: 10.1021/es00141a004. [DOI] [PubMed] [Google Scholar]
- 20.Cameron AJ, Liss PS. The stabilization of “dissolved” iron in freshwaters. Water Research. 1984;18:179–185. [Google Scholar]
- 21.Browman MG, Robinson RB, Reed GD. Silica polymerization and other factors in iron control by sodium silicate and sodium hypochlorite additions. Environmental Science & Technology. 1989;23:566–572. [Google Scholar]
- 22.Robinson RB, Frasier B, Reed GD. Iron and manganese sequestration facilities using sodium-silicate. Journal American Water Works Association. 1992;84:77. [Google Scholar]
- 23.Davis CC, Knocke WR, Edwards M. Implications of aqueous silica sorption to iron hydroxide:□ Mobilization of iron colloids and interference with sorption of arsenate and humic substances. Environmental Science & Technology. 2001;35:3158–3162. doi: 10.1021/es0018421. [DOI] [PubMed] [Google Scholar]
- 24.Zachara JM, Girvin DC, Schmidt RL, Resch CT. Chromate adsorption on amorphous iron oxyhydroxide in the presence of major groundwater ions. Environmental Science & Technology. 1987;21:589–594. doi: 10.1021/es00160a010. [DOI] [PubMed] [Google Scholar]
- 25.Jordan N, Lomenech C, Marmier N, Giffaut E, Ehrhardt J-J. Sorption of selenium(IV) onto magnetite in the presence of silicic acid. Journal of Colloid and Interface Science. 2009;329:17–23. doi: 10.1016/j.jcis.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 26.Sastra VS. Corrosion Inhibitors. Principles and Applications. Wiley; West Sussex, England: 1998. [Google Scholar]
- 27.Rushing JC, McNeill LS, Edwards M. Some effects of aqueous silica on the corrosion of iron. Water Research. 2003;37:1080–1090. doi: 10.1016/s0043-1354(02)00136-7. [DOI] [PubMed] [Google Scholar]
- 28.Hiemstra T, Barnett MO, van Riemsdijk WH. Interaction of silicic acid with goethite. Journal of Colloid and Interface Science. 2007;310:8–17. doi: 10.1016/j.jcis.2007.01.065. [DOI] [PubMed] [Google Scholar]
- 29.Swedlund PJ, Hamid RD, Miskelly GM. Insights into H4SiO4 surface chemistry on ferrihydrite suspensions from ATR-IR, Diffuse Layer Modeling and the adsorption enhancing effects of carbonate. Journal of Colloid and Interface Science. 2010;352:149–157. doi: 10.1016/j.jcis.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Jordan N, Marmier N, Lomenech C, Giffaut E, Ehrhardt J-J. Sorption of silicates on goethite, hematite, and magnetite: Experiments and modelling. Journal of Colloid and Interface Science. 2007;312:224–229. doi: 10.1016/j.jcis.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 31.Swedlund PJ, Webster JG. Adsorption and polymerisation of silicic acid on ferrihydrite, and its effect on arsenic adsorption. Water Research. 1999;33:3413–3422. [Google Scholar]
- 32.Icopini GA, Brantley SL, Heaney PJ. Kinetics of silica oligomerization and nanocolloid formation as a function of pH and ionic strength at 25°C. Geochimica et Cosmochimica Acta. 2005;69:293–303. [Google Scholar]
- 33.Lin S-S, Gurol MD. Catalytic Decomposition of hydrogen peroxide on iron oxide: Kinetics, Mechanism, and Implications. Environmental Science & Technology. 1998;32:1417–1423. [Google Scholar]
- 34.Kwan WP, Voelker BM. Rates of hydroxyl radical generation and organic compound oxidation in mineral-catalyzed Fenton-like systems. Environmental Science & Technology. 2003;37:1150–1158. doi: 10.1021/es020874g. [DOI] [PubMed] [Google Scholar]
- 35.Dzombak DA, Morel FM. Surface complexation modeling: hydrous ferric oxide. John Wiley & Sons; New York: 1990. [Google Scholar]