

Abstract
Background
Activated effector T cells contribute to tissue injury observed in inflammatory bowel disease. T cells are recruited to effector sites after activation in peripheral lymph nodes directs expression of tissue-specific homing receptors. One such mechanism for effector T cell recruitment employs activation-induced fucosylation of P-selectin glycoprotein ligand (PSGL)-1 that mediates binding to endothelial P-selectin. Here, we examine the differential role of PSGL-1 in recruiting effector T cell subsets in colitis.
Methods
C57BL/6 wild type and PSGL-1−/− mice received 2.5% DSS for 6 days and were euthanized 7 and 14 days after the initiation of DSS. Disease activity was monitored throughout. Histologic colitis scores, colonic CD4+ accumulation, and cytokine production were assessed at day 7 and 14. Recruitment of T-helper subsets was assessed by enumerating adoptively transferred Th1 or Th17 CD4+ cells 2 days after transfer to DSS-treated mice.
Results
DSS colitis increases CD4+ T cells in colonic tissue and induces Th1 (IFNγ, TNF) and Th17 (IL-17, IL-22) cytokines. Loss of PSGL-1 attenuates DSS colitis, decreases colonic CD4+ T cell numbers and reduces both Th1 and Th17 cytokine production. Colitis increases recruitment of Th1 (19-fold) and Th17 (2.5-fold) cells. PSGL-1 deficiency in transferred T cells abrogates colonic recruitment of Th1 cells in DSS colitis whereas Th17 recruitment is unaffected.
Conclusions
PSGL-1 selectively controls Th1 recruitment in colitis. Whereas Th17 recruitment is independent of PSGL-1, generation of colonic Th17 cytokine requires initial Th1 recruitment. Therefore, attenuating PSGL-1 binding may prevent colonic recruitment of disease-causing Th1 cells that promote local Th17 generation.
Keywords: PSGL-1, colitis, Th1, Th17, recruitment
Inflammatory bowel disease (IBD) refers to two related disorders known as Crohn's disease (CD) and ulcerative colitis (UC) that affect 1–2 million individuals(1). It occurs in genetically susceptible hosts due to dysregulated mucosal T cell responses to commensal enteric bacteria(2). Mucosal mRNA from IBD patients indicate that T cells in CD produce relatively high levels of T helper type 1 (Th1) and Th17 cytokines whereas cytokine changes consistent with a Th2-type response occur in UC(3–6). Recent genetic linkage studies indicate that dysregulation of components of the mucosal innate immune system (e.g. macrophages, dendritic cells, etc) contribute to dysregulated T cell responses(7).
Traditional therapies for IBD include corticosteroids, mesalamine, immunomodulators, and antibiotics. Unfortunately, nearly one third of the patients prescribed steroids require repeat dosing or persist with refractory disease(8). Biologics that target tumor necrosis factor (anti-TNF) induce rapid remission, but carry the risk of serious infections in 1–4% and rarely fatal T cell lymphoma(9). Therefore, defining alternate targets for therapy is critical. Tissue specific inhibition of T cell recruitment offers an attractive means for attenuating mucosal inflammation without incurring infectious complications associated with systemic immunosuppression.
The complexity of lymphocyte migration allows tissue to selectively recruit cells with specific functional attributes. Naïve T cells do not migrate to inflamed or effector sites until they are activated in lymph nodes. Activation imprints cells with a specific profile of homing receptors needed for recruitment to effector sites (10–12). Thus, activation in the periphery permits the immune system to selectively imprint the proper adhesion molecules on populations of T cells reactive to enteric Ag. Surface molecules expressed on gut-specific memory-like cells (e.g. α4β7 integrin) bind ligands on endothelial villous structures [e.g. mucosal addressin cellular adhesion molecule (MAdCAM)] that facilitate recruitment(13). Inhibition of α4β7/MAdCAM binding has been developed for clinical studies as a means for reducing migration of effector T cells to inflamed tissue in IBD (14). Positive results indicate that blocking intestinal migration offers a unique strategy for attenuating tissue inflammation in IBD without incurring systemic immunosuppression (15, 16).
Relatively limited data exists for adhesion molecules specific for the colon. Data from Austrup et al suggests that in vitro and in vivo differentiated Th1 cells express functional ligands for E- and P-selectin and migrate to acutely inflamed tissues (17). P-selectin glycoprotein ligand (PSGL)-1 is an adhesion molecule up-regulated in T cells under the influence of the Th1 polarizing cytokine, IL-12 (18–20). Data from Granger and colleagues suggest that P-selectin but not E-selectin is expressed constitutively on endothelial cells throughout the intestine (21). As CD is a Th1-mediated inflammation of the intestines, we considered the possibility that PSGL-1 mediated recruitment of Th1 cells activated by enteric antigen in draining lymph nodes is a major determinant of T cell migration in colitis. In studies in our lab, we observed that P-selectin blockade inhibited migration of Th1 cells to the small bowel (SB) lamina propria (LP) (22). Thus, PSGL-1/P-selectin pathways were found to play a significant role in recruitment of differentiated Th1 effector cells to the SB LP.
The potential role of PSGL-1 to mediate trafficking of T helper subset effector cells in IBD prompted us to identify its role in acute colitis. Alex et al demonstrated that acute dextran sodium sulfate (DSS) colitis is dependent on a mixed Th1/Th17-mediated inflammation (23). In the current study, we show that deletion of PSGL-1 attenuated DSS colitis. Specifically, PSGL-1 deficiency attenuated mucosal injury and lymphocytic accumulation in DSS colitis mice.
To examine the role of PSGL-1 in recruitment of peripheral effector T cells in colitis, Th1 and Th17 cells from WT and PSGL-1−/− mice were transferred into mice with active colitis. Compared to uninflamed mice, 19-fold greater numbers of WT Th1 cells migrated to inflamed colons. Colitis also increased Th17 cell recruitment, albeit to a lesser degree that Th1 cells. Interestingly, transferred CD4+ Th1 cells from PSGL-1−/− mice selectively reduced Th1 without affectingTh17 recruitment. Our results suggest a selective role for PSGL-1 in recruitment of Th1 cells to inflamed colon and suggest Th1 recruitment promotes local generation of Th17 cells.
MATERIALS AND METHODS
Mice
C57BL/6 (B6) mice and PSGL-1−/− mice on a B6 background were initially purchased from The Jackson Laboratory (Bar Harbor, MD) and bred within our facility. All mice were used when 6–10 weeks old and housed in the Northwestern University animal care facility under specific pathogen-free conditions. All studies and procedures were approved by the Animal Care and Usage Committee of Northwestern University.
Induction of DSS colitis and clinical disease assessment
Colitis was induced by oral administration of DSS 2.5% (wt/vol, mol. wt. 36,000–50,000, MP Biomedicals, Solon, OH) dissolved in sterile water for 6 days. Starting from the first day of DSS administration, disease activity index (DAI) was recorded every third day, including body weight, stool consistency/diarrhea, and the appearance of occult or gross blood in the stool by a single, trained, blinded observer as previously described by Cooper et al (24).
Histologic colitis assessment and colon length
The entire colon was removed from cecum to anus, measured as an indirect marker of inflammation, and fixed in 10% neutral buffered formalin. 5-μm thick sections were cut, prepared, and stained with hematoxylin and eosin for microscopic examination. Slides were blindly scored as previously described by Dieleman et al utilizing three independent parameters (severity of inflammation 0–3, depth of injury 0–3, and crypt damage 0–4, for a possible total of 10) as well factoring the percentage of tissue involved (multiplying by a factor of 1–4) to give a total histologic score out of 40 (25).
Cell preparation
Spleen and mesenteric lymph node (MLN) were mechanically dissociated and red cells were lysed in ACK lysis buffer (Gibco, Grand Island, NY). Cell suspensions were washed, enumerated and stored in DMEM containing 5% FBS (Cellgro, Herndon, VA) on ice until used. Cells from the colon LP were isolated by removing colons, flushing with cold PBS to remove fecal contents, opening longitudinally, and cut into 1 cm pieces. Colon pieces were digested once for 30 minutes at 37°C in 5 mM EDTA and 10% FBS to release epithelial cells. Supernatents were discarded and the tissue was washed and then minced. The remaining tissue was digested for four 10 minute intervals in a buffer containing 100 U/mL collagenase VIII (Sigma, St. Louis, MO), 25 MM Hepes (Cellgro), 7 mM CaCl2, and 2% FBS. After each digestion, cells released were centrifuged, washed, and stored in 5% DMEM on ice. All aliquots were then pooled, passed through 100 and 40 μm filters, and enumerated prior to washing for flow cytometry.
Colon explant culture ELISA
Colons were removed, flushed with sterile PBS, opened longitudinally, cut into 6 (approximately 1 cm) pieces, shaken at room temperature in RPMI containing 50 μg/mL gentamicin for 20 minutes at 280 rpm and distributed into 24-well plates in 1 mL of RPMI 1640 medium supplemented with 5% fetal bovine serum, 50 μg/mL gentamicin, and 1% antibiotic/antimycotic (penicillin/streptomycin/amphotericin B; GIBCO) for 20 hours at 37°C. Supernatants were collected and stored at −80°C before use for protein quantification with ELISA kits (R&D, Minneapolis, MN).
Real Time PCR
Tissue was isolated and stored in RNAlater (Qiagen, Hilden, Germany), and RNA was isolated using a RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. Real-time PCR analysis was performed using a qScript RT kit (Quanta, Gaithersburg, MD) and QuantiTect SYBR green PCR kit (Quigen) with primers on an ABI 7500 Real Time PCR System. Primers were designed to span genomic DNA intron junctions for specific amplification of mRNA. Samples were normalized using Gapdh. Fold increases were calculated using the ddCT method.
In vitro polarization and adoptive transfer
After mechanical dissociation, splenic CD4+ T cells were isolated by autoMACS (Miltenyi Biotec) using anti-CD4 microbeads. The enriched population was 88–95% positive for CD4 staining. Purified CD4+ T cells were cultured at 1–2 ×106 cells/well on 24-well tissue culture plates coated overnight with 1–2 μg/ml anti-CD3 (BD Biosciences) and 10 μg/ml anti-CD28 (BD Biosciences) for 2 days in the presence of 10 ng/ml rIL-12 (BD Biosciences) and 10 μg/ml anti-IL4 Ab (a gift from C. Reynolds, National Institute of Health, Bethesda, MD), followed by 5 days in a Falcon culture tissue flask with rIL-12, anti-IL4 Ab, and 5 ng/mL rIL-2 (BD Biosciences) to promote Th1 differentiation. For Th17 differentiation, anti-CD3/CD28 stimulated cells were co-cultured with 50 ng/mL rIL-6, 1 ng/mL rTGFβ, and 10 μg/mL anti-IFNγ Ab. The culture medium used was DMEM supplemented with 2 × 10−3 M L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 5 × 10−5 M β-mercaptoethanol, and 10% FCS. Cells were harvested after the seventh day of culture, counted, washed 3 times with sterile PBS, and 107 live cells were transferred intravenously (i.v.) into uninflamed recipient mice or mice who had just completed 6 days of DSS treatment.
Flow cytometry analysis
Cells were isolated from the spleen, MLN, and/or colon LP as described previously. Cells were washed, incubated with anti-CD16/CD32 (Fc blocking Ab), then stained with FITC-, PE-, and APC- conjugated Abs for CD4, CD45.1, CD45.2, and isotype-matched controls (BD Biosciences). In some experiments, cells were stained for functional PSGL-1 expression determined by P-selectin binding using a murine P-selectin/human IgM chimeric Ab followed by a APC-labeled anti-human anti-IgM. Parallel staining with control chimeric CD45-Ig served as controls, as previously described (22). Data were collected and analyzed using a FACSalibur flow cytometer and CELLQuest software (Becton Dickinson).
Statistical analysis
Data were analyzed were analyzed using GraphPad Prism software by means of ANOVA followed by Tukey's range test when more than two groups were compared; otherwise, the Student's t test was used. P-values ≤ 0.05 were considered statistically significant. Unless specified, values represent mean ± standard error of the mean (SEM).
RESULTS
PSGL-1 deletion attenuates acute DSS colitis
To directly assess the role of PSGL-1 in DSS colitis, wild type and PSGL-1−/− mice were given 2.5% DSS in their drinking water for 6 days. Clinical disease activity was monitored from initiation of DSS (day 0), to cessation of DSS (day 6) and through day 21 (Fig. 1). DSS treatment of WT mice induced wasting disease through day 10 that coincided with severe bloody diarrhea that continued through day 17. Disease Activity Index (DAI) peaked at day 10 and returned to baseline near day 21. By comparison, disease severity and duration were attenuated in PSGL-1−/− mice. In PSGL-1−/− mice, DAI was reduced by 40% on day 7 and by 59% at day 10. Weight loss began improving by day 7, and fecal occult blood (FOB)/diarrhea returned to normal by day 14.
FIGURE 1.

Deletion of PSGL-1 reduces the severity and duration of clinical disease activity after treatment with DSS. WT and PSGL-1−/− mice maintained on drinking water alone (control) were compared to mice treated with 2.5% DSS from day 0 through day 6. Body weight, diarrhea, and occult/gross blood were assessed every 3–4 days for 21 days to give individual indices as well as a conglomerate DAI (n = 5–10 per group). *, p < 0.05; #, p < 0.01 vs. untreated control. Data are mean ± SEM and are representative of 3 separate experiments.
Next, we examined mouse colon histology at days 7 and 14 (Fig. 2A). Results indicate that PSGL-1−/− mice developed less colonic shortening (Fig. 2B) and less severe colitis histology scores (6.2 ± 0.4 vs. 3.8 ± 0.9, p<0.05, day 7) (Fig. 2C) due to reductions in ulceration and submucosal leukocyte infiltration. Colitis scores remained high in WT mice through day 14 (5.2 ± 0.7), as chronic leukocyte infiltration expanded and transmural mucosal thickening extended. In contrast, by day 14, PSGL-1−/− mice had significantly better colitis scores (1.6 ± 0.4, p<0.01 vs. WT) with minimal submucosal leukocyte infiltration, surface ulceration and transmural thickening. Together, these data indicated PSGL-1 expression was required for optimal induction of chronic inflammation in DSS colitis mice.
FIGURE 2.

Deletion of PSGL-1 attenuates DSS-induced colitis. (A) Mice were sacrificed at baseline, on day 7 after 6 days of DSS treatment, and on day 14. Colons were stained with H&E to assess histologic colitis. A robust infiltration of leukocytes was seen in WT mice resulting in progressive expansion of the mucosa, submucosa, and muscular thickening from day 7 to day 14. Submucosal infiltration was diminished, and often absent by day 14, in PSGL-1−/− mice despite evidence of DSS-induced mucosal injury. (B) Colon lengths measured from the cecum to anus. (C) Histologic colitis scores. (n = 3–4 per group). *, p < 0.05; #, p < 0.01 vs. WT, same day. Data are mean ± SEM.
PSGL-1 is required for Th1 and Th17 cytokine production in colitis
To assess the role of PSGL-1 expression in the induction of inflammatory mediators in colitis, tissue explants from WT and PSGL-1−/− mice were cultured and supernatant collected. ELISAs from WT explants showed that production of both Th1 (IFNγ and TNF) and Th17 (IL-17, IL-22, IL-6) cytokines increased dramatically during the first week of colitis. Strikingly, PSGL-1 deficiency decreased both Th1 and Th17 cytokine production at relatively early (day 7) and late (day 14) time points (Fig. 3). Data suggests that IFNγ-dependent chemokines play a role in both human IBD and acute DSS colitis (26–28). Analysis of mucosal mRNA showed that induction of IFNγ-dependent chemokines, CXCL9 and CXCL10, were also attenuated (Fig. 4). Together, these data indicate that PSGL-1 expression is required for generation of Th1 and Th17 effector T cells in colitic mice.
FIGURE 3.

Induction of Th1 and Th17 cytokine responses after DSS treatment. Time course of cytokine protein levels from the colons of WT (dark rectangles) and PSGL-1−/− (open rectangles) mice after DSS treatment. Colons were removed at baseline, day 7, and day 14, opened, washed, and the entire colon was cultured in 6 wells of a 24-well plate overnight for direct cytokine analysis of supernatents by ELISA (n = 4 per group). *, p < 0.05; #, p < 0.01 vs. WT, same day. Data are mean ± SEM from duplicate samples.
FIGURE 4.
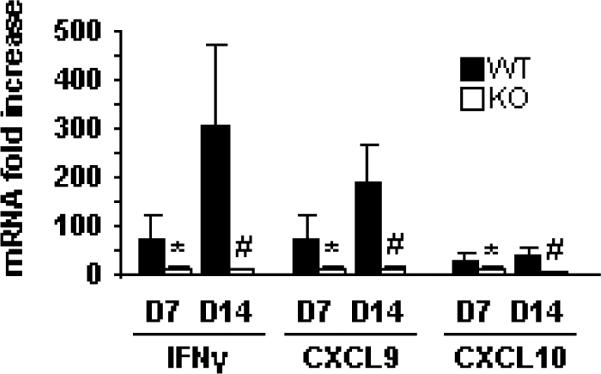
Induction of Th1 cytokine and chemokine responses after DSS treatment. Time course of mRNA levels from the colons of WT (dark rectangles) and PSGL-1−/− (open rectangles) mice after DSS treatment. Colons were removed at baseline, day 7, and day 14, opened, washed, and 0.5 cm of distal colon was preserved in RNAllater for subsequent isolation of mRNA and real time PCR from duplicate samples (n = 4 per group). *, p < 0.05; #, p < 0.01 vs. WT, same day. Data are mean ± SEM.
PSGL-1 is required for T cell recruitment in colitis
Data indicate functional PSGL-1 expression is critical to CD4+ T cell recruitment to inflamed tissues, including the skin and retina (17, 29, 30). To examine the role of PSGL-1 expression in systemic and mucosal lymphocyte trafficking in colitis, numbers of CD4+ T cells were quantified in DSS-treated WT and PSGL-1−/− mice. Data in Fig. 5 show that MLN CD4+ T cell numbers doubled over the two week course of colitis whereas colonic CD4+ T cells increased 8-fold. PSGL-1 deficiency led to reductions of 50% of colonic CD4+ T cells numbers. Surprisingly, the expansion of MLN CD4+ T cell counts seen in WT mice during colitis induction was unabated in PSGL-1−/− mice. Overall, these data were consistent with the notion that reductions in colitis activity seen in PSGL-1−/− mice related to decreased recruitment of T cells after activation in peripheral lymphoid tissue.
FIGURE 5.

Enumeration of CD4+ T cell accumulation in DSS colitis. Cells were isolated at the designated time points and analyzed by flow cytometry to quantify the number of CD4+ T cells.
Th1, but not Th17, recruitment is dependent on PSGL-1
To test the notion that PSGL-1 mediated recruitment of activated T cell subsets from the periphery to the colon, we examined colonic recruitment of adoptively transferred CD4+ T cell subsets from WT and PSGL-1−/− mice into recipients with DSS colitis. Purified CD4+ T cells from CD45.1 WT and PSGL-1−/− donors were activated and polarized into Th1 and Th17 subsets prior to transfer. DSS colitis was induced in WT CD45.2 recipients and 107 polarized CD4+ T cells adoptively transferred. Two days after transfer, MLN and colonic LP cells were isolated from recipient mice, and numbers of CD45.1 donor cells quantified. Data in Fig. 6 show that functional PSGL-1 was expressed by transferred Th1-polarized CD4+ cells but not on Th17 cells.
FIGURE 6.

Th1 polarization induced functional PSGL-1 expression. P-selectin–Ig binding (darker line/open shading) on CD4+ T cells before (unstimulated) and after in vitro polarization in either Th1 or Th17 conditions vs. chimeric control (lighter line/darker shading) showing functional PSGL-1 expression after Th1, but not Th17, polarization.
Transfer data show that both Th1 and Th17 cells migrated efficiently to splenic tissue (Fig. 7, 8). Data also show that that colitis induction had little impact on splenic T cell recruitment. In contrast, colitis increased the relative percentages of transferred Th1 and Th17 WT cells in the colon by 4–5 and nearly two-fold, respectively. More importantly, absolute numbers of donor Th1 and Th17 cells increased by 19- and 7-fold respectively in colons of DSS-treated recipient mice.
FIGURE 7.

PSGL-1 is essential for optimal recruitment of Th1 T cells to the colon during acute colitis. Control WT (uninflamed) and WT DSS-treated (colitis) mice, both expressing WT CD45.2, were adoptively transferred with Th1 CD4+ T cells derived under identical conditions from CD45.1 WT or PSGL-1−/− mice. Mice were euthanized and analyzed 48 hours after adoptive transfer. (A) Cells from spleen and colon LP were stained with anti-CD45.1 mAb–PE, anti–CD45.2 mAb-FITC, as well as anti–CD4 mAb-APC for gating. Numbers on top left quadrant represent the percentages of CD4+ CD45.1+ cells (i.e. adoptively transferred) out of total CD4+ T cells in each compartment. (B) Yields (mean ± SEM) of the transferred CD4+ CD45.1+ cells recovered in spleen and colon LP from each indicated group (n= 3/group) at 48 hours. *, p < 0.01 vs. WT to uninflamed; #, p < 0.01 vs. WT to DSS colitis.
FIGURE 8.

PSGL-1 is not required for recruitment of Th17 T cells to colon during acute colitis. Control WT (uninflamed) and WT DSS-treated (colitis) mice, both expressing WT CD45.2, were adoptively transferred with Th17 CD4+ T cells derived under identical conditions from CD45.1 WT or PSGL-1−/− mice. Mice were euthanized and analyzed 48 hours after adoptive transfer. (A) Cells from spleen and colon LP were stained with anti-CD45.1 mAb–PE, anti–CD45.2 mAb-FITC, as well as anti–CD4 mAb-APC for gating. Numbers on top left quadrant represent the percentages of CD4+ CD45.1+ cells (i.e. adoptively transferred) out of total CD4+ T cells in each compartment. (B) Yields (mean ± SEM) of the transferred CD4+ CD45.1+ cells recovered in spleen and colon LP from each indicated group (n= 3/group) at 48 hours.
To determine the role of PSGL-1 in CD4 T cell recruitment in colitis, Th1 and Th17 cells from CD45.1 PSGL-1−/− mice were transferred into DSS-treated WT recipients. Whereas PSGL-1 deficiency did not affect Th1 donor cell numbers in the spleen, Th1 recruitment to the colon was reduced by 90%. By comparison, Th17 cells transferred from WT and PSGL-1−/− mice migrated equally well to inflamed WT colons. Together, the data suggest that Th1 colonic recruitment utilized PSGL-1 while PSGL-1 was not involved in colonic Th17 recruitment.
DISCUSSION
The role of PSGL-1 in recruitment of lymphocytes, neutrophils, and platelets to peripheral tissue is well established. Regulated migration of T helper populations from the peripheral blood and lymphoid organs to the colon is a critical component in translating innate immune responses into site-specific adaptive immune responses. The importance of P-selectin in mediating T cell recruitment to the intestine is supported by data from Chu et al (31) and Salmi and Jalkanen (32) who found that PSGL-1 was expressed by a substantial subset of LP lymphocytes in colitic mice and in human IBD. Recent studies support the role of PSGL-1 in both experimental colitis (33) and ileitis (34). Additional studies identify a growing repertoire of immune function related to PSGL-1, including recruitment and function of regulatory T cells, dendritic cells, monocytes, neutrophils, and lymphocyte apoptosis (35–38). In this study, evidence is presented that PSGL-1 mediates Th1 recruitment to chronically inflamed colonic tissue. These data support the conclusion that PSGL-1 blockade may be an effective strategy for colitis treatment during chronic disease and/or for maintenance of remission in patients where acute disease has been treated.
Previous studies demonstrated that immunoblockade of PSGL-1 reduces the clinical activity of established, i.e. chronic, DSS colitis, without defining effects on selective T cell subset migration patterns (33). More recent data has shown that chronic DSS colitis is mediated by a mixed T helper population, whereas acute DSS colitis is primarily mediated by Th1 and Th17 populations (23). This is consistent with data presented in this paper, where DSS induced dramatic expression of both Th1 and Th17 cytokine (Fig. 3). PSGL-1deficiency had a more significant therapeutic effect to prevent chronic (day 7 to 21) inflammation as predicted by its impact on Th1 recruitment. Because CD is primarily a Th1/Th17-mediated inflammation, the chronic phase of DSS colitis was chosen to mimic the role of PSGL-1 in Th1 and Th17 recruitment. Furthermore, we demonstrated that CD4+ T cell recruitment increased as DSS colitis developed (Fig. 5). Histologic examination demonstrated a near absence of leukocytes within the submucosa at baseline. Colitis resulted in expansion and filling of the submucosa that extended into the lamina propria (Fig. 2). Examination of T cell subsets indicated that CD4+ T cell numbers increased 8–9 fold over two weeks (Fig. 5). The contribution of Th1 cell recruitment to this effect was highlighted by adoptive transfer studies showing 19-fold greater Th1 recruitment in colitis compared to WT mice (Fig. 7). These data suggest PSGL-1 blockade may best provide a therapeutic effect when used during progression of chronic forms of colitis.
We hypothesized that PSGL-1 is critical to Th1 recruitment and development of colitis. We found that deletion of PSGL-1 attenuated the onset and reduced the duration of DSS colitis. Importantly, both WT and PSGL-1−/− mice had a similar response to the introduction of DSS. Both strains demonstrated similar DAI at day 3 and similar patterns of mucosal injury (ulceration, crypt destruction) during DSS administration suggesting comparable early, innate responses to DSS injury. Both strains had similar CD4+ expansion in MLNs. However, by day 14, WT mice had a dramatic leukocyte infiltration of the submucosa not seen in PSGL-1−/− mice. This infiltration coincided with worsening DAI in WT mice. Disease in PSGL-1−/− mice failed to escalate through DSS colitis induction (i.e. from day 3 through day 7) and rapidly resolved after day 7 when colitis in WT mice progressed. PSGL-1−/− mice had no evidence of clinical disease activity by day 14, suggesting that disease activity and inflammation in WT mice required PSGL-1 expression.
Previous studies indicate that PSGL-1 mediates Th1 recruitment to inflamed tissues outside the intestine (17, 29, 30). In addition, our previous data suggest PSGL-1 was required for optimal recruitment of Th1 effector cells to extralymphoid sites in uninflamed intestine (22). Studies here examined the role of PSGL-1 in T-helper subset recruitment to lymphoid and extra-lymphoid effector sites within inflamed colon. Our data suggest that PSGL-1 deficiency did not affect DSS colitis-induced CD4+ expansion in draining MLNs. However, data clearly show that colitis-induced CD4+ migration to the large intestine was attenuated in PSGL-1−/− mice. Further, data in WT and PSGL-1−/− mice suggested that PSGL-1-mediated intestinal T cell recruitment was required for induction of IFNγ and TNF responses as colitis progressed to a chronic phase (day 14). A more specific role for Th1 CD4+ PSGL-1 expression in intestinal recruitment was suggested by adoptive transfer studies. Comparisons of Th1 and Th17 recruitment to colonic sites during active inflammation showed that Th1 cells required PSGL-1 expression for efficient colonic recruitment. These findings support the conclusion that early DSS-induced mucosal injury is mediated by innate immune responses independent of PSGL-1, whereas development of chronic forms of colitis analogous to human disease require PSGL-1 for recruitment of Th1 cells activated in the periphery. It appears the wave of Th1 lymphocyte recruitment played an important role in development of chronic colitis.
Results shown are consistent with recent findings in relatively early forms of DSS colitis (day 7). In these early stages of colitis, mucosal injury is acute, and tissue injury is mediated by direct effects of DSS on the mucosal barrier. Recent data indicate induction of innate immune responses provide initial protective immunity until more chronic inflammation ensues (39). Interestingly, PSGL-1 deficiency led to more severe colitis in early stages associated with less macrophages, dendritic cells and B cells. Together with data shown here, the two studies suggest an interesting model where PSGL-1 mediates protective innate immunity in the early phase of DSS colitis, then PSGL-1 mediated effector T cell recruitment determines progression to relatively late stages when chronic inflammation is seen.
We found reduced levels of Th17 cytokine, albeit to a lesser degree than seen with Th1 cytokine in PSGL-1−/− mice. The role of Th17 cells in colitis remains poorly understood. Numerous autoimmune models implicate Th17 in inflammatory responses (40–42), and IL-17 expression is increased in both UC and CD (6). However, there is also evidence that Th17 may be a protective/anti-inflammatory pathway in the colon. Prior studies found immunoblockade of IL-17 worsens DSS colitis (43), and adoptive transfer of IL-17−/− CD45RBhigh T cells into Rag−/− recipients increased experimental colitis (44). The enhanced colitis in both models was attributed to increased mucosal expression of Th1 cytokine, suggesting Th17 responses oppose Th1 polarization. Data suggests crosstalk between Th1 and Th17 pathways is critical to maintain regulation of T helper responses is mediated through T-bet's ability to suppress IL-17 production (45–47). Conversely, O'Conner et al provide evidence that the absence of IL-17 results in greater T-bet expression and dysregulated Th1 responses in colitis (44). This data highlights the complexity in the cross talk between Th1 and Th17 populations. Reductions in IL-17 and IL-22 seen here may have been due to reduced recruitment, but data presented show migration of adoptively transferred Th17 cells was not dependent on PSGL-1 (Fig. 8). This was not surprising as surface expression of functional PSGL-1 was low in polarized Th17 cells. This is consistent with data from Yoshizaki et al who showed that Th17 cells expressed PSGL-1 at low levels in experimental scleroderma (while Th1 cells demonstrated high expression) (48). In light of unperturbed recruitment of Th17 cells, we propose reduced Th17 cytokine was due to reduced local production. It is possible that the dramatic Th1 influx seen in WT colitis “prime” either constitutively present colonic Th17 cells or provide a signal for PSGL-1-independent recruitment of peripheral Th17 cells. Data in Fig. 8 show PSGL-1 deletion did not affect Th17 recruitment; we propose PSGL-1 mediates Th1 recruitment in response to epithelial disruption and exposure to commensal antigen and the sequential generation of Th17 cells. Increases in Th17 cytokine may regulate proinflammatory Th1 responses to re-establish homeostasis and control tissue damage. Therefore, Th17 accumulation was reduced in PSGL-1−/− mice as a result of abrogated Th1 recruitment. The modest level of Th17 induction seen PSGL-1−/− mice was adequate to temper tissue destruction resulting from DSS administration, resulting in rapid resolution of disease activity. Our data establishes a specific role for PSGL-1 to control Th1 recruitment without perturbing potentially beneficial Th17 responses in experimental colitis.
As we continue to define clinical phenotypes of human IBD based on immune (i.e. T-helper subtype) responses, understanding how homing pathways selectively recruit specific populations of leukocytes will become more important. Manipulating patterns of T cell recruitment may ultimately allow therapeutic targets to mitigate dysregulated inflammatory responses specific to individual patients.
Acknowledgments
Supported by a Crohn's and Colitis Foundation of America Career Development Award (to J.B.B.), NIH grants DK54778 and DKAI061701 (to T.A.B.) and DK066161(to J.B.B.).
References
- 1.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 4.Parronchi P, Romagnani P, Annunziato F, et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt C, Giese T, Ludwig B, et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 8.Hanauer SB, Dassopoulos T. Evolving treatment strategies for inflammatory bowel disease. Annu Rev Med. 2001;52:299–318. doi: 10.1146/annurev.med.52.1.299. [DOI] [PubMed] [Google Scholar]
- 9.Hoentjen F, van Bodegraven AA. Safety of anti-tumor necrosis factor therapy in inflammatory bowel disease. World J Gastroenterol. 2009;15:2067–2073. doi: 10.3748/wjg.15.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tietz W, Allemand Y, Borges E, et al. CD4+ T cells migrate into inflamed skin only if they express ligands for E- and P-selectin. J Immunol. 1998;161:963–970. [PubMed] [Google Scholar]
- 11.Bradley LM, Watson SR. Lymphocyte migration into tissue: the paradigm derived from CD4 subsets. Curr Opin Immunol. 1996;8:312–320. doi: 10.1016/s0952-7915(96)80118-x. [DOI] [PubMed] [Google Scholar]
- 12.Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. 1990;171:801–817. doi: 10.1084/jem.171.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butcher EC, Williams M, Youngman K, et al. Lymphocyte trafficking and regional immunity. Adv Immunol. 1999;72:209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 14.Briskin M, Winsor-Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151:97–110. [PMC free article] [PubMed] [Google Scholar]
- 15.Targan SR, Feagan BG, Fedorak RN, et al. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672–1683. doi: 10.1053/j.gastro.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 16.Sandborn WJ, Colombel JF, Enns R, et al. Natalizumab induction and maintenance therapy for Crohn's disease. N Engl J Med. 2005;353:1912–1925. doi: 10.1056/NEJMoa043335. [DOI] [PubMed] [Google Scholar]
- 17.Austrup F, Vestweber D, Borges E, et al. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 18.Lim YC, Henault L, Wagers AJ, et al. Expression of functional selectin ligands on Th cells is differentially regulated by IL-12 and IL-4. J Immunol. 1999;162:3193–3201. [PubMed] [Google Scholar]
- 19.Wagers AJ, Waters CM, Stoolman LM, et al. Interleukin 12 and interleukin 4 control T cell adhesion to endothelial selectins through opposite effects on alpha1, 3-fucosyltransferase VII gene expression. J Exp Med. 1998;188:2225–2231. doi: 10.1084/jem.188.12.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Wely CA, Blanchard AD, Britten CJ. Differential expression of alpha3 fucosyltransferases in Th1 and Th2 cells correlates with their ability to bind P-selectin. Biochem Biophys Res Commun. 1998;247:307–311. doi: 10.1006/bbrc.1998.8786. [DOI] [PubMed] [Google Scholar]
- 21.Eppihimer MJ, Wolitzky B, Anderson DC, et al. Heterogeneity of expression of E- and P-selectins in vivo. Circ Res. 1996;79:560–569. doi: 10.1161/01.res.79.3.560. [DOI] [PubMed] [Google Scholar]
- 22.Haddad W, Cooper CJ, Zhang Z, et al. P-selectin and P-selectin glycoprotein ligand 1 are major determinants for Th1 cell recruitment to nonlymphoid effector sites in the intestinal lamina propria. J Exp Med. 2003;198:369–377. doi: 10.1084/jem.20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alex P, Zachos NC, Nguyen T, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 25.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uguccioni M, Gionchetti P, Robbiani DF, et al. Increased expression of IP-10, IL-8, MCP-1, and MCP-3 in ulcerative colitis. Am J Pathol. 1999;155:331–336. doi: 10.1016/S0002-9440(10)65128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egesten A, Eliasson M, Olin AI, et al. The proinflammatory CXC-chemokines GRO-alpha/CXCL1 and MIG/CXCL9 are concomitantly expressed in ulcerative colitis and decrease during treatment with topical corticosteroids. Int J Colorectal Dis. 2007;22:1421–1427. doi: 10.1007/s00384-007-0370-3. [DOI] [PubMed] [Google Scholar]
- 28.Ito R, Shin-Ya M, Kishida T, et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirata T, Merrill-Skoloff G, Aab M, et al. P-Selectin glycoprotein ligand 1 (PSGL-1) is a physiological ligand for E-selectin in mediating T helper 1 lymphocyte migration. J Exp Med. 2000;192:1669–1676. doi: 10.1084/jem.192.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Manivannan A, Jiang HR, et al. Recruitment of IFN-gamma-producing (Th1-like) cells into the inflamed retina in vivo is preferentially regulated by P-selectin glycoprotein ligand 1:P/E-selectin interactions. J Immunol. 2004;172:3215–3224. doi: 10.4049/jimmunol.172.5.3215. [DOI] [PubMed] [Google Scholar]
- 31.Chu A, Hong K, Berg EL, et al. Tissue specificity of E- and P-selectin ligands in Th1-mediated chronic inflammation. J Immunol. 1999;163:5086–5093. [PubMed] [Google Scholar]
- 32.Salmi M, Jalkanen S. Human leukocyte subpopulations from inflamed gut bind to joint vasculature using distinct sets of adhesion molecules. J Immunol. 2001;166:4650–4657. doi: 10.4049/jimmunol.166.7.4650. [DOI] [PubMed] [Google Scholar]
- 33.Rijcken EM, Laukoetter MG, Anthoni C, et al. Immunoblockade of PSGL-1 attenuates established experimental murine colitis by reduction of leukocyte rolling. Am J Physiol Gastrointest Liver Physiol. 2004;287:G115–124. doi: 10.1152/ajpgi.00207.2003. [DOI] [PubMed] [Google Scholar]
- 34.Rivera-Nieves J, Burcin TL, Olson TS, et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med. 2006;203:907–917. doi: 10.1084/jem.20052530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohm AP, Miller SD. Role of ICAM-1 and P-selectin expression in the development and effector function of CD4+CD25+regulatory T cells. J Autoimmun. 2003;21:261–271. doi: 10.1016/s0896-8411(03)00117-3. [DOI] [PubMed] [Google Scholar]
- 36.Shigeta A, Matsumoto M, Tedder TF, et al. An L-selectin ligand distinct from P-selectin glycoprotein ligand-1 is expressed on endothelial cells and promotes neutrophil rolling in inflammation. Blood. 2008;112:4915–4923. doi: 10.1182/blood-2008-04-153866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen SC, Huang CC, Chien CL, et al. Cross-linking of P-selectin glycoprotein ligand-1 induces death of activated T cells. Blood. 2004;104:3233–3242. doi: 10.1182/blood-2003-05-1679. [DOI] [PubMed] [Google Scholar]
- 38.Inoue T, Tsuzuki Y, Matsuzaki K, et al. Blockade of PSGL-1 attenuates CD14+ monocytic cell recruitment in intestinal mucosa and ameliorates ileitis in SAMP1/Yit mice. J Leukoc Biol. 2005;77:287–295. doi: 10.1189/jlb.0204104. [DOI] [PubMed] [Google Scholar]
- 39.Nunez-Andrade N, Lamana A, Sancho D, et al. P-selectin glycoprotein ligand-1 modulates immune inflammatory responses in the enteric lamina propria. J Pathol. doi: 10.1002/path.2850. [DOI] [PubMed] [Google Scholar]
- 40.Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 42.Pernis AB. Th17 cells in rheumatoid arthritis and systemic lupus erythematosus. J Intern Med. 2009;265:644–652. doi: 10.1111/j.1365-2796.2009.02099.x. [DOI] [PubMed] [Google Scholar]
- 43.Ogawa A, Andoh A, Araki Y, et al. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 44.O'Connor W, Jr., Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rangachari M, Mauermann N, Marty RR, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 47.Rutitzky LI, Smith PM, Stadecker MJ. T-bet protects against exacerbation of schistosome egg-induced immunopathology by regulating Th17-mediated inflammation. Eur J Immunol. 2009;39:2470–2481. doi: 10.1002/eji.200939325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshizaki A, Yanaba K, Iwata Y, et al. Cell adhesion molecules regulate fibrotic process via Th1/Th2/Th17 cell balance in a bleomycin-induced scleroderma model. J Immunol. 185:2502–2515. doi: 10.4049/jimmunol.0901778. [DOI] [PMC free article] [PubMed] [Google Scholar]