

Abstract

A rapid synthesis of the tetracyclic core of Kopsia indole alkaloids related to lapidilectine B, grandilodine C, and tenuisine A is reported. Key to the success of this route was an efficient and scalable Ugi four-component coupling to install all the necessary carbons found in the natural products.
Alkaloids isolated from the Kopsia genus of flowering plant, representitive examples of which are shown in Figure 1, demonstrate a wide range of structural diversity as well as pharmacological activities.1 Extracts from Kopsia have been used in traditional treatments for various ailments including rheumatoid arthritis, edema, tonsillitis and hypertension. Lapidilectine B (1), grandilodine C (2)2 and lundurine B (7) show multi-drug resistance (MDR) reversing effects in vincristine-resistant KB cancer cells,3 whereas lundurines A (6) and C (8) show selective cytotoxicity towards B16 melanoma cells. A majority of the synthetic efforts aimed at these compounds have been focused on the lundurine class of molecules.4 The most advanced approach, which arrives at the tetracyclic core of lundurine using creative ring closing metathesis reactions, was recently reported by Martin and coworkers.5
Figure 1.

Selected Kopsia alkaloids
In addition, an elegant total synthesis of lapidilectine B (1) was achieved by Pearson and coworkers in 2001, and featured an inventive use of a 2-azaallyllithium ion in a [3+2] cycloaddition.6 To date, no synthetic efforts have been reported toward the tenuisines7 or grandilodines.
We were drawn to these compounds as synthetic targets because of their unique structures, which offered opportunities for new synthetic tactics, as well as their potential as starting points for new pharmaceuticals. Furthermore, our analysis of the subset of Kopsia alkaloids illustrated in Figure 1 indicated a close structural relationship, which could inform a unified strategy for their syntheses.
In our analysis of the subset of Kopsia alkaloids depicted in Figure 1, it seemed plausible that the fused cyclopropane in the lundurines (see 6–9) could arise, synthetically (or even biosynthetically), from a decarboxylation of a related tenuisine type precursor (e.g., 5).8 Alternatively, the miscroscopic reverse, i.e., carboxylation of the cyclopropane moiety of a lundurine precursor (e.g., 7), could provide an entry to tenuisines such as 5. A similar relationship can be envisioned between fused γ-lactone compounds 1 or 2 and their respective cyclopropane-containing congeners, which are yet to be isolated. Furthermore, a selective carbanion opening of the γ-lactone moiety in 1 or 2 could lead to 3 and 4, respectively.
In order to test aspects of this unified synthetic plan, we sought a versatile synthetic precursor that could give access to either the fused γ-lactone or cyclopropane framework. Fused cyclobutanone 10 (Scheme 2) engendered exciting possibilities for its conversion to a γ-lactone (as in 1 or 2 with R = H; or 5 with R = OMe) using a Baeyer-Villiger type oxidation,9 or to a fused cyclopropane (as in 6–9; R = OMe) using a C-C bond activation/decarbonylation sequence.10 As such, 10 was selected as the penultimate target.
Scheme 2.

Retrosynthetic analysis of cyclobutanone precursor
We envisioned cyclobutanone 10 arising from ester 12 (if R = H) via ketene intermediate 11 by a [2+2] cycloaddition.11 Tetracycle 12 could in turn arise from spiro-fused α,β-unsaturated lactam derivative 13 by cleavage of the cyclohexanone ring and functionalization of the indole C-2 position. The cleavage step could, in principle, be rendered enantioselective by a desymmetrization of ketone 13. Tryptamine-derived lactam 13 could in turn arise from a four-component Ugi coupling12 of 14, 15, 16 and 17.
Our synthetic studies commenced with the Ugi four-component coupling (Scheme 3), which led to adduct 18 in 95% yield. The virtues of this coupling reaction are many fold. First, in this single step, we install all the carbons of the lapidilectine-type Kopsia alkaloid framework. Second, the product precipitates out of the reaction mixture and only requires filtration to be obtained free of impurities. Third, the reaction can be conducted on large scale (> 200 g) without compromising yield.13 The Armstrong isocyanide (16)14 was selected for the Ugi coupling on the basis of the elegant synthetic studies of Sorensen and Seike on FR901483, which showed (as we found) that the Ugi product (18) could easily be converted to the corresponding methyl ester (see 19).15 Thus, treatment of 18 with methanolic HCl provided methyl ester 19, whereby the esterification was attended by a transketalization to the dimethylketal.
Scheme 3.

Synthesis of spiro-lactam 13
Following the Sorensen protocol, subjection of acetamide ester 19 to KOt-Bu in THF effected a Dieckmann condensation. Selective reduction of the ketone group (NaBH4) and elimination of the resulting hydroxy group (activated as the carbonate) provided spiro-fused lactam 13 in excellent yield over three steps (Scheme 3). The accompanying protection of the indole nitrogen made the handling of this compound straightforward. Of note, the conversion of 19 to 13 can be consistently performed on >5g scale without appreciable depression in yield.
With efficient access to 13, we have investigated several strategies for the synthesis of the tetracyclic core of the lapidilectines and structurally related alkaloids (i.e., 12, Scheme 2). These studies began with the conversion of 13 to 20 by TBS silyl enol ether formation (Scheme 4), which proceeded in excellent yield. Oxidative opening of the six-membered ring was effected using the two stage protocol of dihydroxylation (OsO4, TBHP) followed by Pb(OAc)4-mediated cleavage in the presence of methanol, which afforded aldehyde ester 21. In preparation for cyclization studies en route to the tetracyclic core structure (Eqs. 1–3), 21 was converted to acid 22 employing a Pinnick oxidation;16 to C-2 bromo-indole 23 using NBS; and to dimethylacetal 24 under standard acetalization conditions.
Scheme 4.

Oxidative opening of spirocycle 20
As shown in Eqs. 1–3, the lapidilectine-type tetracyclic core, varying only in the oxidation level about the eight-membered ring, can be successfully realized from each of these precursors (i.e., 22, 23 and 24). Tetracycle 25 was directly accessible from crude acid 22 in 43% yield using a polyphosphoric acid mediated Friedel-Crafts acylation (Eq. 1).17 Despite the modest yield of this transformation, the usual ‘two-step’ Friedel-Crafts protocol, involving treatment of the corresponding acid chloride with a Lewis acid (e.g., AlCl3) was less efficient and resulted in significant polymerization. Alternatively, aldehyde 23 was successfully converted to keto-tetracycle 25 (Eq. 2) in 46% yield upon treatment with AIBN and dodecyl thiol (C10H21SH), presumably via an acyl radical intermediate.18 We have found this particular protocol for the formation of 25 to be effective (as compared to starting from acid 22) because it generally leads to less byproduct formation. A Friedel-Crafts type transformation could also be initiated from dimethyl acetal 24 using Amberlyst-15 as the source of acid (Eq. 3) to afford 26 in 54% yield.19 The mass balance of the material was accounted for by aldehyde 21, which likely arises from a competing hydrolyis of the acetal group. Attempts to effect the conversion of 24→26 using Lewis acids or other protic acids resulted only in the hydrolysis of the acetal, which reverts back to aldehyde 21.
 |
(1) |
 |
(2) |
 |
(3) |
In preparation for the exploration of endgame scenarios, tetracycle 26 was converted to 27 (Eq. 4) by cleavage of the carbamate group and selective removal of the alkene group by ionic reduction using methanesulfonic acid and triethylsilane.20 Our current efforts are focused on the selective conversion of the methyl ester group of 27 to an ‘activated ester’ to study our proposed ketene generation/cycloaddition (see 12→10, Scheme 2).
 |
(4) |
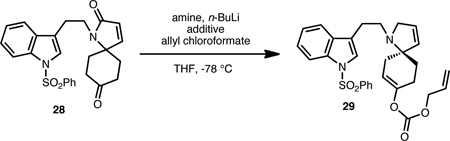
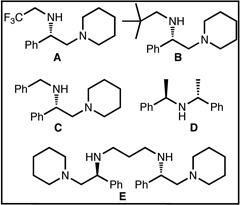
We have also briefly explored opportunities to render our strategy enantioselective. In this regard, our initial efforts have focused on the desymmetrization of spiroketones related to 13 (see 28, Table 1). The control of the single tertiary amine spiro stereocenter would guide the installation of the three additional stereocenters in the target natural products, which would substantially simplify the diastereoselectivity challenge. As a proof of concept for stereoselective desymmetrization, the enantioselective deprotonation of ketone 28 was investigated with a range of chiral, non-racemic kinetic amine bases (see Table 1).21 Our brief survey has revealed that the amide base generated from amine D, in the presence of LiCl (1 equiv),22 yields enol carbonate 29 in 39% ee and 60% yield (entry 5). This preliminary result, albeit in modest ee, could provide the basis for the development of an enantioselective synthesis of lapidilectine and related Kopsia alkaloids.
Table 1.
Enantioselective Desymmetrization of 28
 | |||||
|---|---|---|---|---|---|
| Amines | entry | amine | additive (1 equiv) | % ee | |
 |
1 | A | LiCl | 31 | |
| 2 | A | LiCl, HMPA | 24 | ||
| 3 | B | LiCl | 6 | ||
| 4 | C | LiCl | 24 | ||
| 5 | D | LiCl | 39 | ||
| 6 | D | – | 18 | ||
| 7 | E | LiCl (2 equiv) | 12 | ||
In conclusion, we have developed an effective and scalable synthetic sequence to the tetracyclic core of Kopsia alkaloids related to lapidilectine. The synthetic sequence features a highly enabling Ugi four-component coupling to install all the carbons found in these natural products. The reported strategy holds high potential for providing a unified approach to the subset of Kopsia alkaloids depicted in Figure 1. In addition, preliminary results on enantioselective deprotonation of a spirocyclic ketone provides a proof of concept for the possible enantioselective syntheses of these natural products.
Supplementary Material
Acknowledgement
The authors are grateful to Prof. Erik J. Sorensen (Princeton University) for detailed procedures related to Ref. 15. The authors also thank Prof. Armen Zakarian (UC Santa Barbara) for gifts of diamines A, B and C and Amgen for a gift of tetra-amine E. The NIH (NIGMS RO1 084906) is gratefully acknowledged for financial support of this work. EES and BGP are grateful for an NSF graduate research fellowship and Tobacco Related Disease Research Program (TRDRP) fellowship, respectively. RS and BGP thank Eli Lilly for hosting BGP for a summer internship in 2009.
Footnotes
Supporting Information Available. Experimental details and characterization for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Awang K, Svenet T, Pais M. Nat. Prod. 1993;56:1134–1139. [Google Scholar]
- 2.For the isolation of the grandilodines see: Yap W-S, Gan C-Y, Low Y-Y, Choo Y-M, Etoh T, Hayashi M, Komiyama K, Kam T-S. J. Nat. Prod. 2011;74:1309–1312. doi: 10.1021/np200008g.
- 3.Kam T-S, Lim KH, Yoganathan K. Tetrahedron. 2004;60:10739–10745. [Google Scholar]
- 4.(a) Ferrer C, Amijs CHM, Echavarren AM. Chem.;Eur.J. 2007;13:1358–1373. doi: 10.1002/chem.200601324. [DOI] [PubMed] [Google Scholar]; (b) Donets PA, Van Hecke K, Van Meervelt L, Van der Eycken EV. Org. Lett. 2009;11:3618–3621. doi: 10.1021/ol901356h. [DOI] [PubMed] [Google Scholar]; (c) Ferrer C, Escribano-Cuesta A, Echavarren AM. Tetrahedron. 2009;65:9015–9020. [Google Scholar]
- 5.Orr ST, Tian J, Niggemann M, Martin SF. Org. Lett. 2010;13:5104–5107. doi: 10.1021/ol201980r. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.(a) Pearson WH, Mi Y, Lee IY, Stoy P. J. Am. Chem. Soc. 2001;123:6724–6725. doi: 10.1021/ja016007d. [DOI] [PubMed] [Google Scholar]; (b) Pearson WH, Lee IY, Mi Y, Stoy P. J. Org. Chem. 2004;69:9109–9122. doi: 10.1021/jo048917u. [DOI] [PubMed] [Google Scholar]
- 7.For the isolation of the tenuisines and an account of their structural revision, see ref. 3.
- 8.Kam and co-workers (see ref. 3) isolated the lundurine and tenuisine compounds from the same plant source, lending credence to a biogenentic connection between these natural products.
- 9.Krow GR. Org. React. 1993;43:251–798. [Google Scholar]
- 10.For a review on this type of C-C bond activation: Seiser T, Saget T, Tran DN, Cramer N. Angew. Chem. Int. Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053.
- 11.Snider BB. Chem. Rev. 1988;88:793–811. [Google Scholar]
- 12.Domling A, Ugi I. Angew. Chem. Int. Ed. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 13.Performed by BGP in the Process Group at Eli Lilly, Indianapolis.
- 14.Keating TA, Armstrong RW. J. Am. Chem. Soc. 1996;118:2574–2583. [Google Scholar]
- 15.Seike H, Sorensen EJ. Synlett. 2008:695–701. doi: 10.1055/s-2008-1042813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) For a pertinent discussion on this oxidation, see: Kürti L, Czako B. Strategic Applications of Named Reactions in Organic Synthesis. Elsevier; 2005. pp. 354–355. For seminal publications see: Lindgren BO, Nilsson T. Acta Chemica Scandinavica. 1973;27:888–890. Kraus GA, Roth B. J. Org. Chem. 1980;45:4825–4830. Bal BS, Childers WE, Jr., Pinnick HW. Tetrahedron. 1981;37:2091–2096.
- 17.(a) Owton WM, Brunavs M. Synth. Commun. 1991;21:981–987. [Google Scholar]; (b) Wood JL, Pujanauski BG, Sarpong R. Org. Lett. 2009;11:3128–3131. doi: 10.1021/ol9010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshikai K, Hayama T, Nishimura K, Yamada K, Tomioka K. J. Org. Chem. 2005;70:681–683. doi: 10.1021/jo048275a. (b) For a recent application in a complex molecule synthesis see: Enquist JA, Jr., Stoltz BM. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046.
- 19.Ballini R, Gabrielli S, Palmieri A, Petrini M. Adv. Synth. Catal. 2010;352:2459–2462. [Google Scholar]
- 20.Larson GL, Fry JL. Org. React. 2008:1–737. [Google Scholar]
- 21.O’Brien P. J. Chem. Soc. Perkins Trans. 1. 1998:1439–1457. [Google Scholar]
- 22. Majewski M, Lazny R, Nowak P. Tetrahedron Lett. 1995;36:5465–5468. (b) Of note, diminished enantioselectivity is generally observed in the absence of the LiCl additive (compare, for example, entries 5 and 6).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.