

Abstract
Pathogens may signal through multiple TLRs with synergistic or antagonistic effects on the induction of cytokines, including type I IFN (IFN-I). IFN-I is typically induced by TLR9 but not TLR2. Moreover, we previously reported that TLR2 signaling by Mycobacterium tuberculosis or other TLR2 agonists inhibited TLR9 induction of IFN-I and IFN-I-dependent MHC-I Ag cross processing. The current studies revealed that lipopeptide-induced TLR2 signaling inhibited induction of first-wave IFN-α and IFN-β mRNA by TLR9, whereas induction of second wave IFN-I mRNA was not inhibited. TLR2 also inhibited induction of IFN-I by TLR7, another MyD88-dependent IFN-I-inducing receptor, but did not inhibit IFN-I induction by TLR3 or TLR4 (both TRIF-dependent, MyD88-independent). The inhibitory effect of TLR2 was not dependent on new protein synthesis or intercellular signaling. IRAK1 was depleted rapidly (within 10 min) by TLR2 agonist but not until later (e.g. 2 h) by TLR9 agonist. Since IRAK1 is required for TLR7/9 induced IFN-I production, we propose that TLR2 signaling induces rapid depletion of IRAK1, which impairs IFN-I induction by TLR7/9. This novel mechanism, whereby TLR2 inhibits IFN-I induction by TLR7/9, may shape immune responses to microbes that express ligands for both TLR2 and TLR7/TLR9, or responses to bacteria/virus co-infection.
Keywords: Dendritic cell, type-I interferon, Toll-like receptor, IRAK1
Introduction
The innate immune system recognizes and responds to bacterial exposure with a coordinated cytokine response that must launch host defense to eradicate the offending pathogen yet avoid excessive inflammation that may damage host tissues. To this end, signaling by innate immune pattern recognition receptors (PRRs) controls the identity, quantity and duration of cytokine responses. Many pathogens express agonists of more than one PRR, and the immune system must integrate signaling through multiple PRRs to specify the correct cytokine response. Despite the importance of this integration of signaling pathways to immune homeostasis, the molecular mechanisms of such integration remain unclear. For example, the induction of type I interferon (IFN-I, e.g. IFN-α and IFN-β) is controlled by multiple PRRs that can have positive or negative affects on the expression of IFN-I. Too little IFN-I response may compromise IFN-I-induced immunity to pathogens (1, 2), while excessive or overly prolonged IFN-I may be associated with autoimmunity, e.g. systemic lupus erythematosis (SLE) (3).
IFN-I is induced by agonists of certain Toll-like receptors (TLRs), e.g. by double-stranded RNA via TLR3, by LPS via TLR4, by single-stranded RNA and certain antiviral compounds via TLR7, and by DNA and oligonucleotides (ODNs) containing CpG motifs (e.g. CpG-ODNs) via TLR9. IFN-I induction by TLR3 and TLR4 is dependent on TRIF signaling, while IFN-I induction by TLR7 and TLR9 is dependent on MyD88. Recruitment and activation of IRAK1 is required for induction of IFN-I by TLR7 or TLR9, whereas IRAK1 is not required for induction of pro-inflammatory cytokines by these receptors (4). Initial signaling by PRRs leads to induction of a first wave of IFN-I composed of IFN-β and IFN-α4. First wave IFN-I induces autocrine or paracrine signaling through IFN-I receptor (IFN-IR) to induce a second wave of IFN-I, including all forms of IFN-α and IFN-β, amplifying the IFN-I response. IFN-I signaling results in the induction of a number of IFN-I-stimulated genes (ISGs).
Most pathogens express agonists of multiple PRRs, which may differ in their signaling and regulatory effects. TLR2 and TLR9 induce partially overlapping arrays of cytokines. In contrast to TLR9, TLR2 does not generally induce IFN-I, although signaling by some pathogens through TLR2 may induce IFN-I (5). Simultaneous activation of two or more TLRs, which may mimic physiological situation during host-pathogen interaction, can have synergistic, antagonistic, or additive effects on cytokine responses. However, very little is known about the effect of cross-talk of different TLRs on induction of IFN-I.
We used Mycobacterium tuberculosis (Mtb) as a model, since this pathogen, like many bacteria, expresses agonists of both TLR2 (lipoproteins, glycolipids) and TLR9 (DNA containing CpG motifs) (6–11), and both TLR2 and TLR9 contribute to host resistance to Mtb infection (12). While the impact of IFN-I on tuberculosis pathogenesis remains unclear, Mtb induces IFN-I and ISGs in peripheral blood neutrophils in human tuberculosis (13). Mtb induction of IFN-I is associated with decreased production of other cytokines, e.g. IL-1β (14), TNFα and IL-12 (15). IFN-I increases growth of Mtb in macrophages and increases disease progression (16–18). We recently demonstrated that TLR2 signaling by Mtb or other TLR2 agonists inhibited TLR9 induction of IFN-I and IFN-I-dependent MHC-I Ag cross processing (19). In the current study, we investigated the mechanisms by which TLR2 signaling inhibits induction of IFN-I. Our results show that TLR2 signaling inhibits MyD88-dependent induction of IFN-I through TLR9 or TLR7 by interfering with intracellular signaling through a novel mechanism that includes rapid degradation of IRAK1. This mechanism may shape the role of IFN-I in host-pathogen interactions when both TLR2 and TLR7/9 agonists are present. This mechanism may be exploited by pathogens to evade host defenses. Alternatively, it may be a mechanism for host protection against deleterious effects of IFN-I. Understanding this inhibitory pathway may allow its exploitation to inhibit deleterious effects of IFN-I in other disease settings, e.g. autoimmune disease.
Materials and Methods
Abs and reagents
Triacylated LpqH-lipopeptide containing 15 amino acids of the N-terminal sequence of Mtb LpqH (19-kDa lipoprotein) was purchased from EMC Microcollections (Tübingen, Germany). Mtb lipoprotein LprG was purified as described (20). CpG ODN-A2336 (5′-ggG GAC GAC GTC GTG ggg ggG-3′), CpG ODN-B1668 (5′-tcc atg acg ttc ctg atg ct-3′) were synthesized by Eurofins MWG Operon (Huntsville, AL) or Sigma-Aldrich (St. Louis, MO); lower case letters in ODN sequences refer to nucleotides for which the 3′ internucleotide linkage was phosphorothioate-modified, and upper case letters refer to standard phosphodiester-linked nucleotides. Poly (I:C), LPS (ultrapure Escherichia coli 0111:B4), ssRNA40, and synthetic lipopeptides Pam3CSK4 and FSL-1 were purchased from Invivogen (San Diego, CA). Agonists were dissolved in endotoxin-free (≤0.05 units) PBS (Cambrex, East Rutherford, NJ) or sterile cell-culture water (Sigma-Aldrich). Recombinant murine IFN-β was from PBL Interferonsource (Piscataway, NJ). Cycloheximide (#C7698) was from Sigma. Anti-IRAK1 (#4504), anti-IRAK4 (#4363) and anti-MyD88 (#4283) antibodies were purchased from Cell Signaling Technology (Boston, MA). Anti-β-actin and anti-TRAF6 (#sc-7221) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Murine cell culture and media
Standard medium was RPMI 1640 with L-glutamine, glucose, 10% heat-inactivated FCS (HyClone Laboratories, Logan, UT), 50 μM 2-ME, 1 mM sodium pyruvate and penicillin-streptomycin. DCs were prepared from femur and tibia bone marrow cells of C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME); TLR2−/−, TLR9−/− or MyD88−/− mice (kindly provided by S. Akira, Osaka University, on a C57BL/6 background); IFN-IR−/− A129 mice on a 129/SvEv background (B&K Universal, Grimston, Aldbrough, U.K.); or 129S6/SvEv wild-type mice (Taconic Laboratories, Hudson, NY). Bone marrow cells were cultured at 106 cells/ml in 6-well tissue culture-grade dishes for 8–10 d in recombinant mouse FMS-like tyrosine kinase 3 ligand (Flt3L)-Ig fusion protein (1 μg/ml, BioExpress, Kaysville, UT). Medium and Flt3L-Ig were replenished on days 3 and 6. Alternatively, marrow cells were cultured for 7 d in a 100 mm petri dish with 10 ml J558L cell-conditioned medium (containing GM-CSF) diluted in complete RPMI medium (1:30) to produce “GM-CSF DCs”. Medium and GM-CSF were replenished on days 3 and 6. On day 8 or 9, non-adherent cells were removed, pelleted, resuspended, and counted. Flt3L-derived DCs contain a mixture of mDCs and pDCs, whereas GM-CSF DC cultures contain mDCs (<1% pDCs) (21). Unless otherwise stated, “DCs” refers to Flt3L-derived DCs.
Bacteria
Mtb (strain H37Ra) was from the American Type Culture Collection (Manassas, VA). Bacteria were grown in 7H9 medium (Difco, Lawrence, KS), supplemented with 1% glycerol, 0.05% Tween 80 (Sigma-Aldrich), and 10% albumin/dextrose/catalase (BD Biosciences, Franklin Lakes, NJ). Bacteria were cultured with shaking at 37°C to mid-log phase (~1 wk) and harvested by centrifugation at 5000 × g for 30 min at 4°C. Bacteria were washed in RPMI 1640 medium, resuspended in RPMI 1640 medium supplemented with 10% FCS and 6% glycerol, and flash-frozen in a dry ice/ethanol bath. Prior to use in experiments, bacteria were thawed at 37°C for 30–60 min, declumped with 10 passes through a 23-gauge needle and centrifuged at 200 × g for 2 min to pellet large clumps. The resulting bacteria in suspension were used for experiments. Bacterial CFU values were quantified by culture of serial dilutions of declumped bacteria. Gamma-irradiated Mtb H37Rv whole cell lysate was prepared at Colorado State University, and was received as part of NIH, NIAID Contract No. HHSN266200400091C, entitled “Tuberculosis Vaccine Testing and Research Materials”.
ELISA and quantitative real-time PCR
DCs were cultured at 2 × 105 cells/well in flat-bottom 96-well plates or 3–5 × 106 cells/well in 12-well tissue culture-treated plates for 2–24 h with or without agonists. Plates were centrifuged to pellet cells, and supernatants were diluted and tested by ELISA for mouse IFN-α or IFN-β (PBL InteferonSource) or mouse IL-12p40 or IL-10 (R&D Systems, Minneapolis, MN). For quantitative real-time RT-PCR (qRT-PCR), RNA was isolated from DCs using an RNeasy Plus Mini Kit (Qiagen, Valencia, CA). Total RNA was extracted following on-column DNase digestion using RNeasy Plus mini columns (Qiagen) and collected in RNase-free water. Yield was determined by OD. Oligo(dT)-primed reverse transcription of RNA into cDNA was performed with QuantiTect Reverse Transcription Kit (Qiagen), and 4% of the product was used for each qRT-PCR sample using Bio-Rad iQ SYBR Green Supermix, and the Bio-Rad CFX96 fluorescence detection system (Bio-Rad, Hercules, CA). All conditions were tested in triplicate. Primer pairs from murine gene sequences were for total IFN-α (sense, 5′-ATG GCT AGR CTC TGT GCT TTC CT-3′; antisense, 5′-AGG GCT CTC CAG AYT TCT GCT CTG-3′), IFN-β (sense, 5′-CAT CAA CTA TAA GCA GCT CCA-3′; antisense, 5′-TTC AAG TGG AGA GCA GTT GAG-3′), GAPDH (sense, 5′-AAC GAC CCC TTC ATT GAC-3′; antisense, 5′-TCC ACG ACA TAC TCA GCA C-3′) as described (22) or designed using Clone Manager Suite v7.11 and Primers Designers v5.11 (Scientific & Educational Software, Cary, NC). A BLAST search was performed to verify specificity.
Western blot analysis of whole cell lysates
DCs were incubated with agonists in 12-well plates at 37°C. Subsequent steps were performed at 4°C unless otherwise stated. Cells were washed in PBS with protease inhibitor mixture (P8340, Sigma-Aldrich; 1:200), 1 mM NaF, 1 mM PMSF and 10 nM calyculin A. To prepare whole cell lysates, cells were pelleted and resuspended in 160 μl RIPA lysis buffer (50 mM Tris-Cl, 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 0.5% Na-deoxycholate, and 0.1% SDS [pH 7.4]) with protease inhibitors and mixed on a rotator for 1 h. Samples were centrifuged at 16,000 × g for 10 min, and 150 μl supernatant was collected. Aliquots containing equal quantities of total protein were added to reducing sample buffer (Thermo Fisher Scientific, Waltham, MA; #39000), subjected to electrophoresis on 8% SDS polyacrylamide gels, and transferred onto polyvinylidene difluoride membranes. Membranes were washed in PBS with 0.1% Tween 20 (PBST), incubated for 1 h at room temperature in 5% nonfat milk in PBST, incubated overnight at 4°C with primary Ab in 3% BSA or 5% nonfat milk in PBST, washed, incubated for 1 h at room temperature with HRP-labeled secondary Ab (Cell Signaling Technology), and developed with the ECL detection kit (Thermo Fisher Scientific, #32106).
Results
Mtb and TLR2 agonist lipopeptide inhibit TLR9-induced IFN-I production
To test the effect of TLR2 signaling on TLR9 induction of IFN-I, we incubated DCs with CpG ODN-A2336 (CpG-A ODN) in combination with Mtb strain H37Ra or synthetic LpqH-lipopeptide (containing the N-terminal sequence of Mtb lipoprotein LpqH). CpG-A ODN induced IFN-α and IFN-β production in Flt3L DCs, but this induction was inhibited by Mtb or LpqH-lipopeptide (Fig. 1A, B). The inhibition was dose-dependent, and IFN-I induction was strongly inhibited with 400 nM LpqH-lipopeptide or Mtb H37Ra at an MOI of 1 (Fig. 1). In TLR2−/− DCs, however, CpG-A ODN induction of IFN-α and IFN-β was not significantly inhibited by Mtb or LpqH-lipopeptide, indicating that the inhibitory effect was dependent on TLR2 (Fig. 1A, B). A lysate of Mtb H37Rv (see Methods) inhibited CpG-A ODN induction of IFN-β, similar to the inhibition achieved by LpqH-lipopeptide, confirming that molecules expressed by Mtb recapitulate the inhibition of IFN-I observed with purified TLR2 agonist (Supplemental Fig. 1A). DCs also produce other cytokines in response to TLR signaling, including IL-12p40, IL-10, and others. CpG-A ODN induced higher production of these cytokines than LpqH-lipopeptide in DCs (Fig. 1E, F). When DCs were incubated with both agonists, LpqH-lipopeptide did not significantly inhibit CpG-A ODN-induced IL-12p40 (Fig. 1E) or IL-10 (Fig. 1F) We conclude that DCs incubated with TLR2 agonists are viable and continue to produce IL-10 and IL-12p40, but show inhibition of TLR9-induced IFN-I.
Figure 1. Mtb and LpqH-lipopeptide inhibit TLR9-induced IFN-I in a TLR2-dependent manner.

A, B, Flt3L-derived DCs from wild-type C57BL/6 mice or TLR2−/− mice were cultured for 24 h with CpG-A ODN (300 nM) with or without Mtb H37Ra (MOI=1) (A) or LpqH-lipopeptide (400 nM) (B). C–F, Flt3L-derived DCs were cultured for 24 h with or without CpG-A ODN (300 nM) and various doses of LpqH-lipopeptide. Supernatants were assessed by ELISA for IFN-α (A, C) IFN-β (B, D), IL-12p40 (E) or IL-10 (F). Data represent means and standard deviations of triplicate wells and are representative of 3 or more independent experiments. Student’s t-test was performed to compare results with CpG-A ODN only and CpG-A ODN with Mtb or LpqH-lipopeptide (** = p < 0.01, *** = p < 0.001; n.d., not detected; n.s., not significant).
TLR2 agonist inhibits TLR9-induced IFN-I mRNA expression
IFN-I production can be regulated at multiple levels, including mRNA expression. CpG-A ODN-induced expression of IFN-α and IFN-β mRNA in DCs was significantly inhibited by LpqH-lipopeptide; the inhibition was greater for IFN-β than IFN-α (Fig. 2A, B). In addition to Flt3L-derived DCs, which represent our primary experimental system and comprise a mixture of mDCs and pDCs, the same effect was observed in GM-CSF-derived DCs (Fig. 2C), which are mDCs (21). Kinetic studies revealed significant TLR2-induced inhibition of mRNA for both IFN-α and IFN-β from 2–24 h (Fig. 2D–G). In studies of earlier time points (15–120 min), we found that TLR9 induced IFN-β mRNA at 1 h, and TLR2 inhibition of IFN-β mRNA occurred at this time point (data not shown), suggesting the inhibition occurs rapidly.
Figure 2. LpqH-lipopeptide inhibits TLR9-induced IFN-I mRNA expression.

A–C, Flt3L-derived DCs (A, B) or GM-CSF DCs (C) were cultured for 3 h with medium, LpqH-lipopeptide (400 nM), CpG-A ODN (300 nM) or both agonists. Expression of mRNA for IFN-α and IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as fold-change relative to expression with medium alone. Data represent means and standard deviations for triplicate samples and are representative of three or more independent experiments. D–G, FLt3L-derived DCs were cultured for various times (0, 2, 4, 7.5, 16, 24 h) with CpG-A ODN (300 nM) or CpG-A ODN + LpqH-lipopeptide (400 nM). Expression of mRNA for IFN-α and IFN-β was determined as above. Results are expressed as fold-change relative to expression in the non-treated (0 h) condition on a logarithmic scale (D, E) or as percent of the response to CpG-A ODN alone at each time point (F, G). Data represent means and standard deviations of triplicate samples and are representative of two independent experiments. Student’s t-test was performed to compare results with CpG-A ODN alone to results with CpG-A ODN plus LpqH-lipopeptide (* = p < 0.05, ** = p < 0.01, *** = p < 0.001; #, results are plotted but are too low to be visualized on this graph).
TLR2 agonist inhibits first wave IFN-I induction
We considered two stages of IFN-I mRNA induction at which its expression could be regulated. TLR signaling directly induces first wave IFN-I (restricted to IFN-β and IFN-α4), and the expression of IFN-I species can be substantially amplified by the autocrine/paracrine IFN-I positive feedback loop via the IFN-IR, which induces second-wave IFN-I. Therefore, the TLR2–mediated inhibition of IFN-I expression could involve inhibition of either first- or second-wave IFN-I.
We used two approaches to determine whether TLR2–mediated inhibition of IFN-I expression involves inhibition of the first or second wave of IFN-I induction. In the first approach, we treated DCs with exogenous recombinant mouse IFN-β to induce second wave IFN-I, which we assessed by mRNA expression. Addition of LpqH-lipopeptide did not inhibit IFN-β–induced expression of IFN-β mRNA (Fig. 3A, B), suggesting that mechanisms for induction of second wave IFN-I were not inhibited. We considered that the design of our original experiments with simultaneous addition of LpqH-lipopeptide and CpG-A ODN might activate TLR2 signaling hours prior to the time course of second wave IFN-I induction. Accordingly, in some experiments we pretreated DCs with or without LpqH-lipopeptide (400 nM) for 3 h before adding IFN-β (200 pg/ml) for 6 h to mimic this situation, but this protocol still did not produce inhibition of second wave IFN-β (Fig. 3A). These results indicate that mechanisms intrinsic to the second wave induction of IFN-I are not inhibited by TLR2 signaling.
Figure 3. LpqH-lipopeptide inhibits induction of first wave IFN-I but not second wave IFN-I.
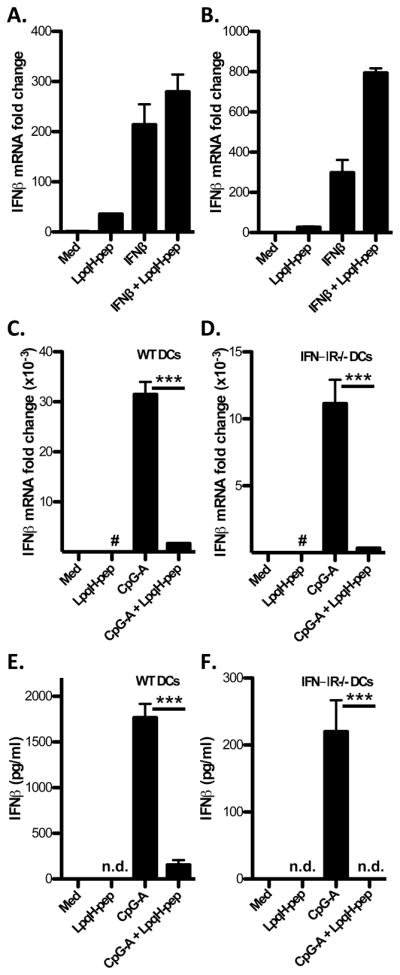
A, Flt3L-derived DCs were left untreated or pretreated with LpqH-lipopeptide (400 nM) for 3 h before a 6-hour incubation with or without IFN-β (200 pg/ml) in the continued presence of LpqH-lipopeptide. B, DCs were simultaneously treated with medium, LpqH-lipopeptide, IFN-β or both agonists for 8 h. C–F, Flt3L-derived DCs from wild-type or IFN-IR−/− mice were cultured with medium, LpqH-lipopeptide (400 nM), CpG-A ODN (300 nM) or both agonists for 3 h (C, D) or 24 h (E, F). A–D, Expression of mRNA for IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as fold-change relative to expression with medium alone. E, F, Supernatants were assessed by ELISA for IFN-β̃ Data in all panels represent means and standard deviations of triplicate samples and are representative of three or more independent experiments. Student’s t-test was performed to compare results with CpG-A ODN alone to results with CpG-A ODN plus LpqH-lipopeptide (*** = p < 0.001; #, results are plotted but are too low to be visualized on this graph; n.d., not detected).
The second approach eliminated second wave IFN-I by using IFN-IR−/− DCs, since the IFN-IR is required for second wave IFN-I. Therefore, all IFN-I production in IFN-IR−/− cells is by the first wave. In IFN-IR−/− DCs, LpqH-lipopeptide inhibited induction of IFN-β mRNA by 3 h (Fig. 3C, D) and IFN-β protein by 4 h (Supplemental Fig. 1B) or 24 h (Fig. 3E, F) after stimulation by CpG-A ODN. These results indicate that the inhibitory mechanism blocks induction of first-wave IFN-I by TLR9 signaling.
In these experiments, wild-type DCs produced higher amounts of IFN-β than IFN-IR−/− cells, which is expected when second wave IFN-I can supplement the first wave. For example, wild-type DCs produced 8–9-fold more IFN-β than IFN-IR−/− DCs after 24 h of stimulation with CpG-A ODN, a time point that allowed a substantial period for first wave to induce the second wave (compare Fig 3E and 3F). After 3 h, when there was less time to induce the second wave, IFN-β mRNA was about 3-fold higher in wild-type DCs compared to IFN-IR−/− DCs. We performed additional experiments to assess IFN-β expression by ELISA at early time points (1 h, 2 h, 4 h and 6 h). IFN-β was not detected after 1–2 h of stimulation with CpG-A ODN (data not shown) and was first detected after 4 h of stimulation, at which time its production was already inhibited by LpqH-lipopeptide (Supplemental Fig. 1B). At 4 h, CpG-A ODN-induced IFN-β was 3-fold higher in wild-type DCs compared to IFN-IR−/− DCs. These results are consistent with the mRNA results at 3 h (Fig. 3C, D). Thus, IFN-β production was less with IFN-IR−/− DCs than wild-type cells at all time points, even early time points, suggesting that in wild-type cells the second wave of IFN-β may be induced so quickly that it overlaps kinetically with the first wave or that knockout cells have a lower baseline expression of signaling or transcription regulatory factors that are required for induction of first wave IFN-β. In summary, LpqH-lipopeptide inhibited IFN-β at the earliest time point at which its production was detected, and it inhibited IFN-β production in IFN-IR−/− DCs that lack second wave IFN-I production. These results indicate that production of first wave IFN-I is inhibited by TLR2 signaling. Since the second wave is dependent on the first wave, this mechanism will result in overall inhibition of IFN-I induction.
TLR2-mediated inhibition of IFN-I induction is not dependent on intercellular signaling or new protein synthesis
TLR2 may inhibit induction of IFN-I either directly through intracellular signaling effects of TLR2 or indirectly through signaling by TLR2-induced cytokines. For example, cytokines such as IL-10 or TNFα inhibit induction of IFN-I (23, 24). To examine this issue, we tested IFN-β mRNA expression at early time points in a mixed culture of TLR9−/− DCs and TLR2−/− DCs. TLR9−/− DCs cannot induce IFN-β mRNA in response to CpG-ODN but can make TLR2-induced cytokines, whereas TLR2−/− DCs cannot respond directly to TLR2 agonists but can respond to TLR2-induced cytokines from other cells. Therefore, this mixed culture would allow indirect inhibition (via TLR2-induced cytokines) but not direct TLR2 signaling inhibition of IFN-I induction by TLR9. LpqH-lipopeptide inhibited CpG-A ODN-induced IFN-β mRNA in wild type DCs alone but not in TLR2−/− DCs alone (Fig. 4). TLR9−/− cells did not recognize CpG-A ODN and thus did not produce any IFN-β mRNA (Fig. 4). When TLR2−/− and TLR9−/− DCs were mixed at a 1:1 ratio, LpqH-lipopeptide did not inhibit CpG-A ODN-induced IFN-β mRNA (Fig. 4). IFN-I production in the mixed cell system was half of that seen in wild type DCs alone, since half of the cells were unable to respond to CpG-A ODN. These results suggest that TLR2 inhibits IFN-I induction directly through intracellular signaling and not indirectly via intercellular signaling by TLR2-induced cytokines.
Figure 4. TLR2-mediated inhibition of IFN-I production occurs by an intracellular mechanism.

Incubations included Flt3L-derived DCs (3–5×106/well) from wild-type, TLR2−/− or TLR9−/− mice, or a 1:1 mixture of TLR2−/− and TLR9−/− DCs (total of 3–5×106 cells). DCs were cultured for 3 h in 12-well plates with medium, LpqH-lipopeptide (400 nM), CpG-A ODN (300 nM), or both agonists. Expression of mRNA for IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as fold-change relative to expression with medium alone. Data represent means and standard deviations of triplicate samples and are representative of three or more independent experiments. Student’s t-test was performed to compare results with CpG-A ODN alone to results with CpG-A ODN plus LpqH-lipopeptide (*** = p < 0.001; #, results are plotted but are too low to be visualized on this graph).
Regulation of IFN-I induction may involve de novo synthesis of negative regulators or the modification (e.g. phosphorylation, ubiquitination) and/or degradation of existing signaling components, which could occur rapidly and without synthesis of new proteins. Accordingly, we used cycloheximide, a protein synthesis inhibitor, to determine whether the inhibitory mechanism requires protein synthesis. Cycloheximide did not block the ability of LpqH-lipopeptide to inhibit CpG-A ODN-induced IFN-β mRNA (Fig. 5). In contrast, the inhibition of IFN-β induction by CpG-B ODN (described previously (22)) was reversed with cycloheximide (and CpG-B ODN became an inducer of IFN-I, like CpG-A ODN, rather than an inhibitor), providing a positive control for cycloheximide function and establishing that TLR2-mediated inhibition occurs by a mechanism distinct from that previously reported for inhibition of IFN-I production by CpG-B ODN. These results indicate that inhibition of IFN-I induction occurs by direct TLR2 intracellular signaling mechanisms that affect the activity of pre-existing signaling components.
Figure 5. TLR2-mediated inhibition is not dependent on new protein synthesis.

Flt3L-derived DCs (3–5×106) were incubated without (A) or with (B) cycloheximide (5 μg/ml) for 1 h before the addition of medium, LpqH-lipopeptide (400 nM) or CpG-B ODN (3 μM) with or without CpG -A ODN (300 nM) for 3 h in the continued presence or absence of cycloheximide. Expression of mRNA for IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as percent of control expression observed with CpG-A ODN alone within each cycloheximide treatment group. Data represent means and standard deviations of triplicate samples and are representative of three independent experiments. Student’s t-test was performed to compare results with CpG-A ODN only to results with CpG-A ODN plus LpqH-lipopeptide or results with CpG-A ODN plus CpG-B ODN (** = p < 0.01, *** = p < 0.001).
TLR2 signaling inhibits induction of IFN-I by TLR7 or TLR9, but not TLR3 or TLR4
TLR9, TLR3, TLR4 and TLR7 can all induce IFN-I. Induction of IFN-I by TLR3 and TLR4 is dependent on TRIF, whereas induction of IFN-I by TLR7 and TLR9 is dependent on MyD88. To dissect the mechanism by which TLR2 signaling inhibits first wave IFN-I induction, we tested the ability of TLR2 signaling to inhibit IFN-I induction via MyD88-dependent vs. TRIF-dependent pathways. LpqH-lipopeptide inhibited IFN-β mRNA induction by CpG-A ODN (TLR9 agonist) and ssRNA40 (TLR7 agonist), but not poly (I:C) (TLR3 agonist) or LPS (TLR4 agonist) (Fig. 6). LpqH-lipopeptide also inhibited TLR7 induction of IFN-β at the protein level (Fig. 6C). These data indicate that TLR2 signaling inhibits MyD88-dependent IFN-I induction, but not TRIF-dependent IFN-I induction.
Figure 6. LpqH-lipopeptide inhibits MyD88-dependent but not TRIF-dependent induction of IFN-I expression.

A, B, Flt3L-derived DCs were cultured for 3 h with medium, poly(I:C) (10 μg/ml), LPS (50 ng/ml), CpG-A ODN (300 nM) or ssRNA (1 μg/ml) with or without LpqH-lipopeptide (400 nM). Expression of mRNA for IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as fold-change relative to expression with medium alone. C, Flt3L-derived DCs were cultured for 24 h with medium, LpqH-lipopeptide, ssRNA or both agonists. Supernatants were assessed by ELISA for IFN-β̃ Data represent means and standard deviations for triplicate samples and are representative of three or more independent experiments. Student’s t-test was performed to compare conditions as indicated (** = p < 0.01, *** = p < 0.001; #, results are plotted but are too low to be visualized on this graph; n.d., not detected).
TLR2-mediated inhibition of IFN-I expression is correlated with rapid depletion of IRAK1
We tested the hypothesis that TLR2 signaling disrupts the TLR9 signaling complex, focusing on IRAK1, since it has an important role in MyD88-dependent induction of IFN-I by TLR9 (4). Upon activation by CpG-A ODN, TLR9 recruits MyD88 and activates IRAK4, IRAK1 and TRAF6, which form an intermediate signaling complex. The complex then phosphorylates IRFs, which translocate into the nucleus to transcribe IFN-I genes. In our studies, CpG-A ODN did not induce IFN-I in IRAK1−/− DCs, confirming that IRAK1 is required for induction of IFN-I (Fig. 7A). Interestingly, IRAK1 was rapidly and significantly degraded within 10 min of stimulation with LpqH-lipopeptide (Fig. 7B). In contrast, TLR9 signaling did not cause significant degradation of IRAK1 until approximately 2 h, and even then to a lesser degree than TLR2 signaling. When added simultaneously with CpG-A ODN, LpqH-lipopeptide also induced IRAK1 degradation (Fig. 7C). IRAK1 degradation occurred with similar kinetics after stimulation with LpqH-lipopeptide alone or LpqH-lipopeptide plus CpG-A ODN (data not shown). IRAK1 degradation was observed in both stimulus conditions as early as 15 min, and IRAK1 levels remained low through the latest time point (2 h). These results suggest that LpqH-lipopeptide inhibits the induction of IFN-I by causing rapid degradation of IRAK1.
Figure 7. LpqH-lipopeptide induces rapid IRAK1 depletion in DCs.

A, Flt3L-derived DCs from wild-type C57BL/6 or IRAK1−/− mice were cultured for 3 h with medium, LpqH-lipopeptide (400 nM), CpG-A ODN (300 nM) or both agonists. Expression of mRNA for IFN-β was determined by qRT-PCR with normalization to GAPDH and is expressed as fold-change relative to expression with medium alone with wild type DCs B, Flt3L-derived DCs were left untreated or treated for various periods with CpG-A ODN (300 nM) or LpqH-lipopeptide (400 nM). C, Flt3L-derived DCs were cultured for 30 min or 120 min with medium, LpqH-lipopeptide (400 nM), CpG-A ODN (300 nM) or both. Cell lysates were prepared and analyzed by Western blot for IRAK1 and β-actin (loading control). Data in panel A represent the means and standard deviations of triplicate wells. Data are representative of 3 or more independent experiments. Student’s t-test was performed to compare conditions as indicated (*** = p < 0.001; #, results are plotted but are too low to be visualized on this graph).
We investigated the levels of several molecules involved in early TLR signaling steps, including TRAF6, MyD88, IRAK4 and IRAK1, in cells treated with CpG-A ODN with or without LpqH-lipoepeptide or other TLR2 agonists. Of these signaling molecules, only IRAK1 was degraded in response to the TLR2 agonists in the time frame tested (90 min) (Supplemental Fig. 1C). In addition to LpqH-lipopeptide, the TLR2 agonists included Mtb lipoprotein LprG and two synthetic lipopeptides, Pam3CSK4 and FSL-1. Pam3-CSK4 induced IRAK1 degradation to an even greater degree than LpqH-lipopeptide; FSL-1 and LprG also induced IRAK1 degradation (perhaps to a lesser degree than Pam3CSK4 or LpqH-lipopeptide, but to a greater degree than CpG-A ODN) (Supplemental Fig. 1C). These data confirm that multiple TLR2 agonists can induce degradation of IRAK1, although different agonists may differ in potency. Thus, among the early TLR signaling components tested, a TLR2-dependent mechanism selectively targets IRAK1 for degradation.
Discussion
Although many TLR agonists drive IFN-I production, TLR2 agonists do not. Moreover, in our previous study we showed that TLR2 signaling inhibited TLR9-induced IFN-I production and IFN-I-dependent MHC-I cross processing (19). In this study, we dissected the mechanisms by which TLR2 signaling inhibits induction of IFN-I. We demonstrate that TLR2 signaling rapidly inhibits MyD88-dependent induction of first-wave IFN-I through TLR9 or TLR7, as early as the initial time point when TLR9-induced IFN-I can be detected (~1 h for mRNA and ~4 h for protein by ELISA). The inhibition involves a protein synthesis-independent intracellular signaling mechanism that affects MyD88-dependent but not TRIF-dependent IFN-I induction. We considered a role for IRAK2, which has been implicated as a negative regulator of IFN-I in other systems (25), but the inhibitory mechanism was still observed in DCs from IRAK2 knockout mice (data not shown). Since IRAK1 is known to be required for MyD88-dependent induction of IFN-I by TLR9 (4), we considered the hypothesis that inhibition or depletion of IRAK1 might explain the inhibition of IFN-I induction. Interestingly, we observed that IRAK1 degradation was rapidly induced by TLR2 agonists but not a TLR9 agonist (which caused lesser IRAK1 degradation only after longer periods). Since IRAK1 is essential for TLR9- or TLR7-induced production of IFN-I but not other cytokines (e.g. IL-12p40) (Supplemental Fig. 1D, E, and reference (4)), we propose that rapid depletion of IRAK1 is a novel mechanism by which TLR2 signaling inhibits IFN-I induction by TLR9 or TLR7.
We investigated the possibility that this inhibition may occur directly through intracellular signaling by TLR2 and TLR9 in the same cell or indirectly via intercellular communication, e.g. through cytokines induced by TLR2 in one cell that act upon another cell. Cytokines such as IL-10 or TNF-α have been reported to be responsible for inhibiting IFN-I induction by TLR9 in some studies (23, 24). Accordingly, we investigated indirect inhibitory mechanisms by mixing TLR2−/− DCs with TLR9−/− DCs. Based on our discovery that the mechanism involves inhibition of first-wave IFN-I, we focused the mixed TLR knockout experiment on IFN-I mRNA expression at a relatively early time point (3 h). This experiment revealed that inhibition is not observed in mixed TLR2−/− and TLR9−/− DCs, indicating that the early rapid inhibition of IFN-I mRNA expression is dependent on direct intracellular signaling and did not occur by intercellular communication that was recapitulated by mixing TLR2−/− with TLR9−/− cells. Furthermore, TLR2-mediated inhibition was not dependent on new protein synthesis, on which the synthesis of potential inhibitory cytokines is dependent. In addition, we found the TLR9 agonist CpG-A ODN induced more IL-10 and TNF-α than the TLR2 agonist lipopeptide, yet CpG-A ODN induces high levels of IFN-I. These observations indicate that IL-10 and TNF-α are not the major drivers of the inhibition of IFN-I production in this system. However, indirect mechanisms could still be partly responsible for inhibition at later time points. In summary, these results indicate a rapid intracellular signaling mechanism by which TLR2 inhibits the induction of IFN-I by TLR9 or TLR7, consistent with the proposed mechanism involving IRAK1 degradation.
TLR2-mediated inhibition of MyD88-dependent IFN-I induction could shape host immune responses in response to stimuli that combine TLR2 agonists with TLR9 or TLR7 agonists. Pathogens that express agonists of both TLR2 as well as TLR9/7 include a wide range of bacteria (e.g. Mtb) and some viruses (e.g. herpes simplex virus, cytomegalovirus, Epstein-Barr virus) (29–31). In addition, pathogens can induce host-derived TLR2 agonists, which may affect responses to pathogen-derived TLR9/7 agonists. Moreover, during bacteria/virus co-infection (e.g. Mtb-HIV co-infection), host cells may be stimulated by both viral TLR7/9 agonists and bacterial TLR2 agonists. In these situations, TLR2 agonists may inhibit TLR9/7-induced IFN-I production. Thus, TLR2-mediated inhibition of TLR9/7-induced IFN-I induction may be important in understanding immune responses to infections and vaccines when both TLR2 and TLR7/TLR9 agonists present, but additional study of specific models is required.
Mechanisms to control IFN-I production may play a role in autoimmune or inflammatory disorders. Dendritic cells and IFN-α play a central role in SLE (32), and neutralizing anti-IFN-α monoclonal antibody therapies are in development or clinical trials (33). IRAK1 has been suggested to play a critical role in the pathogenesis of SLE. Jacob et al. found 5 SNPs spanning the IRAK1 gene that showed disease association in both adult- and childhood-onset SLE (34). Moreover, IRAK1 deficiency abrogated all lupus-associated phenotypes in congenic mouse models bearing the Sle1 or Sle3 disease loci (34). Although more studies are needed to assess the role of these disease-associated SNPs in IRAK1 expression and function, as well as IFN-α production or ISG signature in these patients, impaired IFN-α production caused by IRAK1 deficiency may be a protective factor in pathogenesis of certain IFN-I-driven autoimmune disorders, e.g. SLE and Sjögren’s syndrome. In these scenarios, mechanisms to control IRAK1 expression or activity may have therapeutic potential.
In summary, our studies reveal rapid TLR2-mediated degradation of IRAK1 as a mechanism to inhibit IFN-I induction by TLR9 or TLR7. This mechanism may shape the role of IFN-I in host-pathogen interactions when both TLR2 and TLR7/9 agonists are present. This mechanism may be exploited by pathogens to evade host defenses that depend on IFN-I. On the other hand, we propose that negative regulation of IFN-I expression by TLR2 is part of a host regulatory program that allows IFN-I expression for host defense in viral infections but inhibits IFN-I expression during infections with certain bacteria that trigger TLR2 (in which IFN-I may be harmful rather than helpful to host defense). Moreover, this may be a mechanism for host protection against deleterious effects of IFN-I in autoimmune disease, and understanding of this inhibitory pathway and mechanisms for IRAK1 degradation may reveal therapeutic approaches for these disorders.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI034343, AI035726 and AI069085.
We thank Courtney Niland for help with IRAK1 Western blots and Supriya Shukla for purification of LprG lipoprotein.
Abbreviations
- CpG
deoxycytidyl-deoxyguanosine
- DC
dendritic cell
- mDC
myeloid DC
- pDC
plasmacytoid DC
- dsRNA
double stranded RNA
- Flt3L
FMS-like tyrosine kinase 3 ligand
- IFN-I
type I IFN
- IFN-IR
IFN-I receptor
- IRAK
IL-1R-associated kinase
- IRF
interferon regulatory factor
- ISG
interferon-stimulated gene
- MHC-I
class I MHC
- Mtb
Mycobacterium tuberculosis
- MyD88
myeloid differentiation primary response gene 88
- n.s
not significant
- ODN
oligodeoxynucleotide
- PRR
pattern recognition receptor
- RIG-I
retinoic acid inducible gene I
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- PBST
phosphate buffered saline with 0.1% Tween 20
- PMSF
phenylmethanesulfonylfluoride
- SLE
systemic lupus erythematosus
References
- 1.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7-and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 7.Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nature Rev Microbiol. 2010;8:296–307. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19 kD lipoprotein of Mycobacterium tuberculosis. J Immunol. 2001;167:910–918. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 9.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium tuberculosis LprG (Rv1411c): A novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol. 2004;173:2660–2668. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 10.Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol. 2006;177:422–429. doi: 10.4049/jimmunol.177.1.422. [DOI] [PubMed] [Google Scholar]
- 11.Jung SB, Yang CS, Lee JS, Shin AR, Jung SS, Son JW, Harding CV, Kim HJ, Park JK, Paik TH, Song CH, Jo EK. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun. 2006;74:2686–2696. doi: 10.1128/IAI.74.5.2686-2696.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, Feng CG. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol. 2011;187:2540–2547. doi: 10.4049/jimmunol.1100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res. 2005;25:694–701. doi: 10.1089/jir.2005.25.694. [DOI] [PubMed] [Google Scholar]
- 16.Bouchonnet F, Boechat N, Bonay M, Hance AJ. Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect Immun. 2002;70:3020–3025. doi: 10.1128/IAI.70.6.3020-3025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Telesca C, Angelico M, Piccolo P, Nosotti L, Morrone A, Longhi C, Carbone M, Baiocchi L. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J Infect. 2007;54:e223–226. doi: 10.1016/j.jinf.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmons DP, Canaday DH, Liu Y, Li Q, Huang A, Boom WH, Harding CV. Mycobacterium tuberculosis and TLR2 agonists inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J Immunol. 2010;185:2405–2415. doi: 10.4049/jimmunol.0904005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drage MG, Tsai HC, Pecora ND, Cheng TY, Arida AR, Shukla S, Rojas RE, Seshadri C, Moody DB, Boom WH, Sacchettini JC, Harding CV. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nature Struct Mol Biol. 2010;17:1088–1095. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brawand P, Fitzpatrick DR, Greenfield BW, Brasel K, Maliszewski CR, De Smedt T. Murine plasmacytoid pre-dendritic cells generated from flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol. 2002;169:6711–6719. doi: 10.4049/jimmunol.169.12.6711. [DOI] [PubMed] [Google Scholar]
- 22.Liu YC, Gray RC, Hardy GA, Kuchtey J, Abbott DW, Emancipator SN, Harding CV. CpG-B Oligodeoxynucleotides Inhibit TLR-Dependent and -Independent Induction of Type I IFN in Dendritic Cells. J Immunol. 2010;184:3367–3376. doi: 10.4049/jimmunol.0903079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waibler Z, Anzaghe M, Konur A, Akira S, Muller W, Kalinke U. Excessive CpG 1668 stimulation triggers IL-10 production by cDC that inhibits IFN-alpha responses by pDC. Eur J Immunol. 2008;38:3127–3137. doi: 10.1002/eji.200838184. [DOI] [PubMed] [Google Scholar]
- 24.Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Cormier M, Szabo G. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-{alpha} and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177:6758–6768. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 25.Wan Y, Kim TW, Yu M, Zhou H, Yamashita M, Kang Z, Yin W, Wang JA, Thomas J, Sen GC, Stark GR, Li X. The dual functions of IL-1 receptor-associated kinase 2 in TLR9-mediated IFN and proinflammatory cytokine production. J Immunol. 2011;186:3006–3014. doi: 10.4049/jimmunol.1003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamin TT, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem. 1997;272:21540–21547. doi: 10.1074/jbc.272.34.21540. [DOI] [PubMed] [Google Scholar]
- 27.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 28.Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 29.Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A. 2006;103:17343–17348. doi: 10.1073/pnas.0605102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiola S, Gosselin D, Takada K, Gosselin J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J Immunol. 2010;185:3620–3631. doi: 10.4049/jimmunol.0903736. [DOI] [PubMed] [Google Scholar]
- 31.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascual V, Banchereau J, Palucka AK. The central role of dendritic cells and interferon-alpha in SLE. Curr Opin Rheumatol. 2003;15:548–556. doi: 10.1097/00002281-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Crow MK. Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am. 2010;36:173–186. x. doi: 10.1016/j.rdc.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009;106:6256–6261. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.