

Abstract
IL-13 is a central mediator of airway hyperreactivity and mucus expression, both hallmarks of asthma. IL-13 is found in the sputum of patients with asthma; therefore, IL-13 is an attractive drug target for treating asthma. We have previously shown that IL-13 inhibits Th17 cell production of IL-17A and IL-21 in vitro. Th17 cells are associated with autoimmune diseases, host immune responses, and severe asthma. In this study, we extend our in vitro findings and determine that IL-13 increases IL-10 production from Th17 polarized cells, and that IL-13-induced IL-10 production negatively regulates secretion of IL-17A and IL-21. To determine if IL-13 negatively regulates lung IL-17A expression via an IL-10 dependent mechanism in vivo, we used a model of respiratory syncytial virus (RSV) strain A2 infection in STAT1 KO mice which increases IL-17A and IL-13 lung expression, cytokines not produced during RSV infection in WT mice. To test the hypothesis that IL-13 negatively regulates lung IL-17A expression, we created STAT1/IL-13 double KO (DKO) mice. We found that RSV-infected STAT1/IL-13 DKO mice had significantly greater lung IL-17A expression compared to STAT1 KO mice, and increased IL-17A expression was abrogated by anti-IL-10 antibody treatment. RSV-infected STAT1/IL-13 DKO mice also had increased neutrophil infiltration RSV-infected STAT1 KO mice. Neutralizing IL-10 increased infiltration of inflammatory cells into the lungs of STAT1 KO mice but not STAT1/IL-13 DKO mice. These findings are vital to understanding potential side effects of therapeutics targeting IL-13. Inhibiting IL-13 may decrease IL-10 production and increase IL-17A production, thus potentiating IL-17A-associated diseases.
Introduction
Asthma is a chronic disease which affects 300 million people worldwide (1). CD4+ Th2 cells are a primary driving force of allergic lung inflammation in asthma, and the Th2 cytokine IL-13 is a central mediator of airway hyperreactivity and airway mucus expression, both important hallmarks of asthma (2,3). IL-13 is found in abundance in the sputum of patients with asthma (4), and therefore IL-13 is an attractive drug target for treating asthma (3,5). Currently there are multiple ongoing clinical trials using IL-13 antagonists for asthma therapy (6–12).
IL-13 binds to the IL-13R (also known as the type II IL-4 receptor) which is comprised of 2 subunits, IL-4Rα and IL-13Rα1. We have recently reported that Th17 cells, but not Th0, Th1, or Th2 cells, in both mice and humans express the IL-13Rα1 subunit of the IL-13R (13,14). IL-13Rα1 expression on mouse T cells has been confirmed by Wilson and colleagues (15). IL-13 binds the IL-13R, resulting in phosphorylation and activation of the downstream transcription factor STAT6. Once phosphorylated, STAT6 homodimerizes and translocates to the nucleus for transcription of IL-13-dependent genes. We determined in mouse and human Th17 cells that the addition of IL-13 resulted in STAT6 phosphorylation and attenuated IL-17A and IL-21 production, as well as reduced expression of the Th17 transcription factor ROR-γT (RORC2 in humans) (13,14). The focus of this project is to further define the mechanism by which IL-13 negatively regulates IL-17A production in the lung.
Th17 cells are a subset of CD4+ T cells with distinct properties from Th1, Th2, and regulatory T cells (16,17). Th17 cells are found in the lungs of patients with severe asthma (18,19), and the Th17 cytokine IL-17A is present in the bronchoalveolar lavage (BAL) fluid and bronchial biopsies of patients with moderate to severe asthma (20,21). Th17 cells are also associated with autoimmune and inflammatory disorders, such as the experimental autoimmune encephalitis (EAE) mouse model of multiple sclerosis and rheumatoid arthritis (22), and are critical for the clearance of some extracellular pathogens, (16,23–26). In mice, Th17 cell differentiation occurs when T cells are activated in the presence of TGF-β and IL-6 (26–28), and maintenance and sustainability of Th17 cells requires IL-23 (16,29,30). Th17 cells produce a variety of cytokines, including IL-17A, IL-17F, IL-21, and IL-10 (25). IL-17A and IL-17F have similar structural homology and both increase production of inflammatory cytokines and mucus production (31); however, IL-17A is more biological active than IL-17F (32). IL-21 acts in an autocrine manner to promote IL-17A production, and further inhibits IFN-γ production from Th1 cells (28,33). IL-10 is an anti-inflammatory cytokine which inhibits IL-17A production from Th17 cells (34).
To determine if IL-13 regulates IL-17A production in vivo, we used a mouse model of respiratory syncytial virus (RSV) strain A2 infection in STAT1 KO mice. We previously reported that RSV infection increased lung IL-13 and IL-17A expression in STAT1 KO mice, but not WT BALB/c mice (35). Based on our in vitro findings that IL-13 negatively regulated IL-17A production from Th17 cells, we hypothesized that IL-13 negatively regulates IL-17A production in vivo. To test our hypothesis, we generated STAT1/IL-13 DKO, STAT1/IL-4 DKO and STAT1/STAT6 DKO mice. STAT6 is a downstream transcription factor for IL-13 and IL-4 signaling. The use of STAT1/IL-13 DKO, STAT1/IL-4 DKO, and STAT1/STAT6 DKO mice will determine the independent contribution of IL-13 and IL-4, relative to total STAT6 signaling, in attenuating lung IL-17A protein expression. These studies further define the mechanisms by which IL-13 downregulates IL-17A production, and are important for understanding potential side effects in therapeutics which target IL-13 or IL-13 signaling in lung disease.
Materials and Methods
Mice
Pathogen-free 8 to 10-week old female BALB/c mice were purchased from Charles River Laboratories (Wilmington, MA) (36). STAT1 KO and IL-13KO mice on a BALB/c background were generated as previously described (36,37). IL-4 KO and STAT6 KO mice on BALB/c background were purchased from Jackson laboratories (Bar Harbor, ME). STAT1/IL-13 DKO, STAT1/IL-4 DKO, and STAT1/STAT6 DKO mice were generated by mating and subsequent genotyping. In caring for the animals, investigators adhered to the revised 1996 Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council.
RSV infections
The A2 strain of RSV was provided by R. Chanock (National Institutes of Health, Bethesda, MD). Viral stocks of RSV A2 were generated by infection of HEp-2 cells and titered by plaque assay as previously described (38). Mice were anesthetized with ketamine/xylazine and infected intranasally with 1×106 PFU of RSV A2. Lungs were harvested 6 days post- infection. In select experiments mice were injected intraperitoneally with 100 μg of anti-IL-10 or isotype control antibody (generated from ascites fluid at the University of Michigan) 24 hours prior to RSV infection and 3 days post infection (39).
T cell isolation and Th17 cell polarization and activation
CD4+ T cells were purified from the spleens of mice as previously described (11). T cells were activated using anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) (BD Biosciences) in 96-well plates for 4 days. T cells were differentiated into Th17 cells by adding rmIL-23 (10 ng/ml), rhTGF-β (1 ng/ml), rmIL-6 (20 ng/ml), anti-IFN-γ (10 μg/ml), and anti-IL-4 (10 μg/ml). In select cultures, anti-IL-10 (10 μg/ml), rmIL-13 (10 ng/ml), or rmIL-21 (0.5–5ng/ml) was also added. All Abs and rmIL-13, rmIL-23, and rmIL-21 were purchased from R&D Systems. rmIL-6 and rhTGF-β were purchased from PeproTech (Rocky Hill, NJ).
Cytokine measurements
Cytokine levels were measured from cell culture supernatants or whole lung homogenates with available Duoset or Quantikine ELISA kits (R & D Systems) following the manufacturer’s instructions.
Flow cytometry
Lungs were harvested, minced, and digested for 40 minutes at 37°C in RPMI containing 10% FBS, collegenase type I (0.1%W/V), and DNase (500 μg/ml). The digestion was stopped with 10 μl of 0.5M EDTA, and minced lung was passed through a 70 μm strainer. For staining of intracellular cytokines, cells were stimulated in RPMI 1640, 10% FBS, 1 μM ionomycin (Sigma-Aldrich), 50 ng/ml PMA (Sigma-Aldrich) and 0.07% GolgiStop (BD Pharmingen) for 6 h at 37°C and 5% CO2. Cells were then counted using a hemocytometer and the total number of cells from the lungs of each mouse was recorded. One million cells from the lungs of each mouse were stained for cell surface molecules and intracellular cytokines as previously described (40). We used the following Abs from BD Pharmingen: anti-CD3ε, anti-CD8a, anti-CD4, anti-Ly6G (Gr-1), anti-DX5, anti-γδ T cell receptor, and anti-IL-17A. Anti-CD16/32 Ab (BD Pharmingen) was used to prevent nonspecific staining. Cell samples were analyzed using an LSR II flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star).
Quantitative PCR
A 2-step real-time PCR assay using SYBR green mix (BioRad, Hercules, CA) was used to detect SOCS3 as previously described (41) with GAPDH being used as the housekeeping gene. Primer sequences were as follows: SOCS3: F 5′-GGGTGGCAAAGAAAAGGAG-3′, R 5′-GTTGAGCGTCAAGACCCAGT-3′ and GAPDH: F 5′-GGCCCCTCTGGAAAGCTGTGG-3′, R 5′-CCCGGCATCGAAGGTGGAAGA-3′.
Analysis of inflammatory cell infiltration into bronchoalveolar lavage (BAL) fluid
BAL was performed by instilling 800 μl of saline through a tracheostomy tube and then withdrawing the fluid with gentle suction via syringe. White blood cells were counted on a hemocytometer. Cytologic examination was performed on cytospin preparations (Thermo Shandon). Cytospin slides were fixed and stained using Three-Step Stain (Richard-Allan Scientific). Differential counts were based on counts of 200 cells using standard morphologic criteria to classify the cells as neutrophils, eosinophils, lymphocytes, or other mononuclear leukocytes (alveolar macrophages and monocytes).
Statistical analyses
Data is presented as mean ± SEM. For Figures 1 and 2, data shown represents combined analysis of cell culture wells from 3 independent experiments. For Figures 3–6 data shown represents combined analysis of individual mice. Data were analyzed with ANOVA followed by the Tukey posthoc test or an unpaired 2-tailed T-test using GraphPad Prism version 4 with values being considered significant when p < 0.05.
Figure 1. IL-13 decreases IL-17A production in an IL-21 dependent manner.
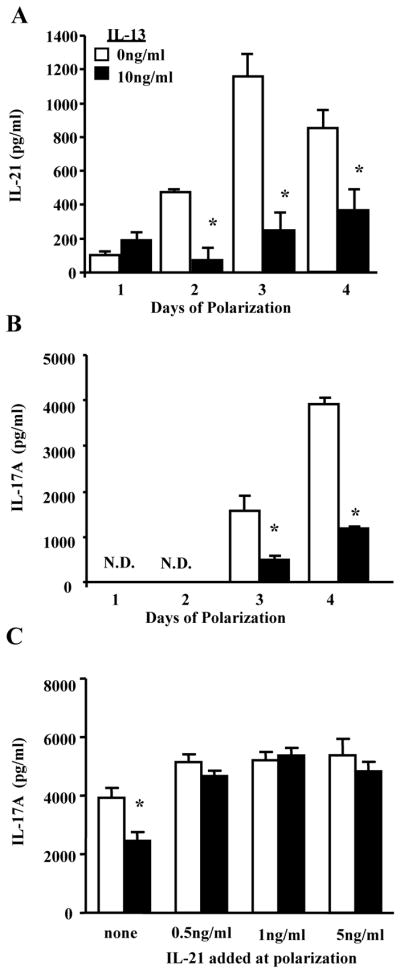
A–B. CD4+ T cells were activated with anti-CD3 and anti-CD28 with Th17 polarizing conditions in the presence of rmIL-13 (0 or 10ng/ml). Cell supernatants were collected 1–4 days after polarization and IL-21 and IL-17A were examined by ELISA. * p<0.05 compared to 0ng/ml of IL-13 for each day, n=4–8 cell culture wells from 3 independent experiments. C. CD4+ T cells were activated and polarized to become Th17 cells in the presence of rmIL-13 (0 or 10ng/ml) and/or rmIL-21 (none, 0.5, 1 or 5ng/ml). Cell supernatants were collected 4 days after polarization and IL-17A was examined by ELISA. * p<0.05 compared to 0ng/ml of IL-13 when no rmIL-21 was added to the culture conditions. n=4–8 cell culture wells from 3 independent experiments.
Figure 2. IL-13 attenuates IL-17A and IL-21 production in an IL-10-dependent manner in mouse CD4+ T cells.
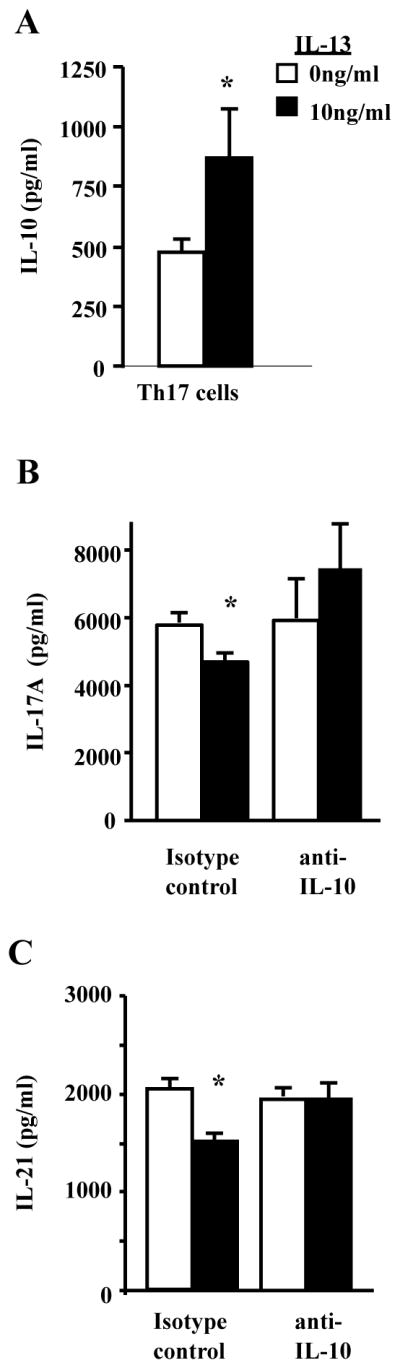
A. CD4+ T cells were activated and polarized to Th17 cells in the presence of IL-13 (0 or10ng/ml). Cell culture supernatants were collected 4 days after polarization and analyzed for IL-10 production. * p<0.05 compared to 0ng/ml of IL-13, n=8 cell culture wells from 3 independent experiments. B–C. CD4+ T cells were activated and polarized to Th17 cells for 4 days with anti-IL10 (10μg/ml) or isotype control in the presence of IL-13. Cell culture supernatants were analyzed for IL-21 and IL-17A production by ELISA 4 days after polarization. * p<0.05 compared to 0ng/ml of IL-13, n=4–5 cell culture wells from 2 independent experiments.
Figure 3. IL-13 and IL-4 independently regulate RSV-induced cytokine production in STAT1 KO mice.

Mice were infected with RSV A2 and lung homogenates were analyzed 6 days post-infection for cytokine production by ELISA. * p<0.05 compared to STAT1 KO mice, ‡ p<0.05 compared to WT mice, n=7–10 mice for each group from 3 independent experiments.
Figure 6. IL-13 deficiency increases IL-10 mediated airway inflammation in STAT1 KO mice which is mediated by IL-10.

Mice were i.p. injected with IgG isotype control or anti-IL10 antibodies one day prior to RSV A2 infection and 3 days after infection. BAL fluid was collected 6 days after infection and infiltration of inflammatory cells was examined. A. Total number of inflammatory cells in BAL fluid. B–E. Neutrophils, eosinophils, macrophages, and lymphocytes in BAL fluid. * p<0.05 compared to STAT1 KO mice treated with isotype control antibody, ‡ p<0.05 compared to isotype control antibody within strain of mice, n=4–5 mice for each group.
Results
IL-13 inhibits IL-21 production, resulting in decreased in IL-17A production in mouse CD4+ T cells
CD4+ T cells isolated from the spleens of WT BALB/c mice were polarized to become Th17 cells with TGF-β, IL-6, IL-23, anti-IL-4, and anti-IFN-γ in the presence of IL-13 (0–10ng/ml). Cell culture supernatants were collected 1–4 days after activation and polarization and were examined for IL-21 and IL-17A cytokine production. IL-21 production was decreased in cells treated with IL-13 starting 2 days after polarization and activation, and IL-13 continued to attenuate IL-21 production at days 3 and 4 of T cell polarization (Figure 1A). IL-17A production was decreased 3 days after polarization with maximal IL-17A production occurring 4 days after polarization (Figure 1B). To determine if decreased IL-21 production results in inhibited IL-17A production, naïve CD4+ T cells were activated and polarized in the presence of recombinant IL-21 (0–5ng/ml), as well as IL-13 (0–10ng/ml). IL-17A production was measured 4 days after polarization, and recombinant IL-21 restored IL-17A production in the presence of IL-13 (Figure 1C). Taken together, these data suggest that IL-13 attenuates IL-17A production by decreasing IL-21 production.
IL-13 attenuates IL-17A and IL-21 production in an IL-10-dependent manner in mouse CD4+ T cells
IL-10 downregulates IL-17A production from Th17 cells (42), and IL-10 produced by Th17 cells attenuatsEAE severity in mice (34). Therefore, we hypothesized that IL-13 increases IL-10 production in Th17 cells, and that inhibiting IL-10 restores IL-17A production in the presence of IL-13. As hypothesized, IL-10 levels were increased in mouse Th17 cells polarized in the presence of IL-13 (Figure 2A). Also as hypothesized, IL-10 neutralization in the presence of IL-13 restored IL-17A and IL-21 production to the levels seen in Th17 cells not exposed to IL-13 (Figure 2B–C). These data suggest that IL-13 negatively regulates IL-17A and IL-21 production in an IL-10 dependent mechanism.
IL-13 negatively regulates RSV-induced IL-17A production in the lung
We have previously shown IL-13 negatively regulates IL-17A production from Th17 cells in both mice and humans (13,14), however, whether IL-13 regulates Th17 cytokine production in the lung is unknown. We hypothesized that IL-13 negatively regulates lung Th17 cytokine production, and we capitalized on a model of RSV A2 infection in STAT1 KO mice. As we have previously shown, RSV A2 infection of STAT1 KO mice resulted in significant increases of lung IL-17A and IL-13 expression whereas IL-17A and IL-13 were not detected in RSV-infected WT mice (Figure 3A and D) (35). To determine the specific role of IL-13 and IL-13 signaling on RSV-induced IL-17A production in STAT1 KO mice, we generated STAT1/IL-13 DKO, STAT1/IL-4 DKO, and STAT1/STAT6 DKO mice. Both IL-13 and IL-4 activate STAT6, and negatively regulate Th17 cytokine secretion in vitro (13,14,17). Six days after RSV A2 infection, the known peak of lung IL-17A in protein expression in STAT1 KO mice (35), lungs were harvested and analyzed for cytokine production (Figure 3A–D). RSV-infected WT mice did not have detectable levels of IL-17A or IL-17F. RSV-infected STAT1 KO mice had significant increases in IL-17A, IL-17F, IL-10, and IL-13 expression compared to RSV-infected WT mice. RSV-infected STAT1/IL-13 DKO and STAT1/IL-4 DKO mice had significant increases in IL-17A and IL-17F expression, and a significant decrease in IL-10 expression compared to RSV-infected STAT1 KO mice. IL-17A and IL-17F production from RSV-infected STAT1/STAT6 DKO mice was approximately 40% greater compared to RSV-infected STAT1/IL-13 DKO or STAT1/IL-4 DKO mice, but this increase was not statistically significant. IL-13 was not detected in RSV-infected STAT1/IL-13 DKO mice, but IL-13 levels were not different between RSV-infected STAT1 KO, STAT1/IL-4 DKO, and STAT1/STAT6 DKO mice (Figure 3D). We did not find a difference in IL-21 production in any group examined (data not shown). The peak of IL-21 production likely occurs at an earlier time point as supported by our in vitro findings (Figure 1A). Taken together, these data show that both IL-13 and IL-4 negatively regulate IL-17A and IL-17F production in whole lung homogenates of RSV-infected STAT1 KO mice. Based on our previous studies with IL-13 regulating IL-17A production in vitro, we decided to focus on the role of IL-13 in RSV-induced IL-17A production for the remainder of the study.
RSV-induced IL-17A is expressed by CD3+ CD4+ CD8− T cells and CD3+ CD4− CD8− cells
IL-17A is produced from numerous cell types. Therefore to determine the cellular source of IL-17A and investigate the role of IL-13, cells were isolated from lungs of STAT1 KO, STAT1/IL-13 DKO and STAT1/STAT6 DKO mice 6 days after RSV infection and were restimulated with PMA and ionomycin in the presence of golgi-stop for analysis by flow cytometry. Cells were stained for antibodies to identify CD4+ T cells, CD8+ T cells, γδ T cells, Gr-1+ cells, and DX5+ cells (Figure 4A and data not shown). Lungs from RSV-infected STAT1 KO mice had IL-17A-expressing CD3+CD4+CD8− cells as well as CD3+CD4−CD8− cells (Figure 4A), but RSV-infected STAT1/IL-13 DKO mice had significant increases in IL-17A-expressing CD3+CD4+CD8− cells compared to RSV-infected STAT1 KO mice (Figure 4B). RSV-infected STAT1/STAT6 DKO mice also significantly increased IL-17A expression by CD3+CD4+CD8− cells as well as CD3+CD4−CD8− cells compared to RSV-infected STAT1 KO mice (Figure 4B). RSV-infected CD3+CD4−CD8− cells were not DX5+ cells, γδ T cells, or Gr-1+ cells as no IL-17A expression was detected in cells expressing these markers (Figure 4A and data not shown). These data suggest IL-13 negatively regulates IL-17A expression in CD3+CD4+CD8− Th17 cells.
Figure 4. IL-17A is expressed by CD3+CD4+CD8− and CD3+ CD4−CD8− cells in STAT1 KO, STAT1/IL-13 DKO, and STAT1/STAT6 DKO mice after RSV infection.

Mice were infected with RSV A2 and lungs were harvested 6 days post- infection. A single cell suspension from lungs was restimulated with PMA, ionomycin, and golgi-plug. Surface staining was performed with antibodies against CD3, CD4, CD8, and γδ T cells. Intracellular cytokine staining was performed with antibodies against IL-17A. A. Representative dot plots which are CD3-gated. B. Total number of cells which are CD3+CD4+CD8− or CD3+CD4−CD8−. Total cell numbers were calculated by multiplying the percentage of IL-17A+ CD3+CD4+ or IL-17A+CD3+CD4− cells by the total cell number determined by counting cells using a hemocytometer. * p<0.05 compared to STAT1 KO mice, n=7–10 mice for each group from 3 independent experiments.
IL-10 neutralization decreases RSV-induced IL-17A production in STAT1 KO mice
Based on our in vitro results showing IL-13 negatively regulated IL-17A production in an IL-10 dependent manner (Figure 2), we hypothesized neutralizing IL-10 in vivo would increases RSV-induced IL-17A production in STAT1 KO mice, but has no effect on IL-17A production in RSV-infected STAT1/IL-13 DKO mice. To test these hypotheses we i.p. injected mice with IL-10 antibody or isotype control one day prior to RSV infection and 3 days post-infection. Mice were RSV-infected and lungs were harvested 6 days post-infection and examined for cytokine production. IL-10 neutralization decreased detection of secreted IL-10 in lung homogenates of STAT1 KO and STAT1/IL-13DKO mice (Figure 5A). IL-10 neutralization significantly increased IL-17A and IL-17F lung production in RSV-infected STAT1 KO mice, but had no effect on IL-17A or IL-17F lung production in RSV-infected STAT1/IL-13 DKO mice (Figure 5B and C). IL-10 neutralization had no effect on IL-13 production (Figure 5D).
Figure 5. IL-10 neutralization increases RSV-induced IL-17A production in STAT1 KO mice.

Mice were i.p. injected with IgG isotype control or anti-IL10 antibodies one day prior to RSV A2 infection and 3 days after infection. A–D. Lungs were harvested 6 days after infection and analyzed for cytokine production by ELISA. E. SOCS3 relative expression normalized to GAPDH and WT isotype control treatment. * p<0.05 compared to STAT1 KO mice treated with isotype control antibody, ‡ p<0.05 compared to isotype control antibody within strain of mice, n=5 mice for each group.
IL-10 binds to the IL-10R and has previously been shown to decrease IL-17A production from Th17 cells by increasing SOCS3 expression (43). Based on our findings showing that neutralizing IL-10 increases IL-17A in RSV-infected STAT1 KO mice, but not RSV-infected STAT1/IL-13 DKO mice, we examined the relative expression levels of SOCS3 by real-time PCR from lungs of mice 6 days after RSV infection. RSV-infected STAT1/IL-13 DKO mice treated with isotype control antibody had significantly decreased SOCS3 expression compared to RSV-infected STAT1 KO mice treated with isotype control antibody (Figure 5E). IL-10 neutralization significantly decreased SOCS3 relative expression in RSV-infected STAT1 KO mice, but had no effect on SOCS3 relative expression in RSV-infected STAT1/IL-13 DKO mice. Taken together these results reveal that IL-13 negatively regulates IL-17A and IL-17F production increasing SOCS3 expression in an IL-10 dependent manner.
IL-13 deficiency increases IL-10 mediated airway neutrophilia in RSV-infected STAT1 KO mice
RSV infection increases infiltration of inflammatory cells into the lungs of STAT1 KO mice (35). Increased IL-17A expression increases neutrophil infiltration into target tissue (44). Therefore, we hypothesized that IL-13 deficiency would increase neutrophil infiltration into the lungs of RSV-infected STAT1 KO mice. BAL fluid was collected 6 days after RSV infection and examined for infiltration of inflammatory cells. In mice treated with isoptye antibody, RSV-infected STAT1/IL-13 DKO mice had a statistically significant 2.5-fold increase in the infiltration of neutrophils compared to RSV-infected STAT1 KO mice (Figure 6B). Neutralization of IL-10 increased the total number of inflammatory cells in BAL fluid of RSV-infected STAT1 KO mice, as well as the number of neutrophils, eosinophils, and macrophages (Figure 6). Neutralization of IL-10 did not alter the number of inflammatory cells in RSV-infected STAT1/IL-13 DKO. Taken together these data show that RSV-infected STAT1/IL-13 DKO mice have increased infiltration of neutrophils and that neutralizing IL-10 increases infiltration of inflammatory cells in RSV-infected STAT1 KO mice.
Discussion
Our previous in vitro studies revealed that mouse CD4+ Th17 cells expressed a functional IL-13Rα1 (13,14). IL-13 signaling through this receptor inhibited Th17 cytokine production (13,14), but the mechanism of action remained unknown. In this study we determined that IL-13 increases IL-10 production, which attenuates IL-17A and IL-21 expression in vitro. Adding recombinant IL-21 at the time of Th17 polarization restored IL-17A production in the presence of IL-13. IL-10 neutralization prevented IL-13 from attenuating IL-17A and IL-21 production in Th17 cells in vitro. These data show IL-13 inhibits IL-17A production by increasing IL-10 expression which in turn decreases IL-21 expression. This is the first report detailing that the mechanism by which IL-13 negatively regulates Th17 cytokine production is mediated by IL-10.
Based on these in vitro results, we hypothesized that IL-13 attenuates IL-17A production in vivo, also by an IL-10 dependent mechanism. To test this hypothesis, we utilized the model of RSV A2 infection in STAT1 KO mice, which induces lung IL-13 and IL-17A protein expression, whereas neither of these cytokines is produced in WT mice with RSV A2 infection (35). In the in vivo experiments reported in this manuscript, we found that there was a significant two-fold increase in IL-17A and IL-17F detected in STAT1/IL-13 DKO mice compared to STAT1 KO mice. This is the first report of IL-13 modulating IL-17A expression in the lung and corresponds with our in vitro data. IL-4, which also activates STAT6, negatively regulated IL-17A and IL-17F expression (13,14,17) in vivo. We found that IL-13 and IL-4 have similar inhibitory effects on IL-17A expression in RSV-infected STAT1 KO micel and when viewed in the context of RSV-infected STAT1/STAT6 DKO mice, there appears to be an additive effect of these cytokines in negatively regulating lung IL-17A protein expression. Our results suggest that both IL-13 and IL-4 are critical mediators in IL-17A inhibition by the STAT6 signaling pathway.
IL-17A expression was detected in CD3+ CD4+ CD8− cells as well CD3+ CD4−CD8− cells. In this model, cells expressing DX5, GR-1, or γδ T cells did not produce IL-17A. DX5 is a marker which stains NK cells and a small subset of T-lymphocytes, which are likely NK T cells (45). Gr-1 is expressed on granulocytes, monocytes during differentiation, and weakly on plasmacytoid dendritic cells. CD3+ double negative cells (CD4− and CD8−) have previously been shown to produce both IL-17A and IFN-γ after Francisella tularensis and L. monocytogenes infections in mice (24,46). These double negative cells were required for clearance of L. monocytogenes (24), and IL-17A was also vital for clearance of Francisella tularensis and L. monocytogenes infections in mice (24,46). IL-17A expression by CD3+ double negative cells was significantly increased in STAT1/STAT6 DKO mice compared to STAT1 KO and STAT1/IL-13 DKO mice. These data suggest that IL-17A production from CD3+ double negative cells is negatively regulated by IL-4, but not IL-13.
In vitro, IL-10 production was significantly increased in Th17 cells polarized in the presence of IL-13, and IL-10 was required for IL-13 inhibition of IL-17 and IL-21 production. In vivo, we showed IL-10 production was significantly increased in STAT1 KO mice compared to WT mice, but in the absence of IL-13 (in STAT1/IL-13DKO mice) IL-10 production was significantly decreased. Further, when IL-10 was neutralized prior to RSV infection, IL-17A and IL-17F production was significantly increased in STAT1 KO mice but not in STAT1/IL-13 DKO mice. Combined, our in vitro and in vivo studies show IL-13 increases IL-10 production, and IL-10 in turn decreases IL-17A and IL-17F production.
IL-10 has anti-inflammatory properties and IL-10Rα was expressed on IL-17A producing CD4+ cells in the small intestines (47). IL-10 inhibited IL-17A production from Th1/Th17 cells, which produce IFN-γ and IL-17A, as well as Th17 cells, which only produce IL-17A, verifying that IL-10 can directly affect Th17 cells (47). These data show IL-10R is expressed on Th17 cells and IL-10 attenuates IL-17A production. McGeachy and colleagues showed that IL-10 is produced by Th17 cells and TGF-β and IL-6 were required for IL-10 production (34). Further, IL-10 neutralization in the EAE model increased disease severity in mice receiving adoptive transfer of CD4+ T cells stimulated with TGF-β and IL-6 (34). We previously determined in human T cells that IL-13Rα1 expression on T cells required the Th17 differentiating cytokines TGF-β, IL-6, and IL-23 (14). Therefore, TGF-β and IL-6 are required for IL-13Rα1 expression, allowing for IL-13 signaling, as well as IL-10 production by Th17 cells.
IL-10 binds to the IL-10R and induces STAT3 and SOCS3 expression (43). While STAT3 is a required transcription factor for Th17 differentiation and cytokine production, IL-10 negatively regulates Th17 cytokine production by increasing SOCS3 expression which downregulates IL-6/gp130 signaling (43). In this study we showed SOCS3 expression was decreased when IL-10 was neutralized in RSV infected STAT1 KO mice, but not STAT1/IL-13 DKO mice. Taken together these studies define how IL-13 regulates Th17 cell cytokine secretion by upregulating IL-10 production and SOCS3 expression, resulting in the attenuation of IL-17A and IL-17F production.
IL-13 is a central mediator of airway hyperreactivity and mucus production, and IL-13 is increased in sputum of patients with asthma (4). Therefore, therapeutics which target IL-13 and IL-13 signaling are attractive in treating allergic airway diseases including asthma, and several therapeutics are currently undergoing clinical trials (6–11). Therapeutics targeting IL-13 and IL-13 signaling pathways have a strong safety profile. However most studies do not recruit patients with moderate to severe asthma (6,11,48), and none of the studies measured IL-17A levels (6–11). Patients with moderate to severe asthma have increased IL-17A+ cells compared to mild asthmatics and healthy controls (20), and IL-17A is increased in the BAL fluid of patients with moderate to severe asthma (20,21). Therefore, patients with severe asthma or patients with Th17-mediated diseases, such as multiple sclerosis, rheumatoid arthritis, and colitis, may have adverse side effects associated with IL-13 targeted therapeutics. Corren and colleagues reported that the anti-IL-13 antibody, lebrikizumab, increased FEV1 in uncontrolled asthmatics compared to placebo control (6). However, patients receiving lebrikizumab had a significant increase in adverse events involving the musculoskeletal system, some of which are diseases associated with Th17 cells(6). In conclusion, we have further defined the mechanism by which IL-13 regulates IL-17A production both in vitro and in vivo. IL-13 decreases IL-17A production in an IL-10-dependent pathway in STAT1 KO mice. These findings are vital to understanding potential side effects of therapeutics targeting IL-13 or IL-13 signaling in patients with Th17-mediated diseases and potentially severe asthma.
Acknowledgments
We would like to acknowledge the Richard King Mellon Foundation Institute for Pediatric Research, Children’s Hospital of Pittsburgh.
Declaration of all sources of funding: National Institute of Health R01 HL 090664, R01 AI 070672, R01 AI 059108, GM 015431, R21 HL106446, U19AI095227, K12HD043483-08 and Veteran Affairs (1I01BX000624).
Abbreviations
- AHR
airway hyperreactivity
- BAL
bronchoalveolar lavage
- EAE
experimental autoimmune encephalomyelitis
- RSV
respiratory syncytial virus
References
- 1.Ruth L. A breath of hope. Researchers are combining their efforts to study the genetic and environmental factors in asthma. EMBO Rep. 2007;8:319–321. doi: 10.1038/sj.embor.7400952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–690. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 3.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 4.Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A, May R, Monk P, Bradding P, Wardlaw AJ, Pavord ID, Brightling CE. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–691. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bree A, Schlerman FJ, Wadanoli M, Tchistiakova L, Marquette K, Tan XY, Jacobson BA, Widom A, Cook TA, Wood N, Vunnum S, Krykbaev R, Xu X, Donaldson DD, Goldman SJ, Sypek J, Kasaian MT. IL-13 blockade reduces lung inflammation after Ascaris suum challenge in cynomolgus monkeys. J Allergy Clin Immunol. 2007;119:1251–1257. doi: 10.1016/j.jaci.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 7.Gauvreau GM, Boulet LP, Cockcroft DW, Fitzgerald JM, Carlsten C, Davis BE, Deschesnes F, Duong M, Durn BL, Howie KJ, Hui L, Kasaian MT, Killian KJ, Strinich TX, Watson RM, YN, Zhou S, Raible D, O’Byrne PM. Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2011;183:1007–1014. doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson GC, Kariyawasam HH, Tan AJ, Hohlfeld JM, Quinn D, Walker C, Rodman D, Westwick J, Jurcevic S, Kon OM, Barnes PJ, Krug N, Hansel TT. The effects of an anti-IL-13 mAb on cytokine levels and nasal symptoms following nasal allergen challenge. J Allergy Clin Immunol. 2011;128:800–807. doi: 10.1016/j.jaci.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Singh D, Kane B, Molfino NA, Faggioni R, Roskos L, Woodcock A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10:3. doi: 10.1186/1471-2466-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 11.Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, Wenzel SE, Chon Y, Dunn M, Weng HH, Lin SL. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med. 2010;181:788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- 12.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 13.Newcomb DC, Zhou W, Moore ML, Goleniewska K, Hershey GK, Kolls JK, Peebles RS., Jr A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;182:5317–5321. doi: 10.4049/jimmunol.0803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newcomb DC, Boswell MG, Zhou W, Huckabee MM, Goleniewska K, Sevin CM, Khurana Hershey GK, Kolls JK, Peebles RS., Jr Human T(H)17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J Allergy Clin Immunol. 2011;127:1006–1013. doi: 10.1016/j.jaci.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson MS, Ramalingam TR, Rivollier A, Shenderov K, Mentink-Kane MM, Madala SK, Cheever AW, Artis D, Kelsall BL, Wynn TA. Colitis and intestinal inflammation in IL10−/− mice results from IL-13Ralpha2-mediated attenuation of IL-13 activity. Gastroenterology. 2011;140:254–264. doi: 10.1053/j.gastro.2010.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 18.Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, Querci V, Angeli R, Matucci A, Fambrini M, Liotta F, Parronchi P, Maggi E, Romagnani S, Annunziato F. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol. 2010;125:222–230. doi: 10.1016/j.jaci.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, Bernstein JA, Huston DP, Liu YJ. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010;207:2479–2491. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 21.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 22.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowley SC, Meierovics AI, Frelinger JA, Iwakura Y, Elkins KL. Lung CD4−CD8− double-negative T cells are prominent producers of IL-17A and IFN-gamma during primary respiratory murine infection with Francisella tularensis live vaccine strain. J Immunol. 2010;184:5791–5801. doi: 10.4049/jimmunol.1000362. [DOI] [PubMed] [Google Scholar]
- 25.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 26.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L, I, Ivanov I, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 29.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 30.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008 doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi-Joannopoulos K, Carreno BM, Collins M, Wolfman NM. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. 2007;282:13447–13455. doi: 10.1074/jbc.M700499200. [DOI] [PubMed] [Google Scholar]
- 33.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto K, Durbin JE, Zhou W, Collins RD, Ho SB, Kolls JK, Dubin PJ, Sheller JR, Goleniewska K, O’Neal JF, Olson SJ, Mitchell D, Graham BS, Peebles RS., Jr Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J Allergy Clin Immunol. 2005;116:550–557. doi: 10.1016/j.jaci.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 36.Durbin JE, Johnson TR, Durbin RK, Mertz SE, Morotti RA, Peebles RS, Graham BS. The role of IFN in respiratory syncytial virus pathogenesis. J Immunol. 2002;168:2944–2952. doi: 10.4049/jimmunol.168.6.2944. [DOI] [PubMed] [Google Scholar]
- 37.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, Murray R, Grencis R, McKenzie AN. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–432. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 38.Graham BS, Perkins MD, Wright PF, Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol. 1988;26:153–162. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 39.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol. 1999;162:392–399. [PubMed] [Google Scholar]
- 40.Peebles RS, Jr, Sheller JR, Collins RD, Jarzecka AK, Mitchell DB, Parker RA, Graham BS. Respiratory syncytial virus infection does not increase allergen-induced type 2 cytokine production, yet increases airway hyperresponsiveness in mice. J Med Virol. 2001;63:178–188. [PubMed] [Google Scholar]
- 41.Zhou W, Blackwell TS, Goleniewska K, O’Neal JF, FitzGerald GA, Lucitt M, Breyer RM, Peebles RS., Jr Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J Leukoc Biol. 2007;81:809–817. doi: 10.1189/jlb.0606375. [DOI] [PubMed] [Google Scholar]
- 42.Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J, Bromberg J, Chen SH, Mayer L, Unkeless JC, Xiong H. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38:1807–1813. doi: 10.1002/eji.200838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, Skoogh BE, Linden A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 45.Arase H, Saito T, Phillips JH, Lanier LL. Cutting edge: the mouse NK cell-associated antigen recognized by DX5 monoclonal antibody is CD49b (alpha 2 integrin, very late antigen-2) J Immunol. 2001;167:1141–1144. doi: 10.4049/jimmunol.167.3.1141. [DOI] [PubMed] [Google Scholar]
- 46.Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, Waisman A, Kuchroo VK, Glimcher LH, Oukka M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J Immunol. 2010;184:1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber S, Gagliani N, Esplugues E, O’Connor W, Jr, Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, Roncarolo MG, Battaglia M, Flavell RA. Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wenzel SE, Busse WW. Severe asthma: lessons from the Severe Asthma Research Program. J Allergy Clin Immunol. 2007;119:14–21. doi: 10.1016/j.jaci.2006.10.025. [DOI] [PubMed] [Google Scholar]